Organocatalytic Michael Addition of 1,3-Dicarbonyl Indane Compounds to Nitrostyrenes

Abstract
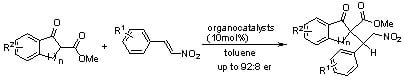
:
1. Introduction
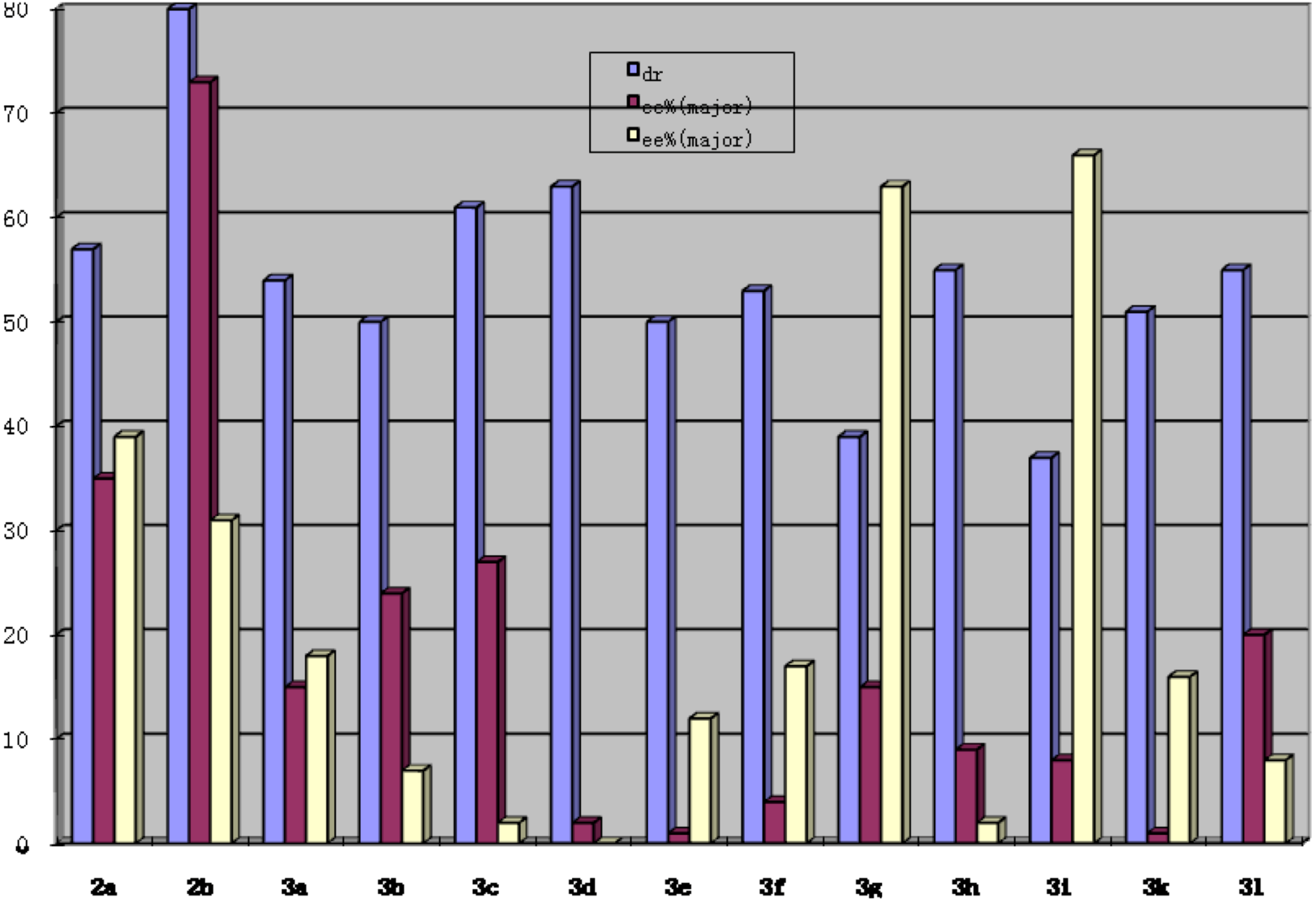
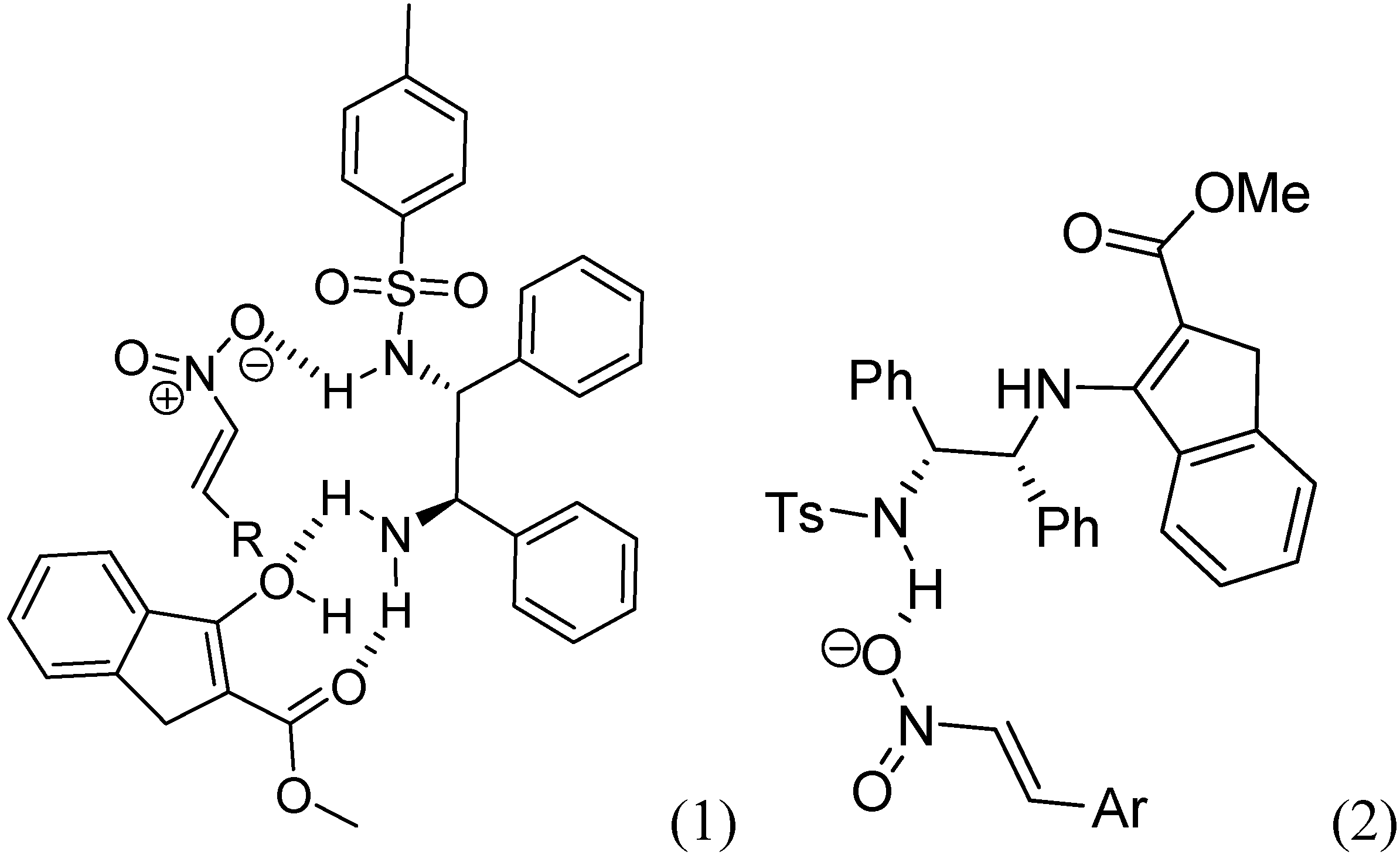
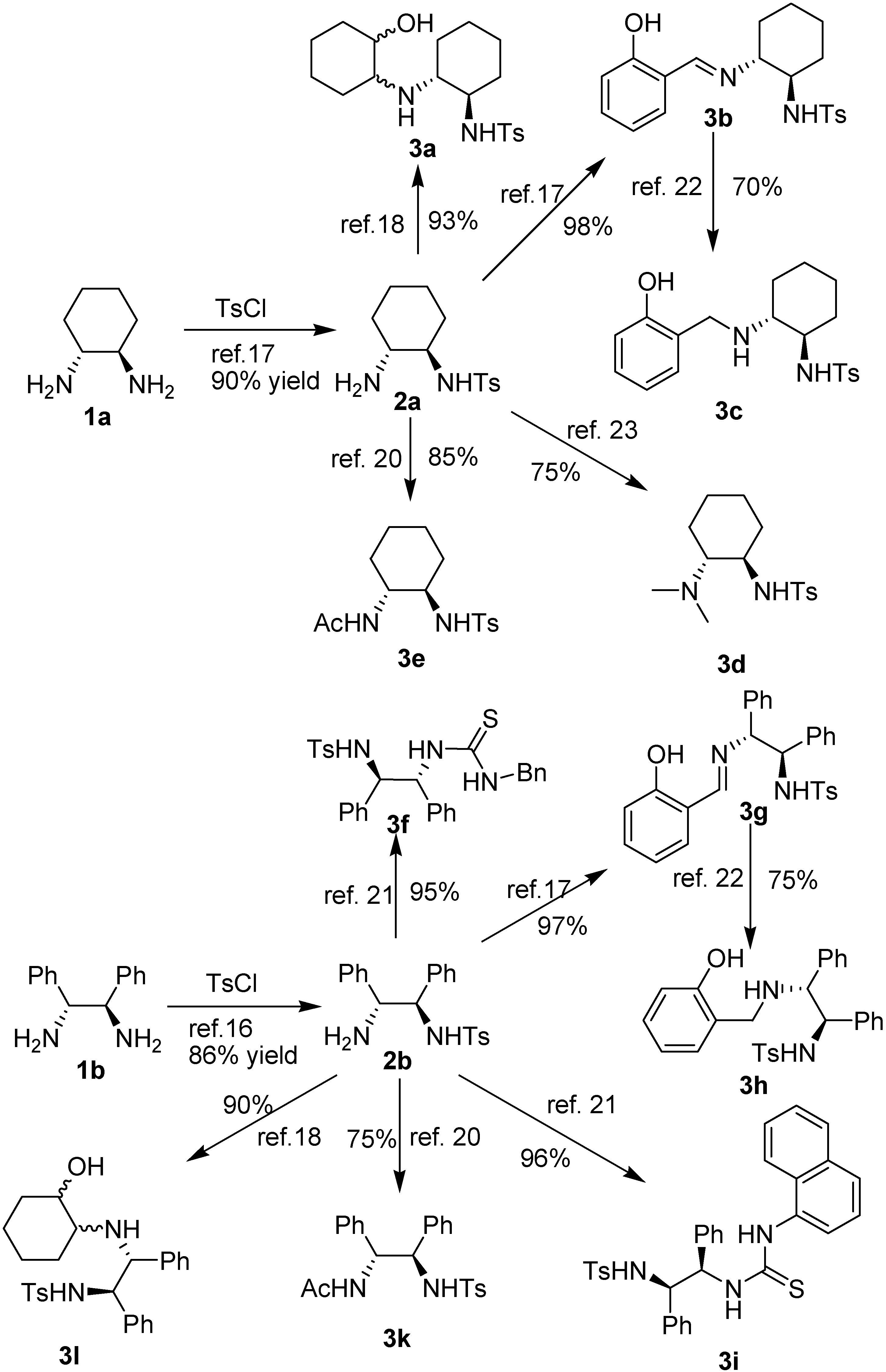
2. Results and Discussion
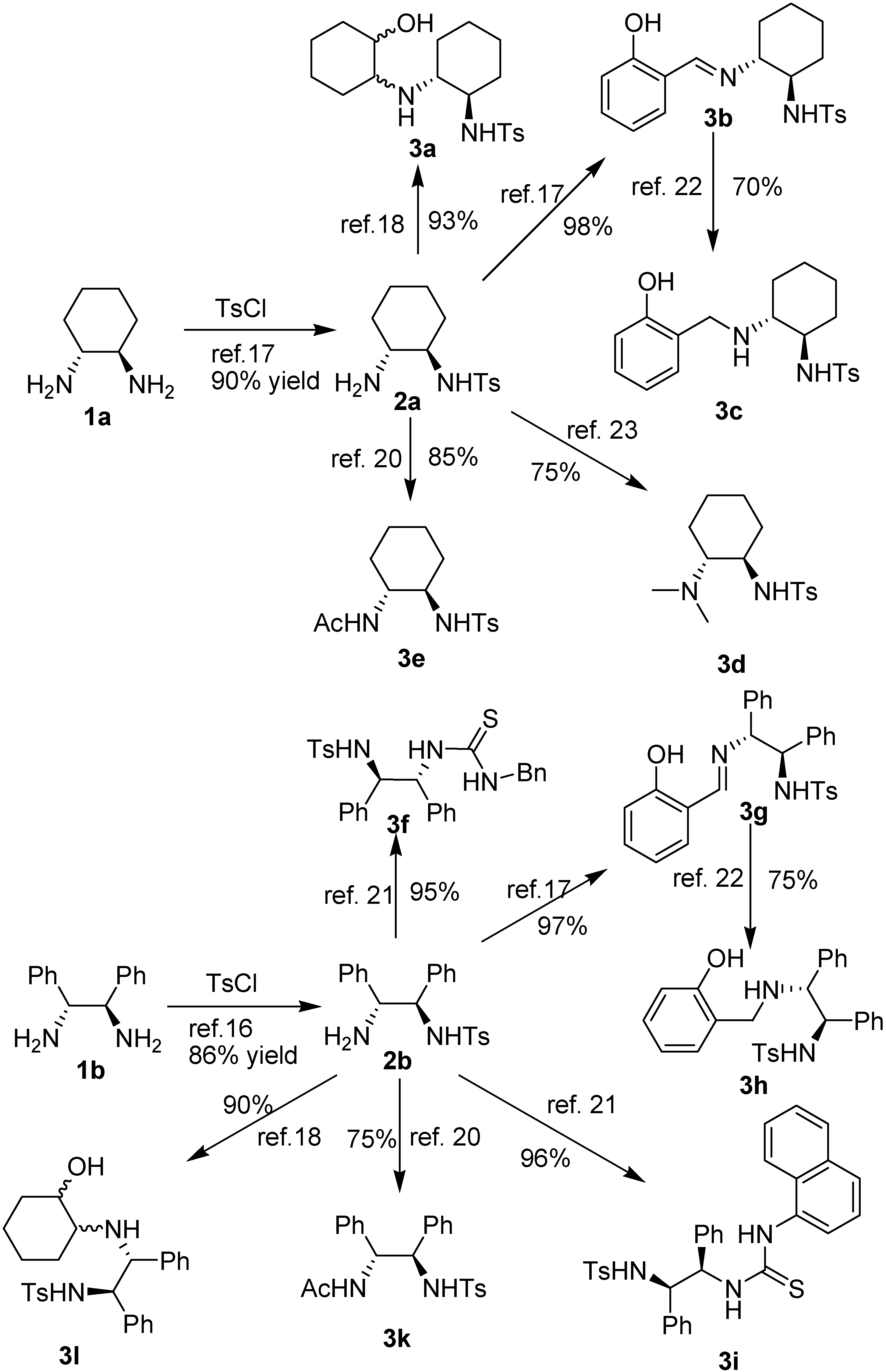

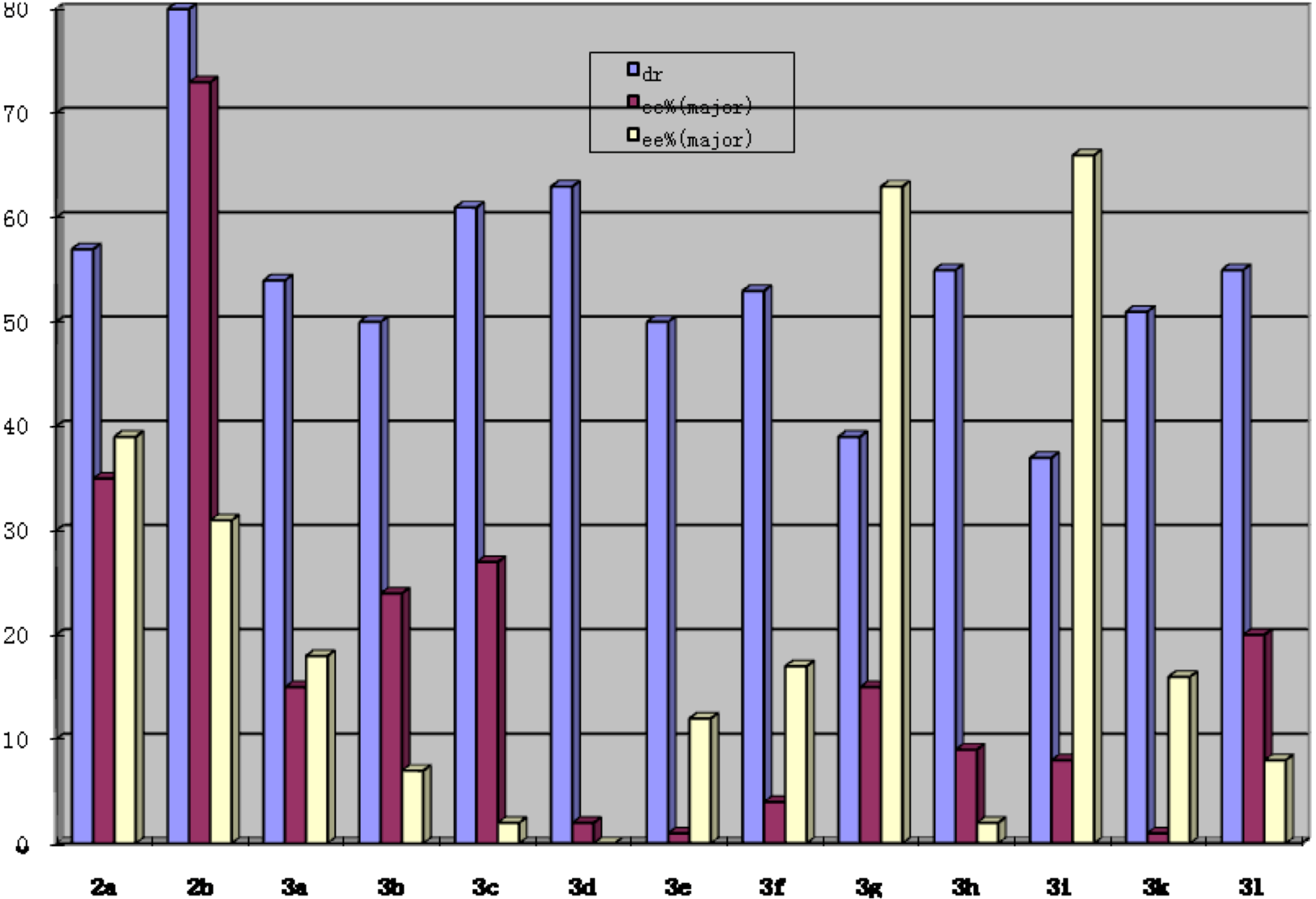

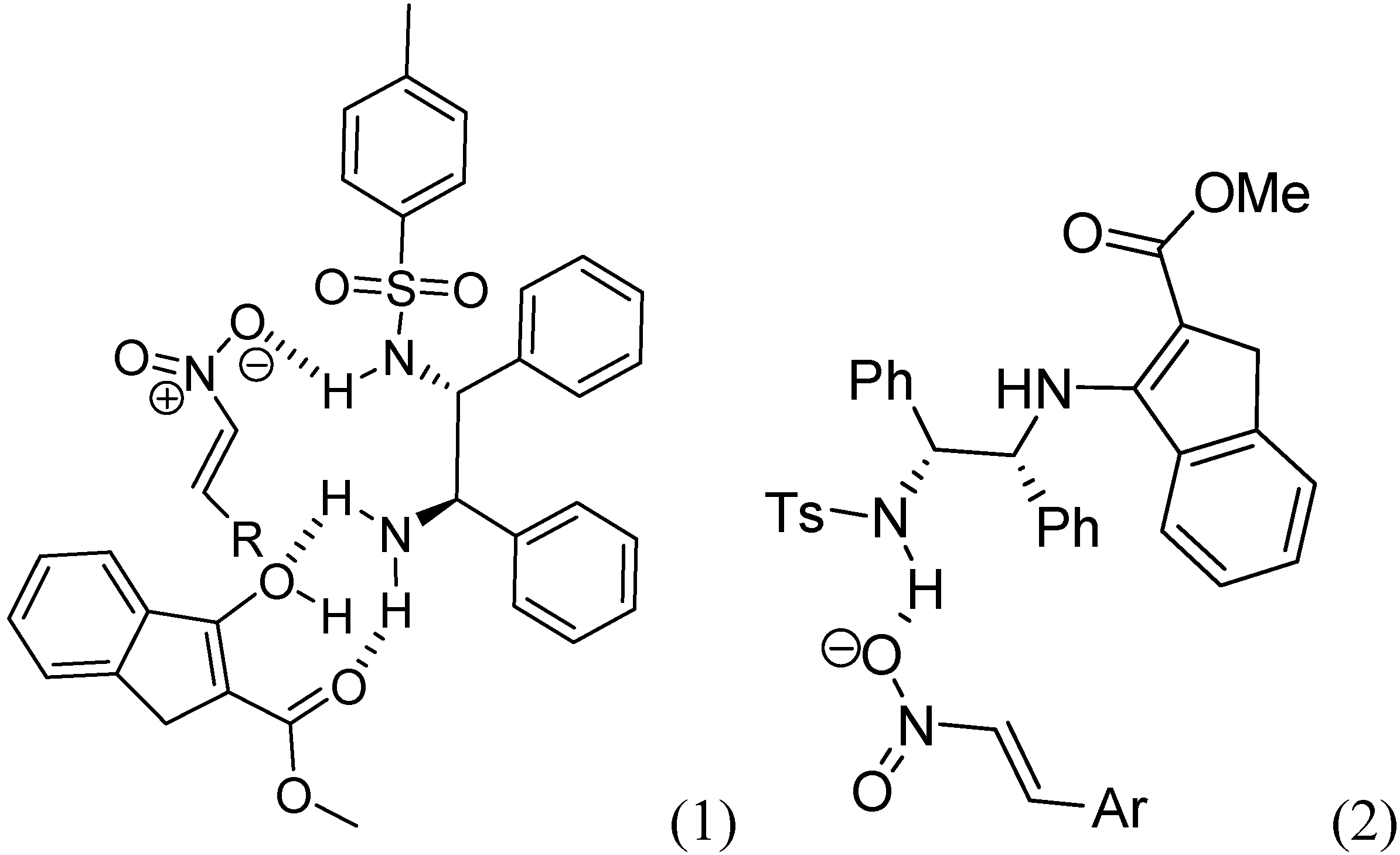
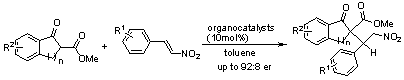{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

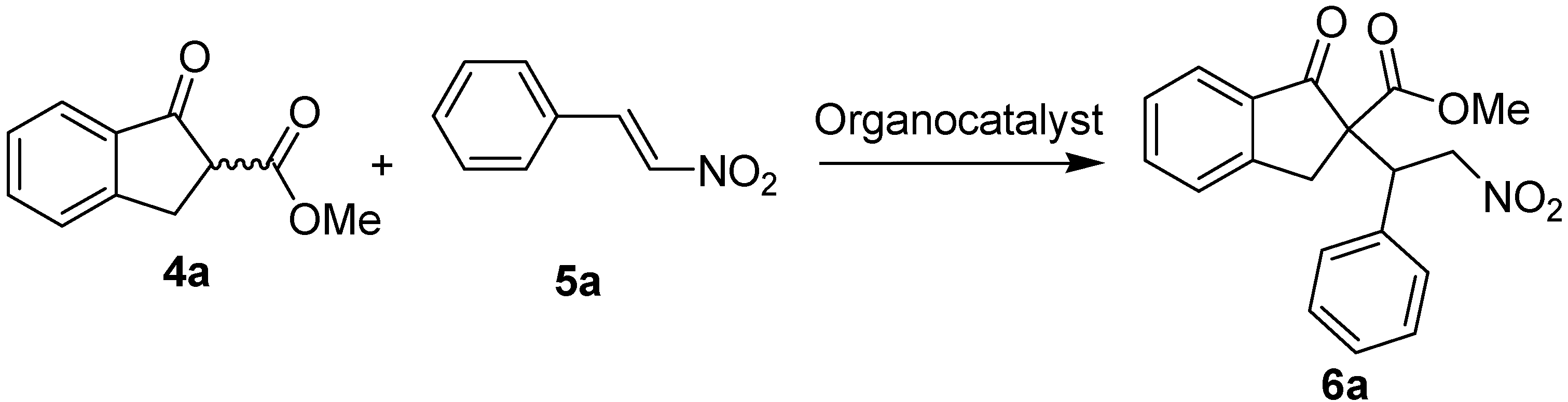
| Entrya | R1 | R2, n | Time (h) | Yield%b | Drc | er (minor)d | er (major)d |
|---|---|---|---|---|---|---|---|
| 1 | H | H, n = 1 | 24 | 6a: 87 | 80/20 | 66:34 | 87:13 |
| 2 | p-Me | H, n = 1 | 24 | 6b: 85 | 76/24 | 60:40 | 84:16 |
| 3 | o-Cl | H, n = 1 | 24 | 6c: 81 | 85/15 | 64:36 | 90:10 |
| 4 | p-Br | H, n = 1 | 24 | 6d: 72 | 82/18 | 61:39 | 87:17 |
| 5 | p-OMe | H, n = 1 | 24 | 6e: 78 | 75/25 | 57:43 | 86:14 |
| 6 | p-Cl | H, n = 1 | 24 | 6f: 81 | 83/17 | 63:37 | 86:14 |
| 7 | 2,4-Cl | H, n = 1 | 24 | 6g: 79 | 82/18 | 52:48 | 85:15 |
| 8 | p-F | H, n = 1 | 24 | 6h: 85 | 83/17 | 65:35 | 89:11 |
| 9 | o-Br | H, n = 1 | 24 | 6i: 77 | 77/23 | 82:18 | 92:8 |
| 10 | p-CN | H, n = 1 | 24 | 6k: 76 | 85/15 | 65:35 | 84:16 |
| 11 | o-Br | 4-Me, n = 1 | 48 | 6l: 72 | 73/27 | 71:29 | 86:14 |
| 12 | o-Br | 4-OMe, n = 1 | 48 | 6m: 87 | 82/18 | 75:25 | 87:13 |
| 13 | o-Br | 5-Br, n =1 | 48 | 6n: 78 | 75/25 | 50:50 | 84:26 |
| 14 | o-Cl | 4-Me, n = 1 | 48 | 6o: 83 | 76/24 | 78:22 | 80:20 |
| 15 | o-Cl | 4-OMe, n =1 | 48 | 6p: 85 | 84/16 | 62:38 | 85:15 |
| 16 | o-Cl | 5-Br, n =1 | 48 | 6q: 87 | 74/26 | 50:50 | 82:18 |
| 17 | o-Cl | H, n =2 | 48 | trace | - | - | - |
 | p-F | H, n = 1 | 24 | 6h: 93 | 57/43 | 51:49 | 77/23 |
3. Experimental Section
3.1. General
3.2. General Procedure for Michael Addition of 1,3-Dicarbonyl Indane Compounds to Nitroolefins
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds available from authors.
References and Notes
- Jung, M.E. Stabilized Nucleophiles with electron deficient alkenes and alkynes. In Comprehensive Organic Synthesis, 1st; Trost, B.M., Fleming, I., Eds.; Pergamon: Oxford, UK, 1991; Volume IV, pp. 1–67. [Google Scholar]
- Yamaguchi, M.; Yokota, N.; Minami, T. The Michael addition of dimethyl malonate to α,β-unsaturated aldehydes catalysed by proline lithium salt. J. Chem. Soc. Chem. Commun. 1991, 1088–1089. [Google Scholar]
- Perlmutter, P. Conjugate Addition Reactions in Organic Synthesis; Pergamon Press: Oxford, UK, 1992. [Google Scholar]
- Ji, J.G.; Barnes, D.M.; Zhang, J.; King, S.A.; Wittenberger, S.J.; Morton, H.E. Catalytic enantioselective conjugate addition of 1,3-dicarbonyl compounds to nitroalkenes. J. Am. Chem. Soc. 1999, 121, 10215–10216. [Google Scholar]
- Vicario, J.L.; Badia, D.; Carrillo, L. Organocatalytic enantioselective Michael and hetero-Michael reactions. Synthesis 2007, 2065–2092. [Google Scholar]
- Hallan, N. Aburel, P.S.; Jørgensen, K.A. Highly enantio- and diastereoselective organocatalytic asymmetric domino Michael-aldol reaction of β-ketoesters and α,β-unsaturated ketones. Angew. Chem. Int. Ed. 2004, 43, 1272–1277. [Google Scholar]
- Tsogoeva, S.B. Recent advances in asymmetric organocatalytic 1,4-conjugate additions. Eur. J. Org. Chem. 2007, 1701–1716. [Google Scholar]
- Almaşi, D.; Alonso, D.A.; Nájera, C. Organocatalytic asymmetric conjugate additions. Tetrahedron Asymmetry 2007, 18, 299–365. [Google Scholar] [CrossRef]
- Långstöm, B.; Bergson, G. Asymmetric induction in a Michael-type reaction. Acta Chem. Scand. 1973, 27, 3118–3119. [Google Scholar] [CrossRef]
- Yamaguchi, M. Conjugate addition of stabilized carbanions. In Comprehensive Asymmetric Catalysis I-III; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: Berlin, Germany, 1999; Volume 3, pp. 1121–1139. [Google Scholar]
- Connon, S.J. Asymmetric catalysis with bifunctional cinchona alkaloid-based urea and thiourea organocatalysts. Chem. Commun. 2008, 2499–2510. [Google Scholar] [CrossRef]
- Ono, N. The Nitro Group in Organic Synthesis; Wiley-VCH: New York, NY, USA, 2001. [Google Scholar]
- Barnes, D.M.; Ji, J.G.; Fickes, M.G.; Fitzgerald, M.A.; King, S.A.; Morton, H.E.; Plagge, F.A.; Preskill, M.; Wagaw, S.H.; Wittenberger, S.J.; Zhang, J. Development of a catalytic enantioselective conjugate addition of 1,3-dicarbonyl compounds to nitroalkenes for the synthesis of endothelin-A antagonist ABT-546. Scope, mechanism, and further application to the synthesis of the antidepressant rolipram. J. Am. Chem. Soc. 2002, 124, 13097–13105. [Google Scholar]
- Almasi, D.; Alonso, D.A.; Gomez-Bengoa, E.; Najera, C. Chiral 2-Aminobenzimidazoles as Recoverable Organocatalysts for the Addition of 1,3-Dicarbonyl Compounds to Nitroalkenes. J. Org. Chem. 2009, 74, 6163–6168. [Google Scholar]
- Li, H.; Wang, Y.; Tang, L.; Wu, F.; Liu, X.; Guo, C.; Foxman, B.M.; Deng, L. Stereocontrolled Creation of Adjacent Quaternary and Tertiary Stereocenters by a Catalytic Conjugate Addition. Angew. Chem. Int. Ed. 2005, 44, 105–108. [Google Scholar]
- Wang, J.; Li, H.; Duan, W.; Zu, L.; Wang, W. Organocatalytic asymmetric Michael addition of 2,4-pentadione to nitroolefins. Org. Lett. 2005, 7, 4713–4716. [Google Scholar]
- Wang, C.J.; Zhang, Z.H.; Dong, X.Q.; Wu, X.J. Chiral amine-thioureas bearing multi hydrogen bonding donors: highly efficient organocatalysts for asymmetric Michael additions of acetylacetone to nitroolefins. Chem. Commun. 2008, 1431–1433. [Google Scholar]
- Han, X.; Luo, C.; Lu, Y. Asymmetric generation of fluorine-containing quaternary carbons adjacent to tertiary stereocenters: uses of fluorinated methines as nucleophiles. Chem. Commun. 2009, 2044–2046. [Google Scholar]
- Jiang, X.; Zhang, Y.; Liu, X.; Zhang, G.; Lai, L.; Wu, L.; Zhang, J.; Wang, R. Enantio- and diastereoselective asymmetric addition of 1,3-dicarbonyl compounds to nitroalkenes in a doubly stereocontrolled manner catalyzed by bifunctional rosin-derived amine thiourea catalysts. J. Org. Chem. 2009, 74, 5562–5567. [Google Scholar]
- Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.N.; Takemoto, Y. Enantio- and diastereoselective Michael reaction of 1,3-dicarbonyl compounds to nitroolefins catalyzed by a bifunctional thiourea. J. Am. Chem. Soc. 2005, 127, 119–125. [Google Scholar]
- Gao, P.; Wang, C.G.; Wu, Y.; Zhou, Z.H.; Tang, C.C. Sugar-derived bifunctional thiourea organocatalyzed asymmetric Michael addition of acetylacetone to nitroolefins. Eur. J. Org. Chem. 2008, 4563–4566. [Google Scholar]
- McCooey, S.H.; Connon, S.J. Urea- and thiourea-substituted cinchona alkaloid derivatives as highly efficient bifunctional organocatalysts for the asymmetric addition of malonate to nitroalkenes: Inversion of configuration at C9 dramatically improves catalyst performance. Angew. Chem. Int. Ed. 2005, 44, 6367–6370. [Google Scholar] [CrossRef]
- Terada, M.; Ube, H.; Yaguchi, Y. Axially chiral guanidine as enantioselective base catalyst for 1,4-addition reaction of 1,3-dicarbonyl compounds with conjugated nitroalkenes. J. Am. Chem. Soc. 2006, 128, 1454–1455. [Google Scholar]
- Yu, Z.; Liu, X.; Zhou, L.; Lin, L.; Feng, X. Bifunctional guanidine via an amino amide skeleton for asymmetric Michael reactions of β-ketoesters with nitroolefins: A concise synthesis of bicyclic β-amino acids. Angew. Chem. Int. Ed. 2009, 48, 5195–5198. [Google Scholar]
- Tan, B.; Chua, P.J.; Li, Y.; Zhong, G. Organocatalytic asymmetric tandem Michael-Henry reactionsL a highly stereoselective synthesis of multifunctionalized cyclohexanes with two quaternary stereocenters. Org. Lett. 2008, 10, 2437–2440. [Google Scholar] [CrossRef]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral Squaramide Derivatives are Excellent Hydrogen Bond Donor Catalysts. J. Am. Chem. Soc. 2007, 130, 14416–14417. [Google Scholar]
- Ju, Y.D.; Xu, L.W.; Li, L.; Lai, G.Q.; Qiu, H.Y.; Jiang, J.X.; Lu, Y. Noyori’s Ts-DPEN ligand: An efficient bifunctional primary amine-based organocatalyst in enantio- and diastereoselective Michael addition of 1,3-dicarbonyl indane compounds to nitroolefins. Tetrahedron Lett. 2008, 49, 6773–6777. [Google Scholar] [CrossRef]
- Luo, J.; Xu, L.W.; Hay, R.A.S.; Lu, Y. Asymmetric Michael additions of ketoesters to nitroolefins catalyzed by a novel cinchona-derived bifunctional catalyst. Org. Lett. 2009, 11, 437–440. [Google Scholar]
- Ikariya, T.; Hashiguchi, S.; Murata, K.; Noyori, R. Preparation of optically active (R,R)-hydrobenzoin from benzoin or benzil. Org. Synth. 2005, 82, 10–14. [Google Scholar]
- Balsells, J.; Mejorado, L.; Phillips, M.; Ortega, F.; Aguirre, G.; Somanathan, R.; Walsh, P.J. Synthesis of chiral sulfonamide/Schiff base ligands. Tetrahedron Asymmetry 1998, 9, 5134–5142. [Google Scholar]
- Zhao, P.Q.; Xu, L.W.; Xia, C.G. Transition-metal-based Lewis acid catalyzed ring opening of epoxides using amines under solvent-free conditions. Synlett 2004, 846–850. [Google Scholar]
- Liu, B.; Liu, J.; Jia, X.; Huang, L.; Li, X.; Chan, A.S.C. The synthesis of chiral N-tosylatedaminoimine ligands and their application in enantioselective addition of phenylacetylene to imines. Tetrahedron Asymmetry 2007, 18, 1124–1128. [Google Scholar] [CrossRef]
- Saravanan, P.; Singh, V.K. An efficient method for acylation reactions. Tetrahedron Lett. 1999, 40, 2611–2614. [Google Scholar] [CrossRef]
- Wei, S.; Yalalov, D.A.; Tsogoeva, S.B.; Schmatz, S. New highly enantioselective thiourea-based bifunctional organocatalysts for nitro-Michael addition reactions. Catal. Today 2007, 121, 151–157. [Google Scholar]
- Kubota, K.; Hamblett, C.L.; Wang, X.L.; Leighton, J.L. Strained silacycle-catalyzed asymmetric Diels-Alder cycloadditions: the first highly enantioselective silicon Lewis acid catalyst. Tetrahedron 2006, 62, 11397–11401. [Google Scholar]
- Luo, S.Z.; Xu, H.; Li, J.; Zhang, L.; Cheng, J.P. A simple primary-tertiary diamine-Brønsted acid catalyst for asymmetric direct aldol reactions of linear aliphatic ketones. J. Am. Chem. Soc. 2007, 129, 3074–3075. [Google Scholar]
- List, B.; Reisinger, C. Noyori ligand as organocatalyst. Synfacts 2008, 12, 1328–1328. [Google Scholar]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric enamine catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef]
- Deutsch, J.; Niclas, H.J.; Ramm, M. Studies on diastereoselective additions of 2-substituted cyclopentanones to β-nitrostyrene. J. Prakt. Chem. 1995, 337, 23–28. [Google Scholar] [CrossRef]
- Brunner, H.; Kimel, B. Asymmetric Catalysis, CIII [1]: Enantioselective Michael addition of 1,3-dicarbonyl compounds to conjugated nitroalkenes. Monatsh. Chem. 1996, 127, 1063–1072. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, Z.-Y.; Yang, H.-M.; Ju, Y.-D.; Li, L.; Luo, M.-X.; Lai, G.-Q.; Jiang, J.-X.; Xu, L.-W. Organocatalytic Michael Addition of 1,3-Dicarbonyl Indane Compounds to Nitrostyrenes. Molecules 2010, 15, 2551-2563. https://doi.org/10.3390/molecules15042551
Jiang Z-Y, Yang H-M, Ju Y-D, Li L, Luo M-X, Lai G-Q, Jiang J-X, Xu L-W. Organocatalytic Michael Addition of 1,3-Dicarbonyl Indane Compounds to Nitrostyrenes. Molecules. 2010; 15(4):2551-2563. https://doi.org/10.3390/molecules15042551
Chicago/Turabian StyleJiang, Zhen-Yu, Hua-Meng Yang, Ya-Dong Ju, Li Li, Meng-Xian Luo, Guo-Qiao Lai, Jian-Xiong Jiang, and Li-Wen Xu. 2010. "Organocatalytic Michael Addition of 1,3-Dicarbonyl Indane Compounds to Nitrostyrenes" Molecules 15, no. 4: 2551-2563. https://doi.org/10.3390/molecules15042551
APA StyleJiang, Z.-Y., Yang, H.-M., Ju, Y.-D., Li, L., Luo, M.-X., Lai, G.-Q., Jiang, J.-X., & Xu, L.-W. (2010). Organocatalytic Michael Addition of 1,3-Dicarbonyl Indane Compounds to Nitrostyrenes. Molecules, 15(4), 2551-2563. https://doi.org/10.3390/molecules15042551