The Hydroarylation Reaction—Scope and Limitations


Abstract
:1. Introduction
2. Results and Discussion
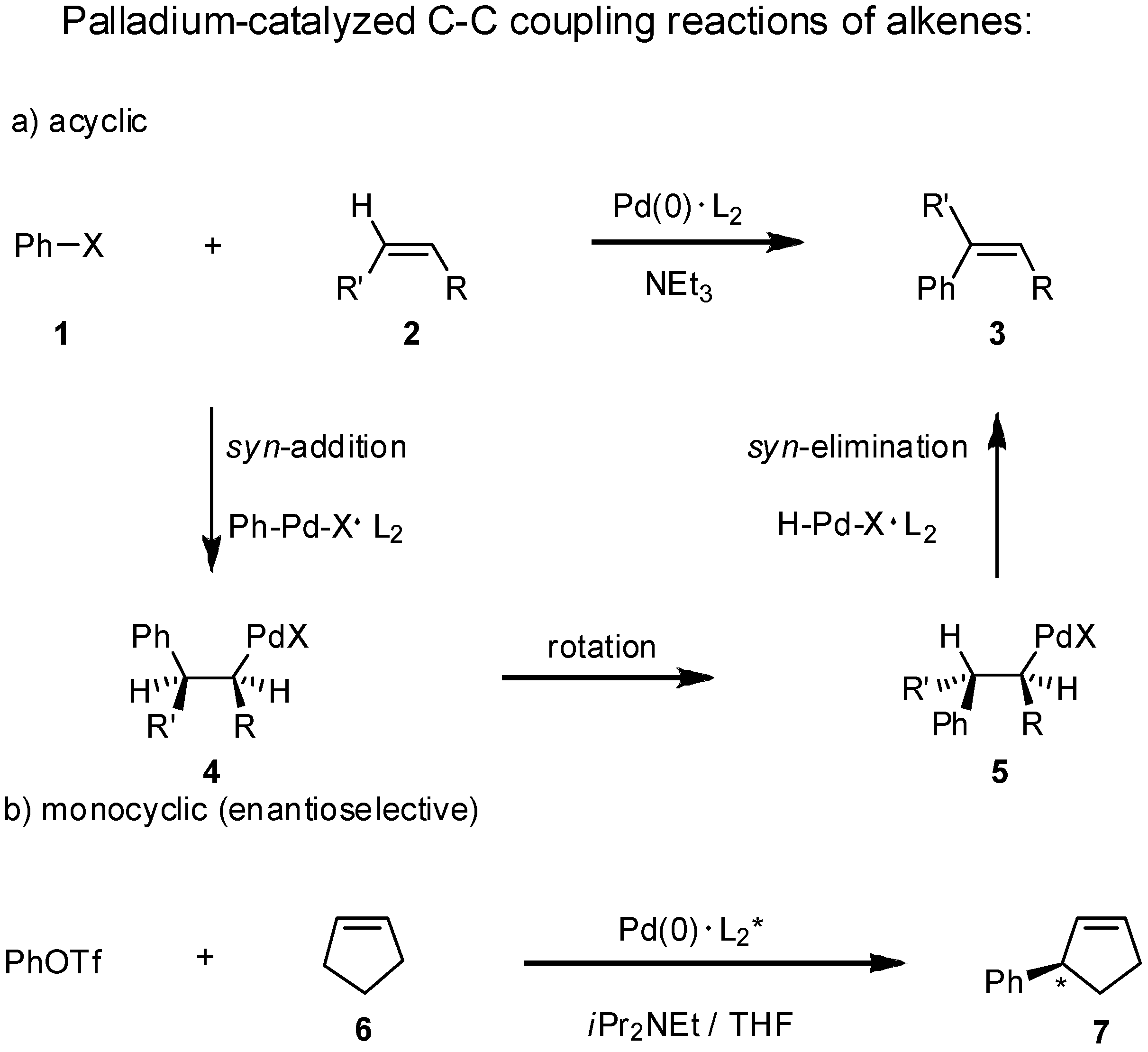


{kind=link}
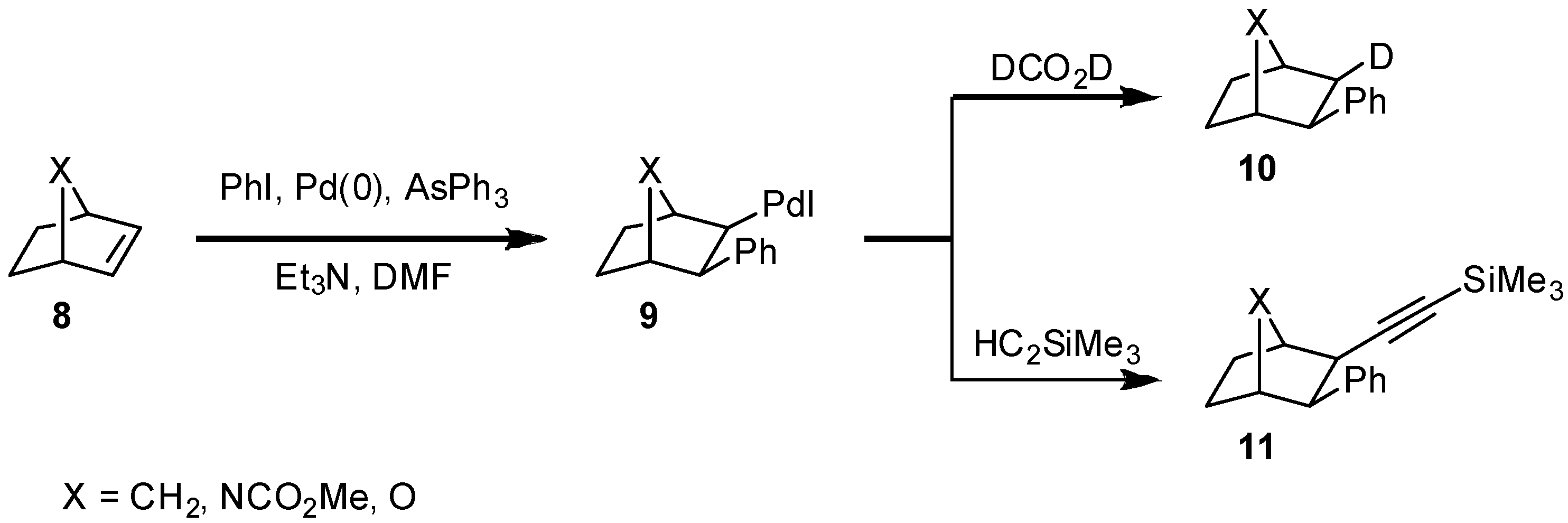{kind=link}
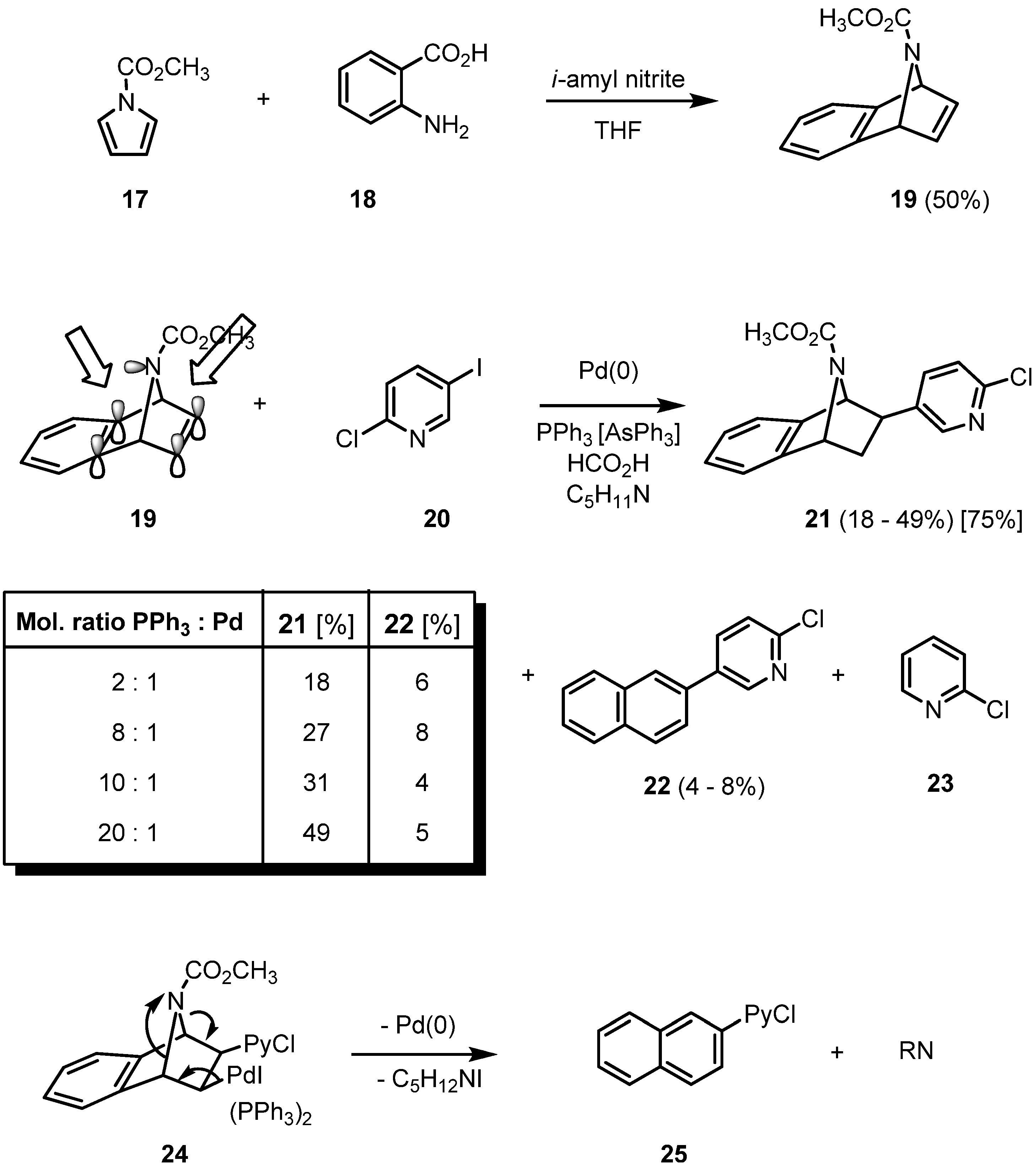{kind=link}
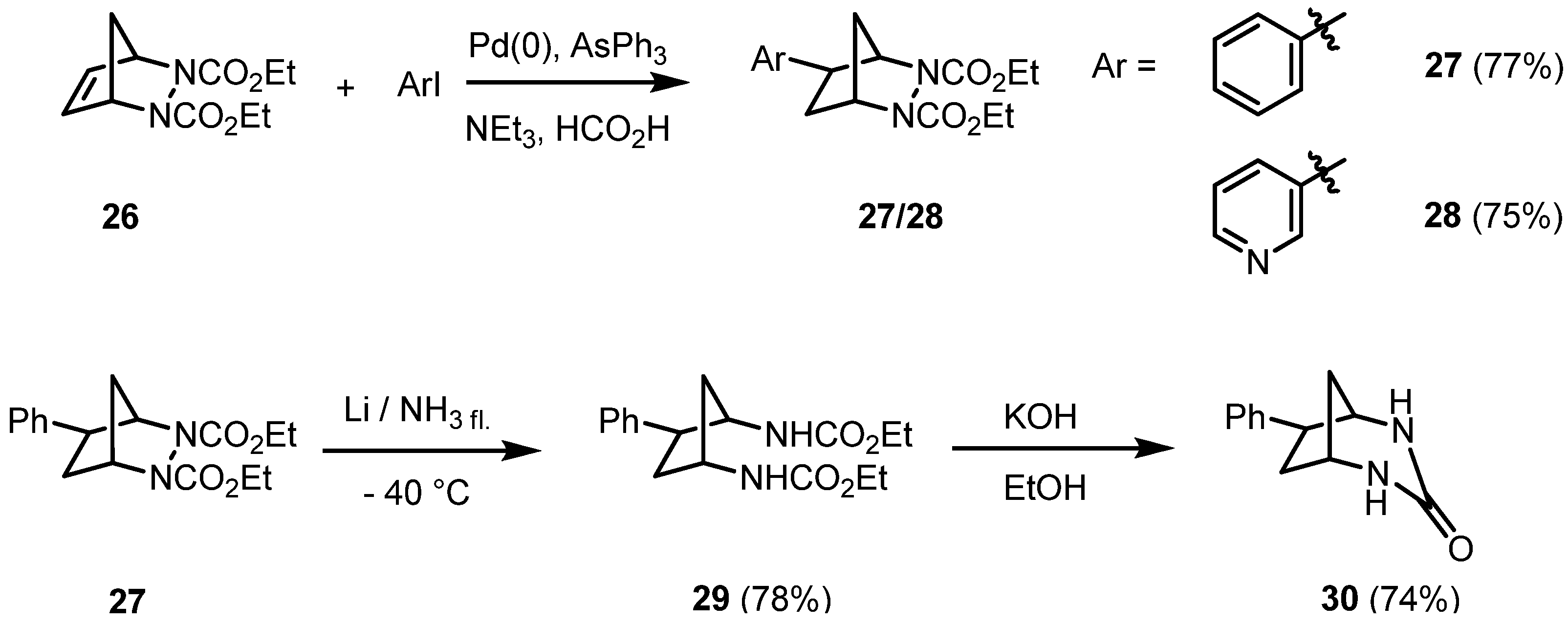
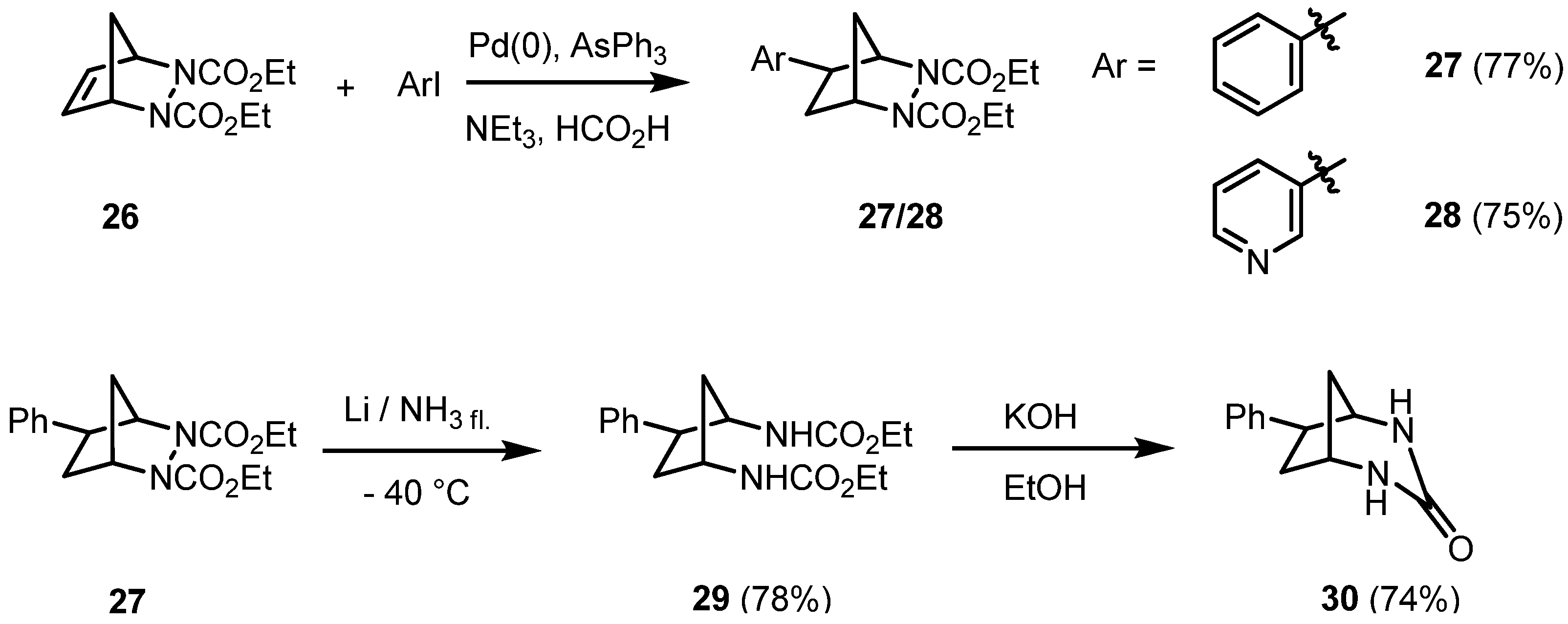{kind=link}
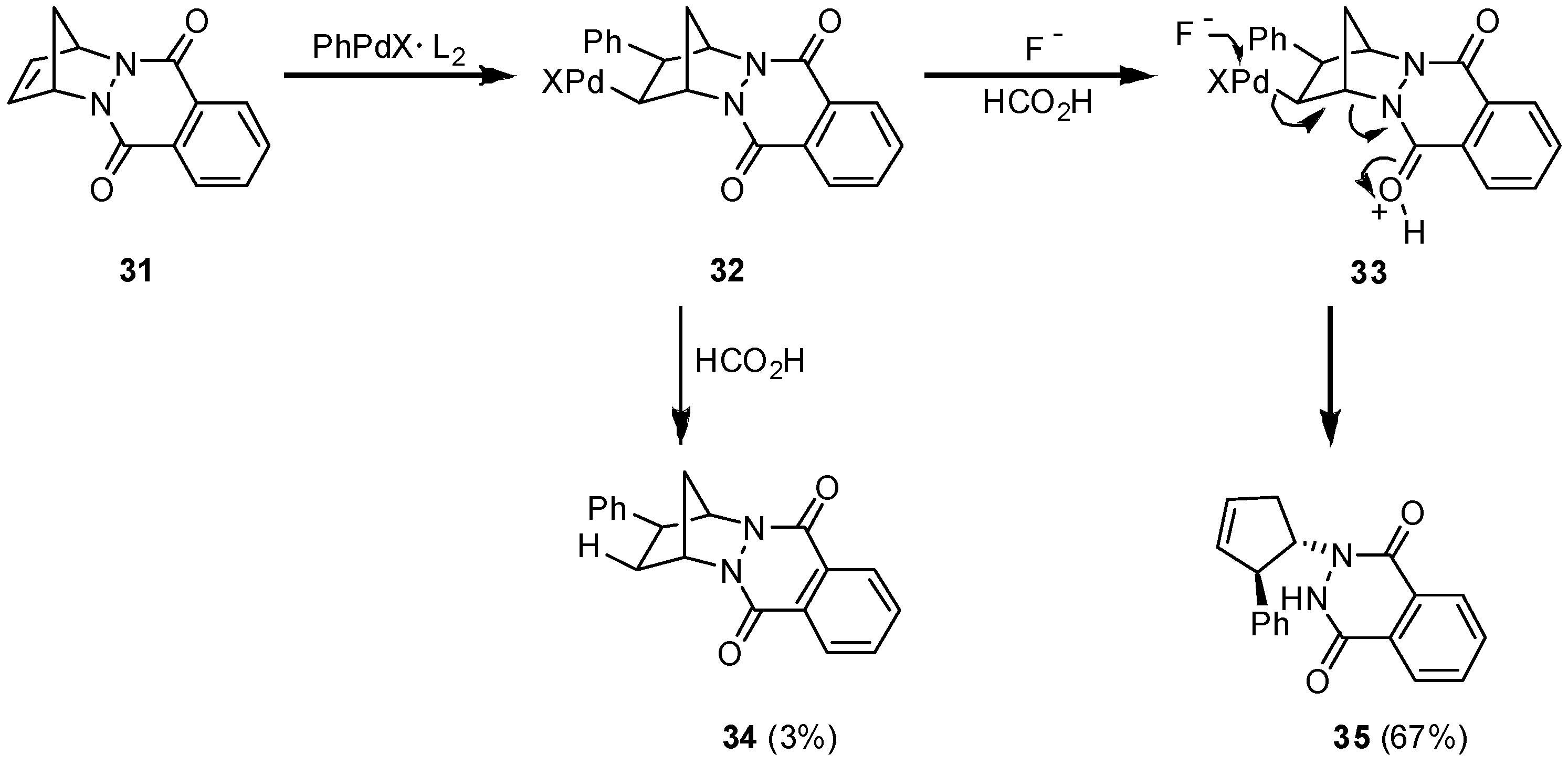{kind=link}
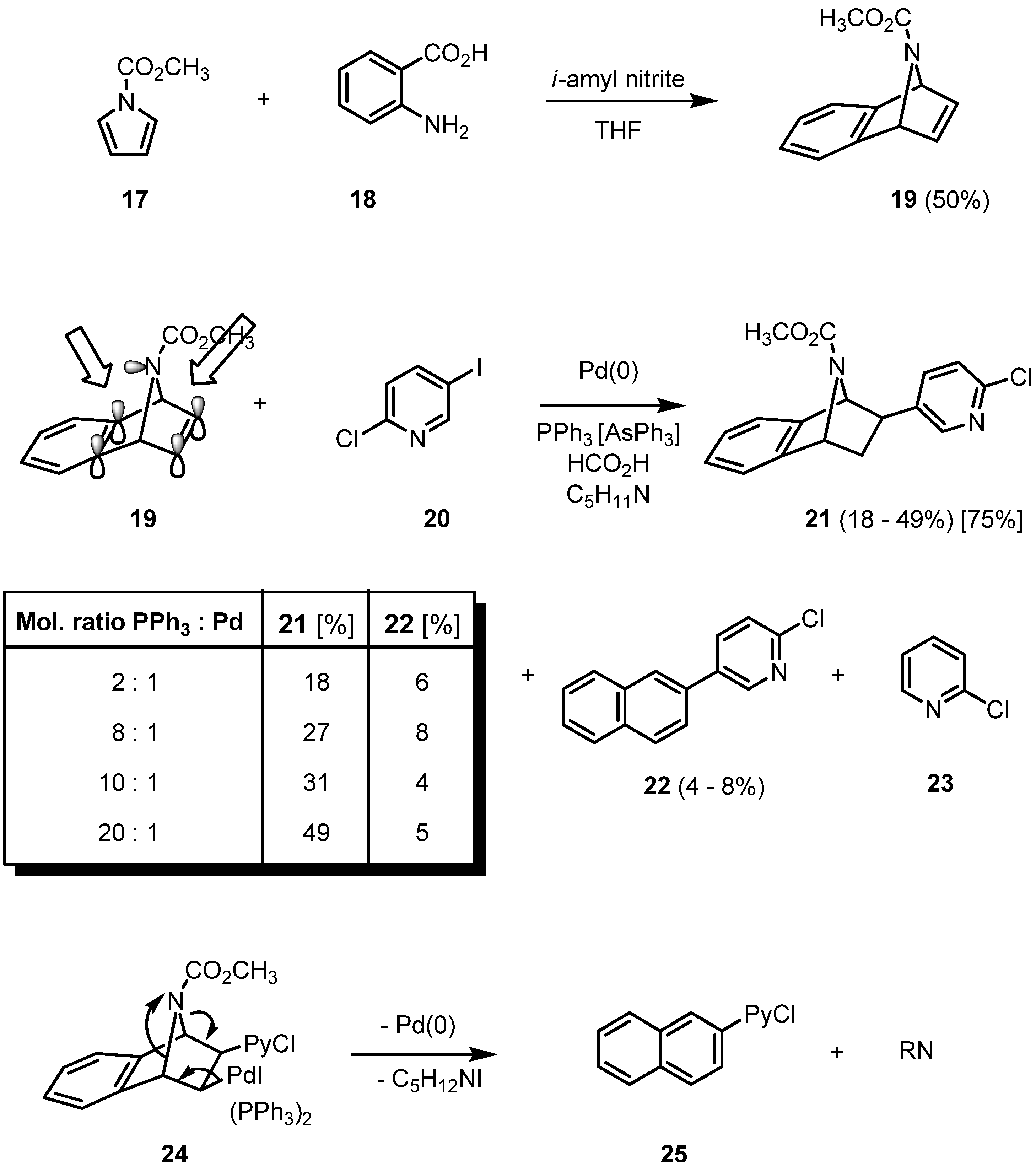
{kind=link}
{kind=link}
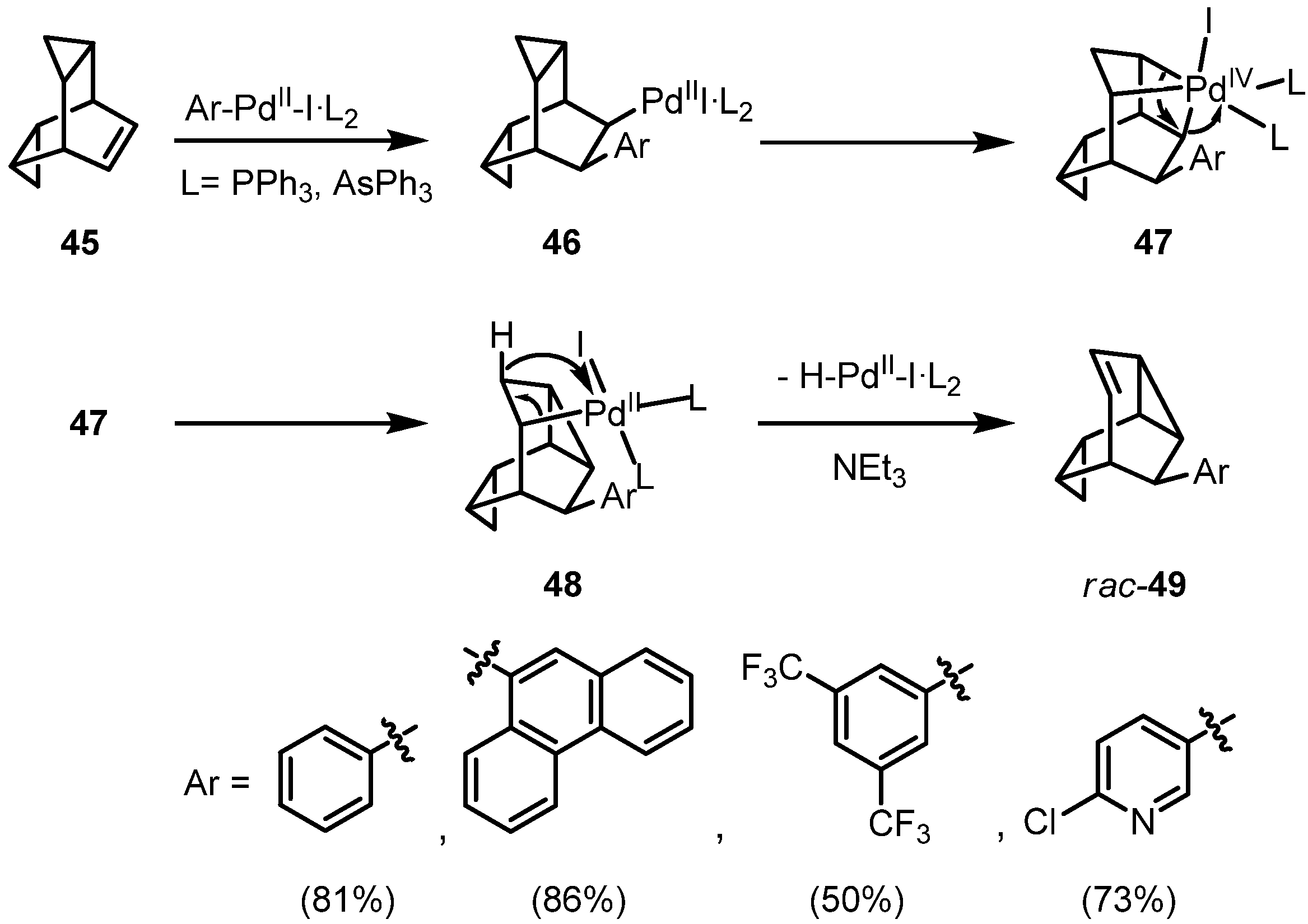{kind=link}
 |

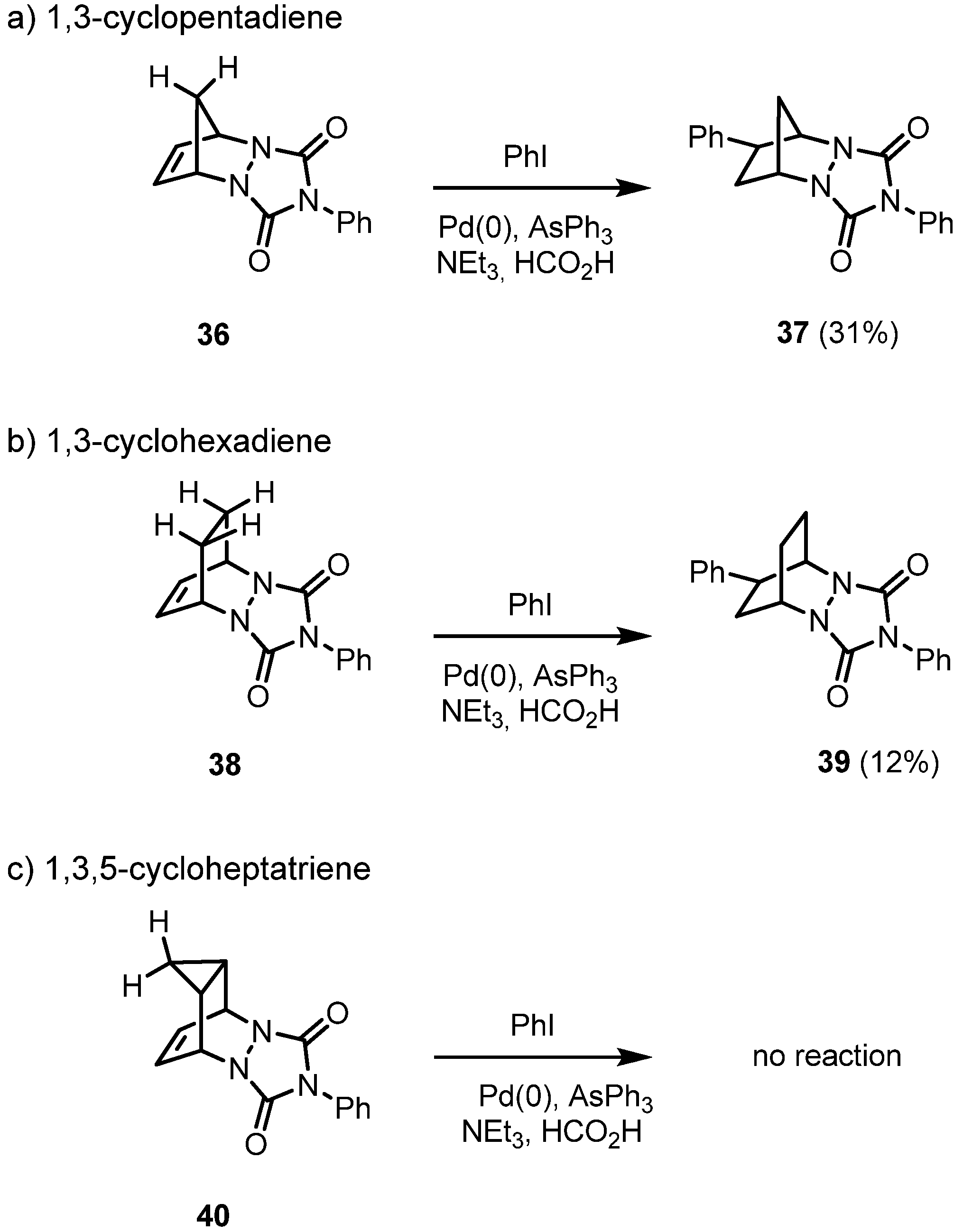
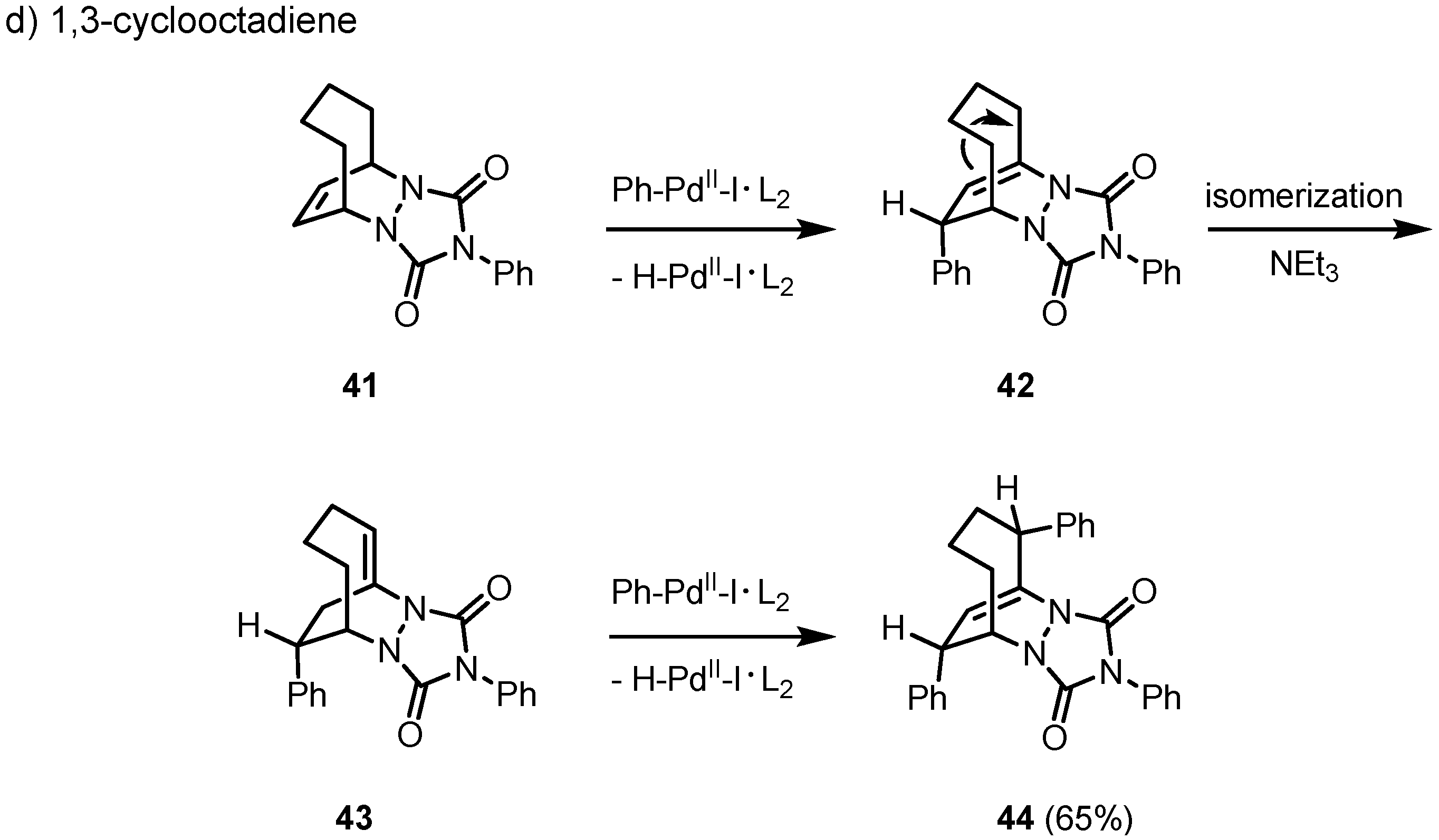
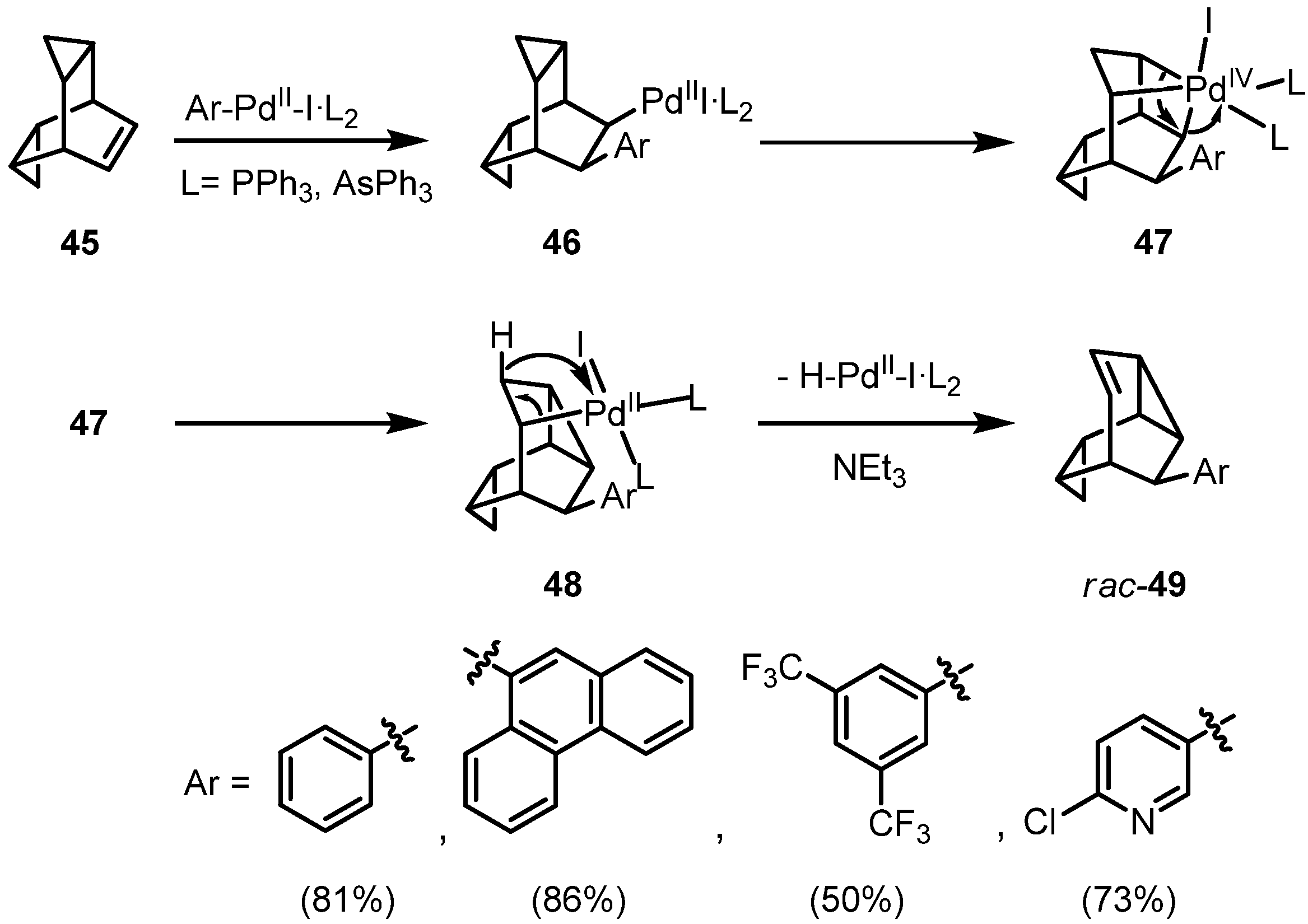
3. Conclusions
References
- Heck, R.F.; Nolley, J.P., Jr. Palladium-catalyzed vinylic hydrogen substitution reaction with aryl, benzyl, and styryl halides. J. Org. Chem. 1974, 37, 2320–2322. [Google Scholar]
- Mizoroki, T.; Mori, K.; Ozaki, A. Arylation of olefin with aryl iodide catalyzed by palladium. Bull. Chem. Soc. Jpn. 1971, 44, 581. [Google Scholar] [CrossRef]
- Heck, R.F. Palladium-Catalyzed Vinylation of Organic Halides. Org. React. 1982, 27, 345–390. [Google Scholar]
- Heck, R.F. Transition Metal-Catalyzed Reactions of Organic Halides with CO, Olefins, and Acetylenes. Adv. Catal. 1977, 26, 323–349. [Google Scholar] [CrossRef]
- Heck, R.F. Palladium-catalyzed reactions of organic halides with olefins. Acc. Chem. Res. 1979, 12, 146–151. [Google Scholar] [CrossRef]
- Meijere, A. de; Meyer, F.E. Fine Feathers Make Fine Birds: The Heck Reaction in Modern Garb. Angew. Chem. Int. Ed. 1995, 33, 2379–2411. [Google Scholar] [CrossRef]
- Beletskaja, I.; Cheprakov, A.V. The Heck reaction as a sharpening stone of palladium catalysis. Chem. Rev. 2000, 100, 3009–3066. [Google Scholar] [CrossRef]
- Negishi, E.; de Meijere, A. Handbook of Organopalladium Chemistry for Organic Synthesis, 1st ed; Wiley-Interscience: New York, NY, USA, 2002; pp. 1123–1448. [Google Scholar]
- Catellani, M. Catalytic Multistep Reactions via Palladacycles. Synlett 2003, 298–312. [Google Scholar] [CrossRef]
- Arcadi, A.; Marinelli, F.; Cacchi, S.; Bernocchi, E.; Ortar, G. Palladium-catalyzed preparation of exo-aryl derivatives of the norbornane skeleton. J. Organomet. Chem. 1989, 368, 249–256. [Google Scholar]
- Larock, R.C; Johnson, P.L. Palladium-catalysed intermolecular arylation and alkenylation of bicyclic alkenes. J. Chem. Soc. Chem. Commun. 1989, 1368–1370. [Google Scholar]
- Brunner, H.; Kramler, K. Asymmetric Catalysis 72. Enantioselective Hydroarylation of Norbornene and Norbornadiene with Palladium(II) Acetate/Phosphine Catalysts. Synthesis 1991, 1121–1124. [Google Scholar] [CrossRef]
- Sakuraba, S.; Awano, K.; Achiwa, K. Asymmetric Heck-type Hydroarylation of Norbornene with Phenyl Triflate Catalyzed by Palladium Complexes of chiral (ß-N-Sulfonylaminoalkyl)-phosphines. Synlett 1994, 291–292. [Google Scholar]
- Namyslo, J.C.; Kaufmann, D.E. Chemistry in the Ambient Field of the Alkaloid Epibatidine, 1: 2-(Hetaryl)-Substituted 7-Azabicyclo[2.2.1]heptane Systems. Eur. J. Org. Chem. 1998, 1997–2001. [Google Scholar]
- Namyslo, J.C.; Kaufmann, D.E. Asymmetric Synthesis of Both Enantiomers of N-Protected Epibatidine via Reductive Heck-Type Hetarylation. Synlett 1999, 804–806. [Google Scholar]
- Göksu, G.; Gül, M.; Öcal, N.; Kaufmann, D.E. Hydroarylation of of Bicyclic, Unsaturated Dicarboximides: Access to Aryl-Substituted, Bridged Hydroisoindolines. Tetrahedron Lett. 2008, 49, 2685–2688. [Google Scholar] [CrossRef]
- Mata, Y.; Pàmies, O.; Diéguez, M. Screening of a Modular Sugar-Based Phosphite-Oxazoline Ligand Library in Asymmetric Pd-Catalyzed Heck Reactions. Chem. Eur. J. 2007, 13, 3296–3304. [Google Scholar] [CrossRef]
- Namyslo, J.C.; Kaufmann, D.E. Triphenylarsine as an Efficient Ligand in the Pd-Catalyzed Synthesis of Epibatidin and Analogs. Synlett 1999, 114. [Google Scholar]
- Spande, T.F.; Garaffo, H.M.; Edwards, M.W.; Yeh, H.J.C.; Pannel, L.; Daly, J.W. Epibatidine: a novel (chloropyridyl)azabicycloheptane with potent analgesic activity from an Ecuadoran poison frog. J. Am. Chem. Soc. 1992, 114, 3475–3478. [Google Scholar] [CrossRef]
- Klinge, J. Neuartige, stark aktivierende Liganden für übergangsmetallkatalysierte Reaktionen. PhD Thesis, Clausthal University of Technology, Clausthal-Zellerfeld, Germany, 2008. [Google Scholar]
- Storsberg, J.; Nandakumar, M.V.; Sankaranarayanan, S.; Kaufmann, D.E. Stereoselective Palladium-Catalyzed C-C-Coupling Reactions with a Diazabicyclo[2.2.1]heptene. Adv. Synth. Catal. 2001, 343, 177–180. [Google Scholar] [CrossRef]
- Yao, M.-L.; Adiwidjaja, G.; Kaufmann, D.E. Two Step, Stereoselective Hydrazidoarylation of 1,3-Cyclopentadiene. Angew. Chem. Int. Ed. 2002, 41, 3375–3378. [Google Scholar] [CrossRef]
- Storsberg, J.; Yao, M.-L.; Öcal, N.; de Meijere, A.; Adam, A.E.W.; Kaufmann, D.E. Palladium-catalyzed, stereoselective rearrangement of a tetracyclic allyl cyclopropane under arylation. Chem. Commun. 2005, 5665–5666. [Google Scholar]
- Sample Availability: Samples of compounds 27, 28, 30, 34, 35, 36, 38, 48 are available from the authors.
© 2010 by the authors;
Share and Cite
Namyslo, J.C.; Storsberg, J.; Klinge, J.; Gärtner, C.; Yao, M.-L.; Ocal, N.; Kaufmann, D.E. The Hydroarylation Reaction—Scope and Limitations. Molecules 2010, 15, 3402-3410. https://doi.org/10.3390/molecules15053402
Namyslo JC, Storsberg J, Klinge J, Gärtner C, Yao M-L, Ocal N, Kaufmann DE. The Hydroarylation Reaction—Scope and Limitations. Molecules. 2010; 15(5):3402-3410. https://doi.org/10.3390/molecules15053402
Chicago/Turabian StyleNamyslo, Jan C., Jörg Storsberg, Jens Klinge, Christian Gärtner, Min-Liang Yao, Nuket Ocal, and Dieter Eckhard Kaufmann. 2010. "The Hydroarylation Reaction—Scope and Limitations" Molecules 15, no. 5: 3402-3410. https://doi.org/10.3390/molecules15053402
APA StyleNamyslo, J. C., Storsberg, J., Klinge, J., Gärtner, C., Yao, M.-L., Ocal, N., & Kaufmann, D. E. (2010). The Hydroarylation Reaction—Scope and Limitations. Molecules, 15(5), 3402-3410. https://doi.org/10.3390/molecules15053402