Synthesis and Conformational Study of a Novel Macrocyclic Chiral(Salen) ligand and its Uranyl and Mn Complexes

 ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
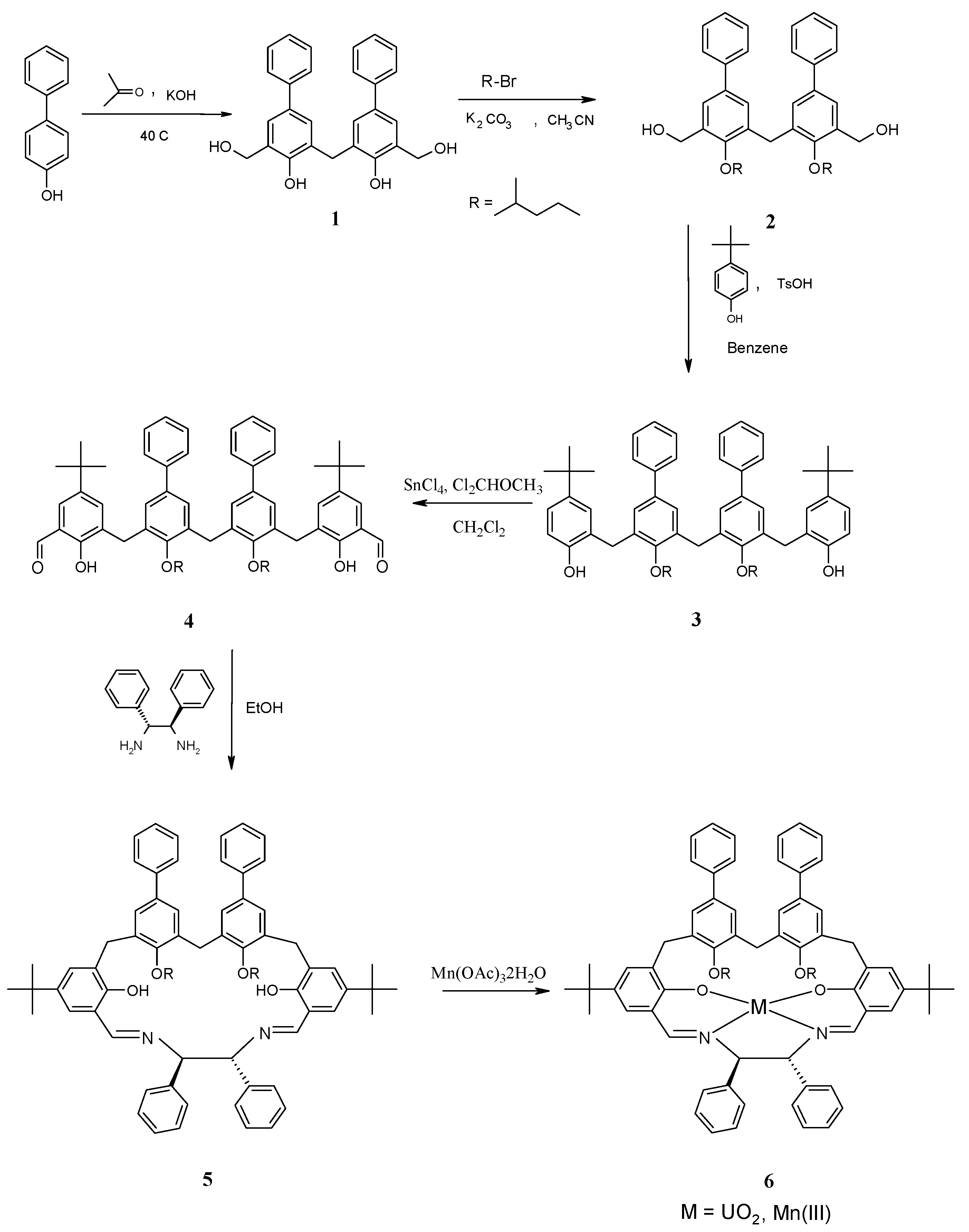
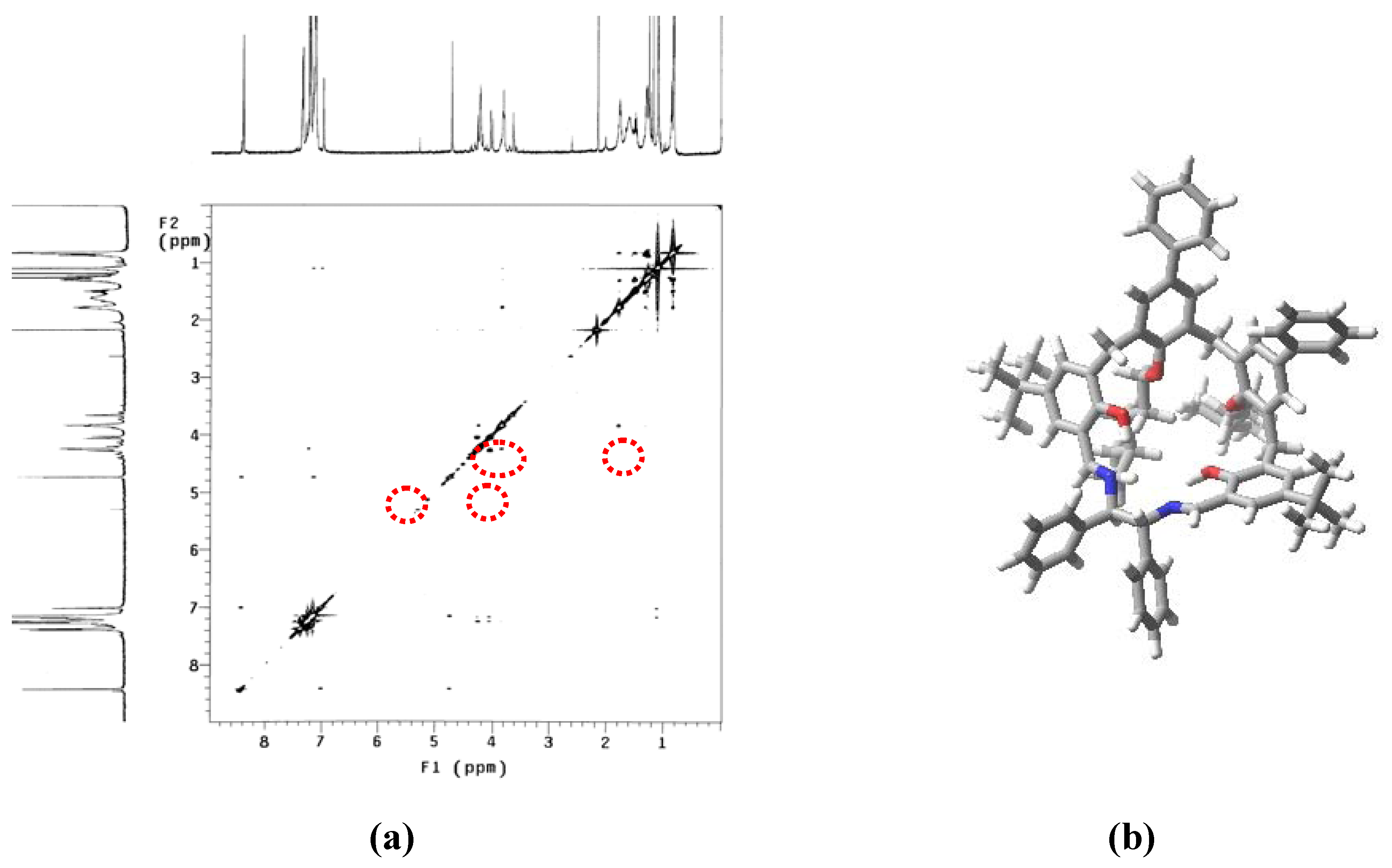
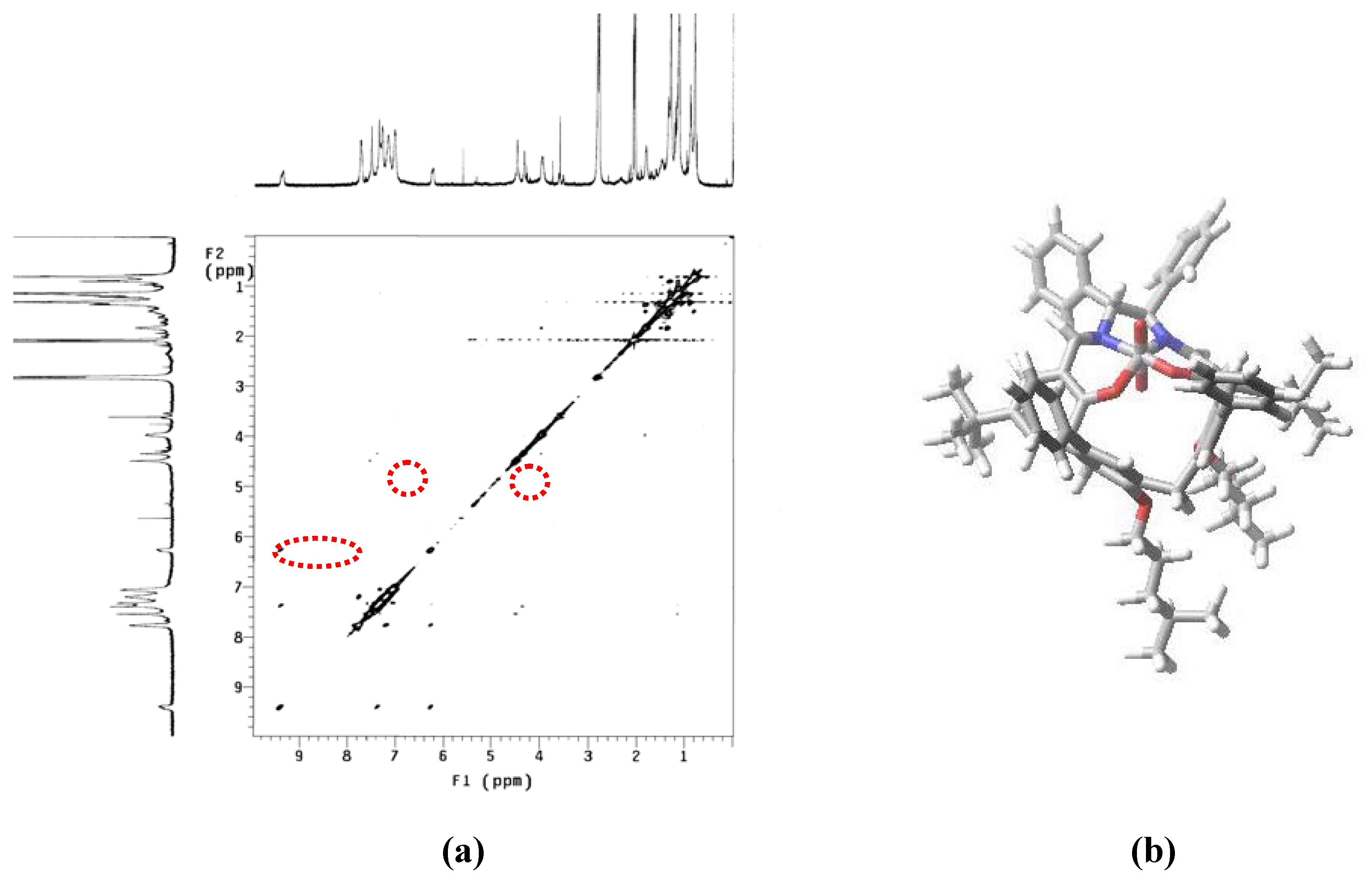
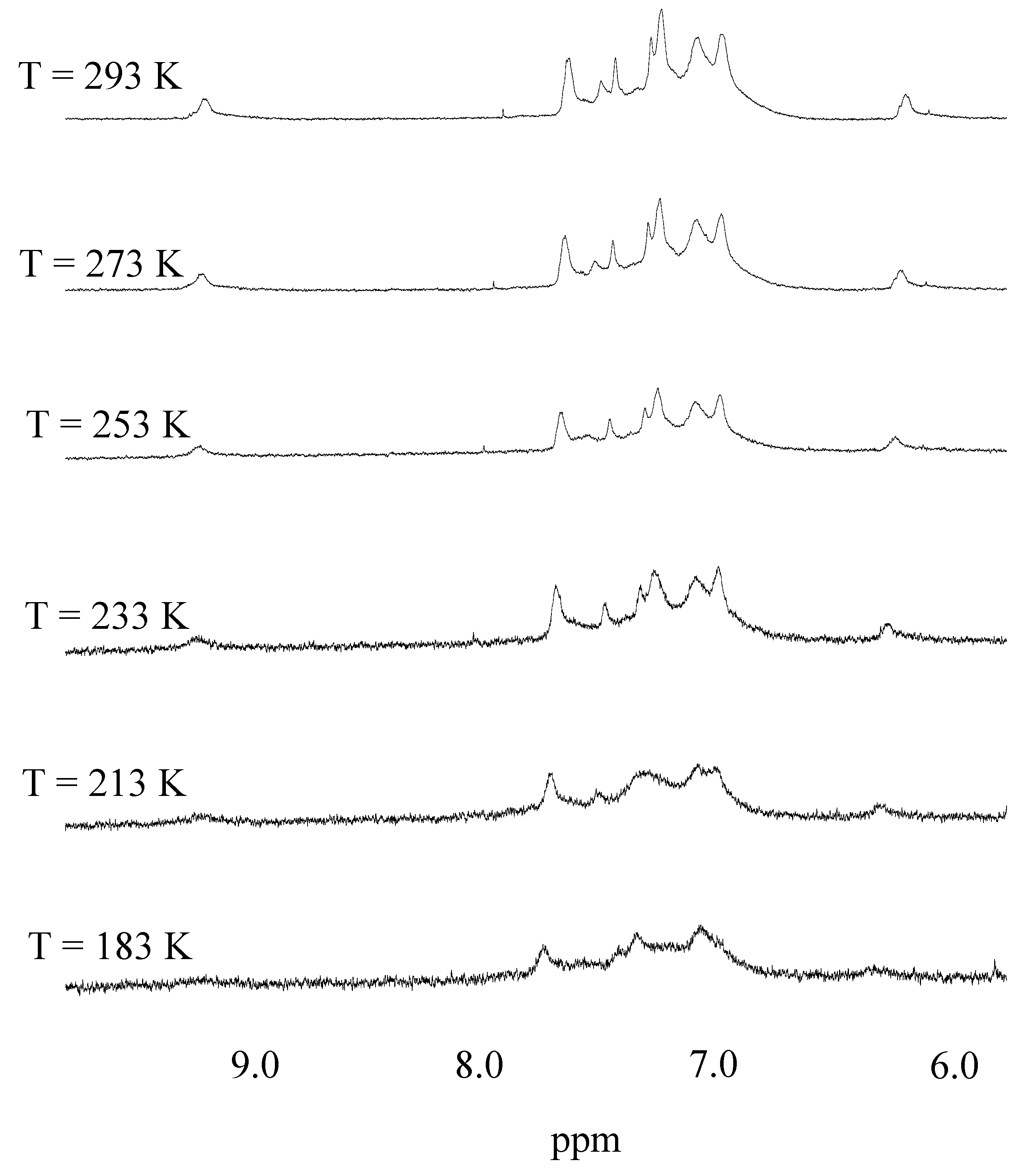

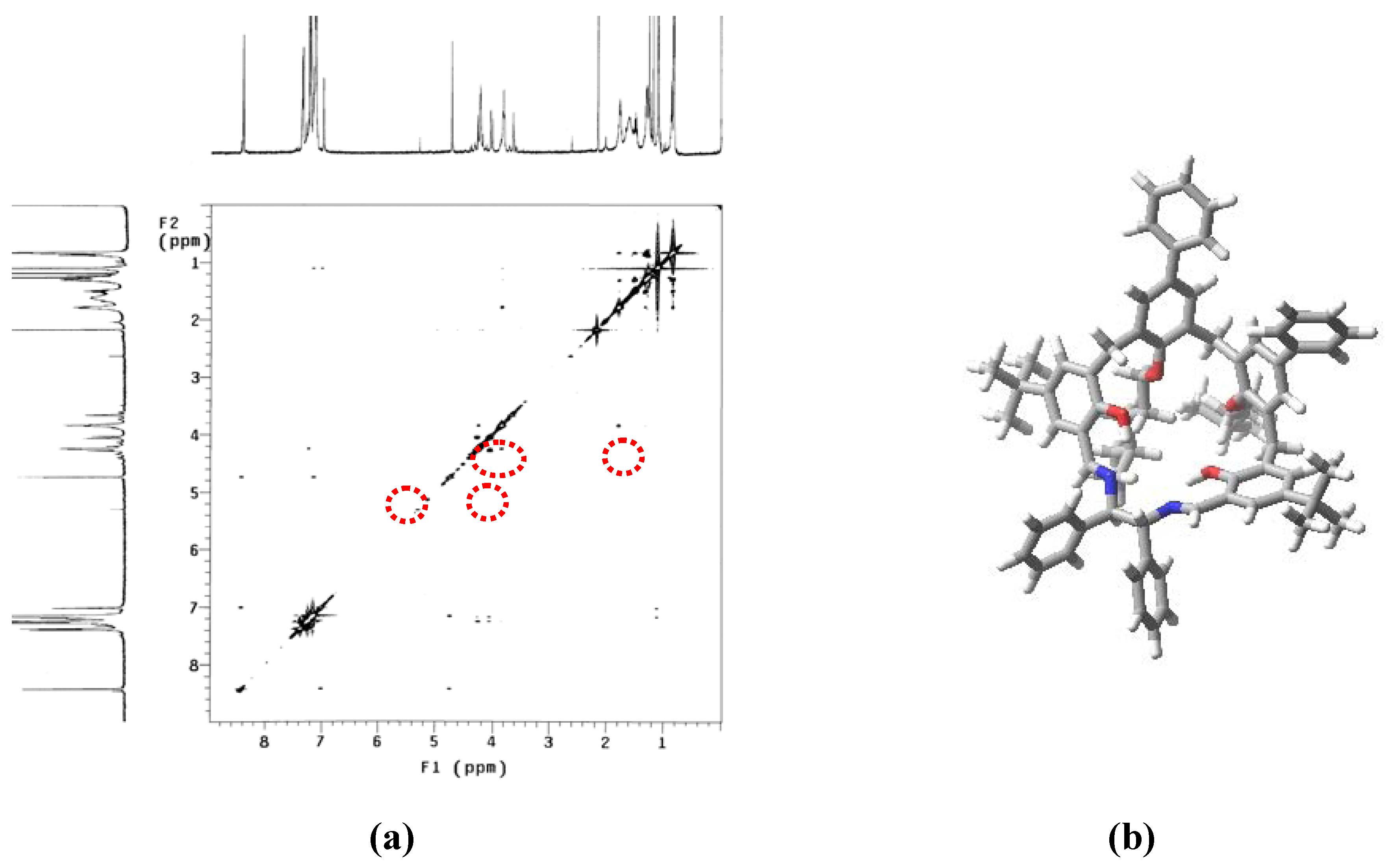{kind=link}
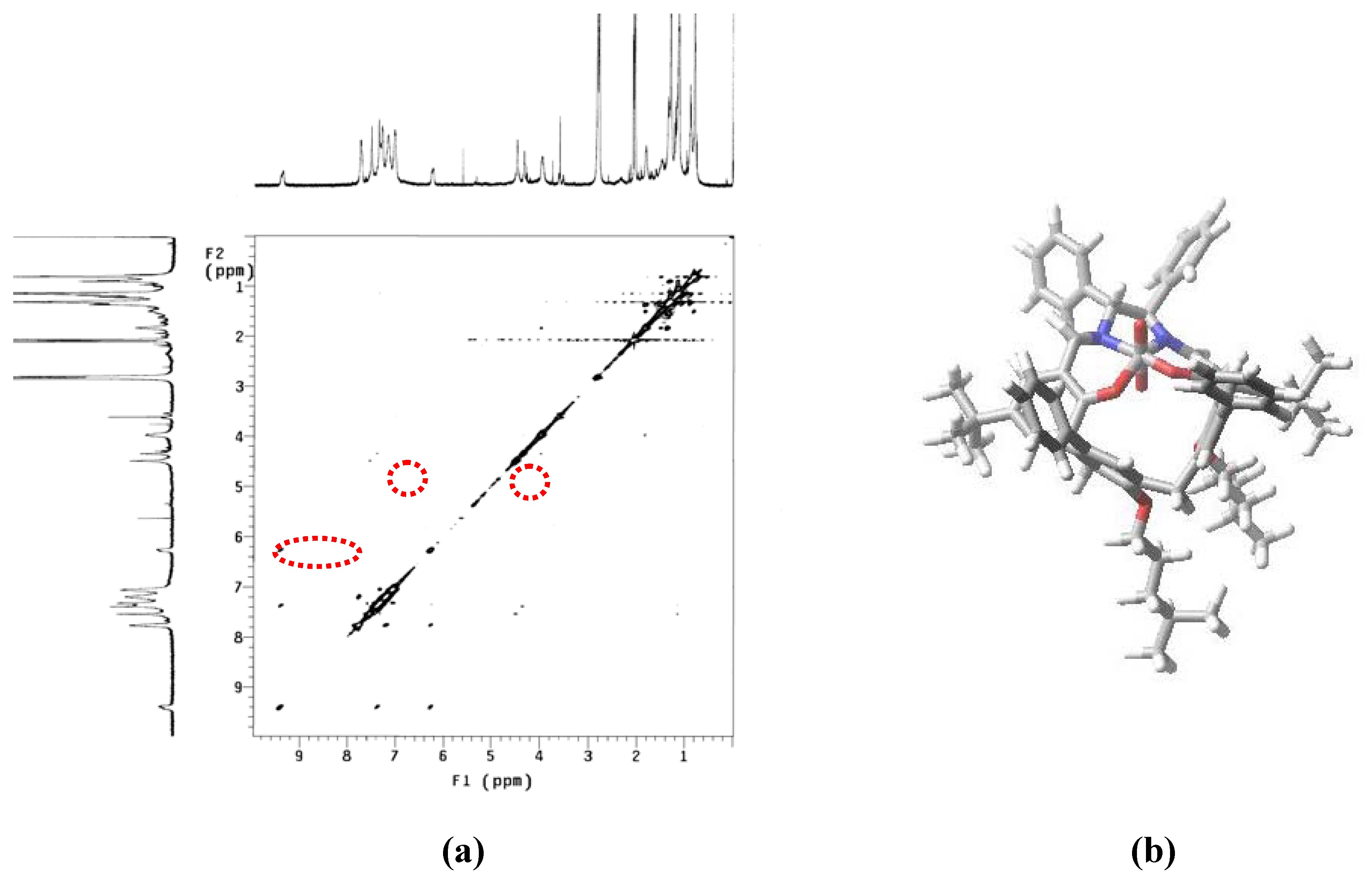{kind=link}
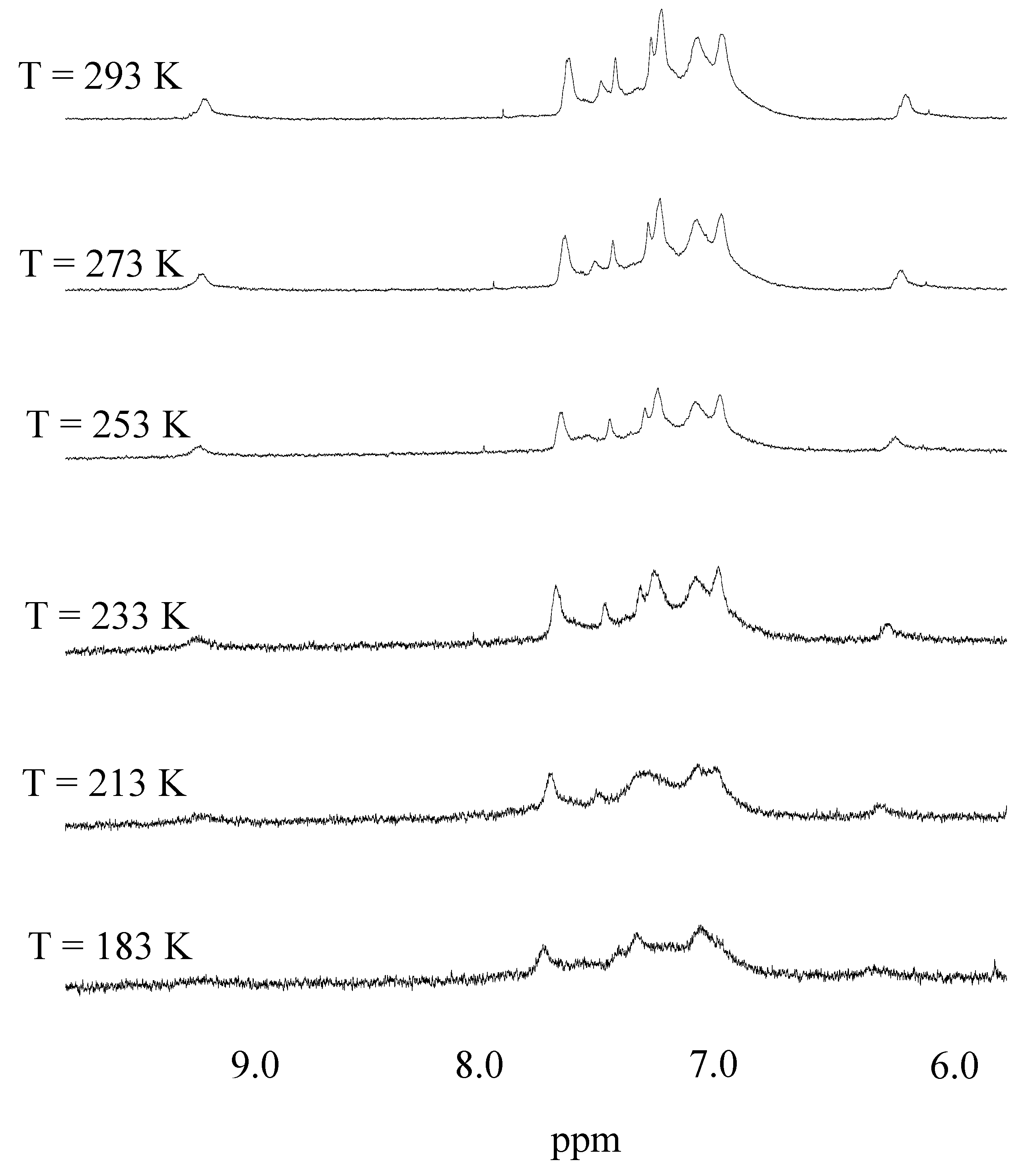{kind=link}
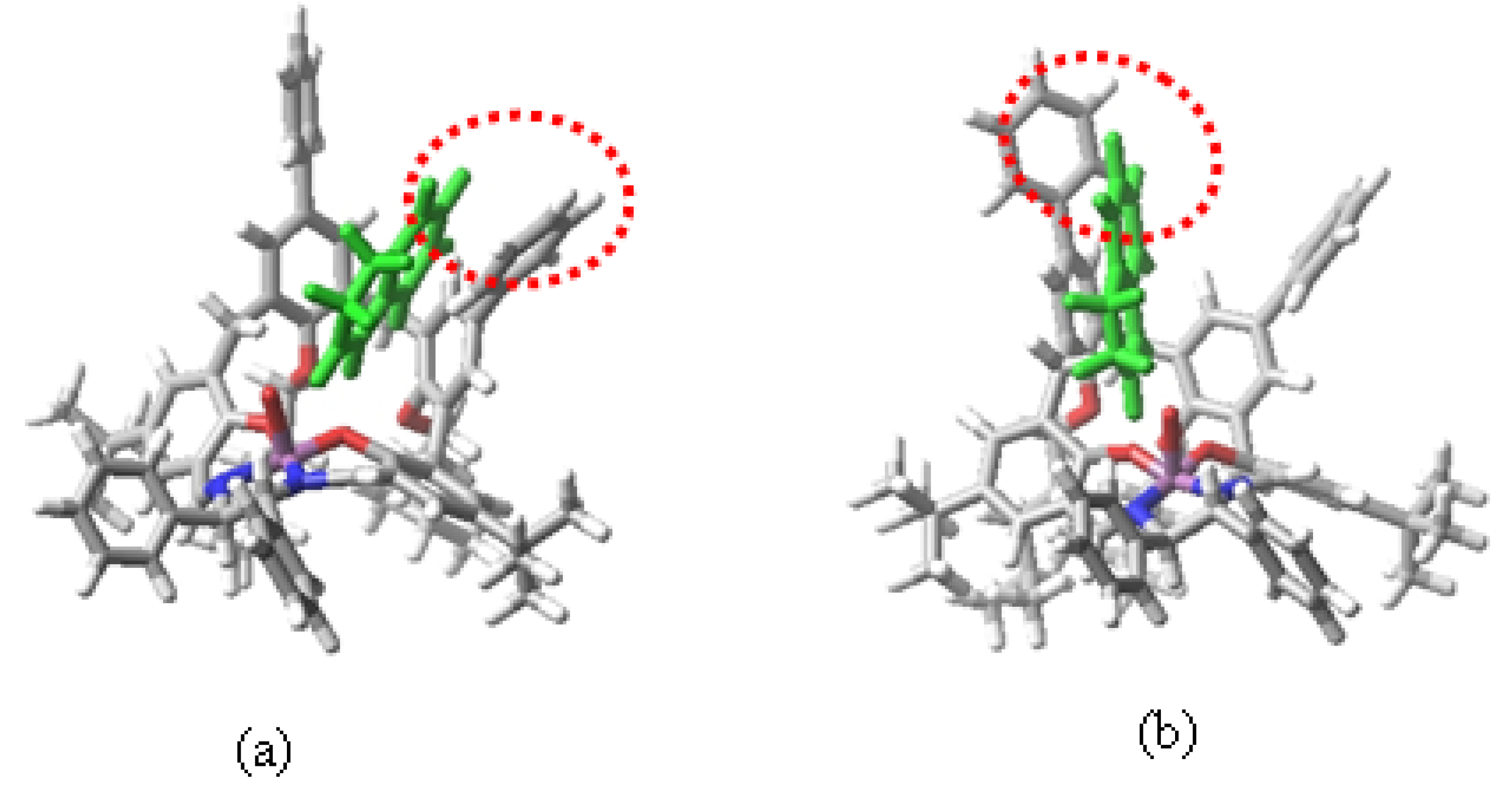{kind=link}
{kind=link}
| Entry | Alkene | Conv. (%) | Yield (%) | ee (%) | Conf.c |
|---|---|---|---|---|---|
| 1 | 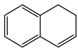 | 50d | 90b | 30b | 1R,2S |
| 2 | | 80 | 90b | 50b | 1R,2S |
| 3 | 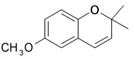 | 90e | 100e | 63f | 3R,4R |
| 4 | 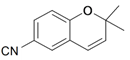 | 90e | 100e | 50f | 3R,4R |
| 5 |  | 50e | 100e | 52f | 3R,4R |
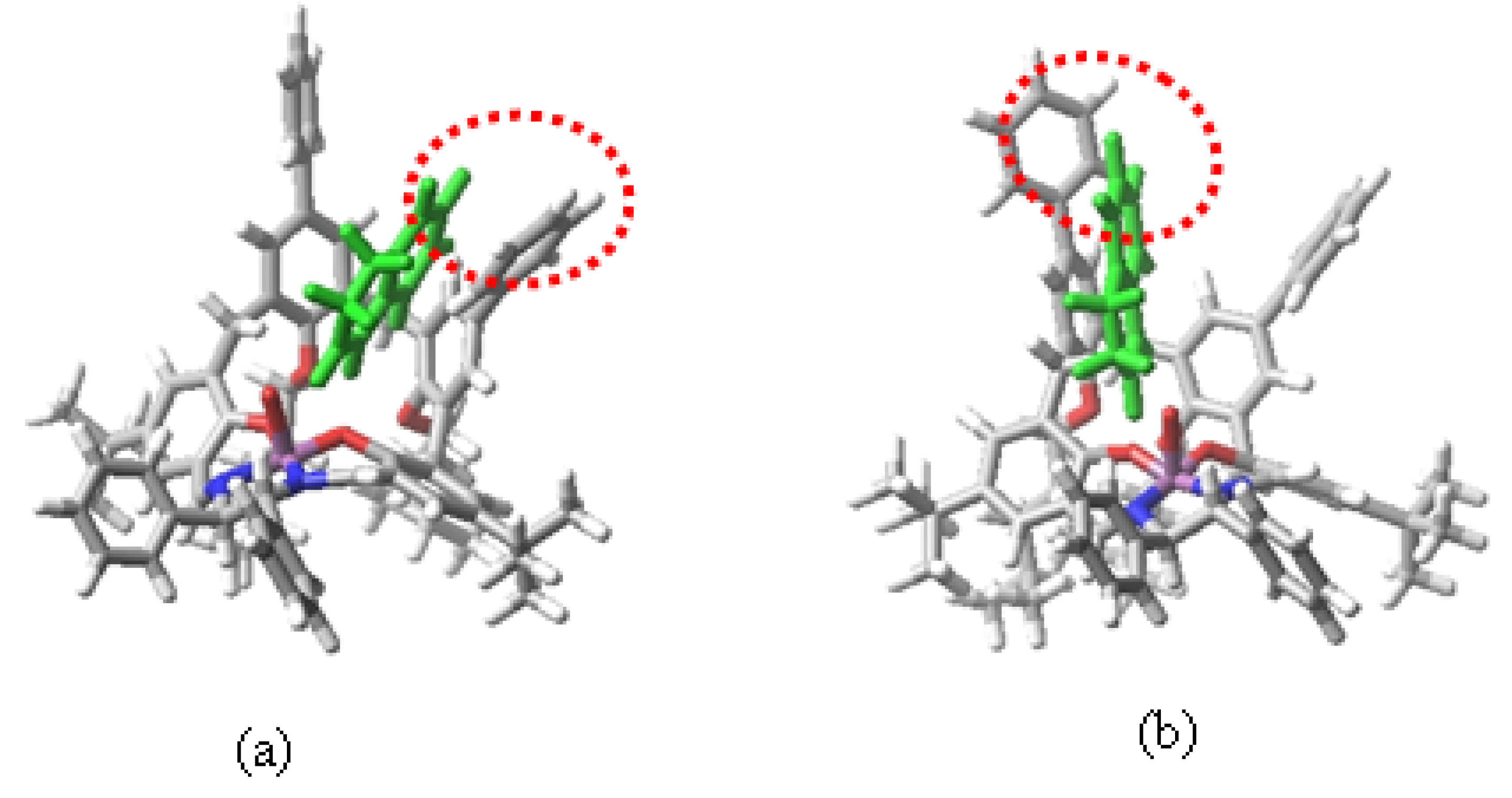
3. Experimental
3.1. General
3.2. General Procedures for the Epoxidation Reactions
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References and Notes
- Steed, J.W.; Atwood, J.L. Supramolecular Chemistry; John Wiley & Sons: Chichester, UK, 2000. [Google Scholar]
- Katsuki, T. Catalytic Asymmetric Synthesis, 2nd ed.; Ojima, I., Ed.; Wiley-VCH: New York, 2000; pp. 287–325. [Google Scholar]
- Katsuki, T. Catalytic asymmetric oxidations using optically active (salen) manganese(III) complexes as catalysts. Coord. Chem. Rev. 1995, 140, 189–214. [Google Scholar] [CrossRef]
- Katsuki, T. Some recent advances in metallosalen chemistry. Synlett 2003, 3, 281–297. [Google Scholar] [CrossRef]
- Irie, R.; Noda, K.; Ito, Y.; Matsumoto, N.; Katsuki, T. Catalytic asymmetric epoxidation of unfunctionalized olefins. Tetrahedron Lett. 1990, 31, 7345–7384. [Google Scholar] [CrossRef]
- Egami, H.; Irie, R.; Sakai, K.; Katsuki, T. Enantioselective epoxidation of conjugated Z-olefins with newly modified Mn(salen) complex. Chem. Lett. 2007, 36, 46–47. [Google Scholar] [CrossRef]
- Matsumoto, K.; Katsuki, T. Asymmetric Synthesis–The Essentials, 2nd ed.; Christmann, M., Bräse, S., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 123–127. [Google Scholar]
- Zhang, W.; Loebach, J.L.; Wilson, S.R.; Jacobsen, E.N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by salen manganese complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar]
- Jacobsen, E.N.; Zhang, W.; Guler, L.M. Electronic tuning of asymmetric catalysts. J. Am. Chem. Soc. 1991, 113, 6703–6704. [Google Scholar] [CrossRef]
- Jacobsen, E.N. Catalytic Asymmetric Synthesis; Ojima, I., Ed.; VCH: Weinheim, Germany, 1993; Chapter 4.2. [Google Scholar]
- Palucki, M; Finney, N.S.; Pospisil, P. J.; Guler, M. L.; Ishida, T.; Jacobsen, E.N. The mechanistic basis for electronic effects on enantioselectivity in the (salen)Mn(III)-catalyzed epoxidation reaction. J. Am. Chem. Soc. 1998, 120, 948–954. [Google Scholar]
- Yoon, T. P.; Jacobsen, E. N. Privileged chiral catalysis. Science 2003, 299, 1691–1693. [Google Scholar]
- Amato, M.E.; Ballistreri, F.P.; Pappalardo, A.; Sciotto, D.; Tomaselli, G.A.; Toscano, R.M. Synthesis and conformational aspects of 20- and 40-memered macrocyclic mono and dinuclearuranyl complexes incorporating salen and (R)-BINOL units. Tetrahedron 2007, 63, 9751–9757. [Google Scholar]
- Amato, M.E.; Ballistreri, F.P.; Pappalardo, A.; Tomaselli, G.A.; Toscano, R.M.; Williams, D.J. Novel chiral Mn(III)-salen complexes containing a calix[4]-arene unit as enantioselective epoxidation catalysts. Eur. J. Org. Chem. 2005, 3562–3570. [Google Scholar]
- Gutsche, C.D. Calixarenes. 8. Short, stepwise synthesis of p-phenylcalix[4]arene and p-phenyl-p-tert-butylcalix[4]arene and derived products. J. Org. Chem. 1982, 47, 2713–2719. [Google Scholar]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C. Macromodel - an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar]
- Ballistreri, F.P.; Patti, A.; Pedotti, S.; Tomaselli, G.A.; Toscano, R.M. Synthesis of novel chiral “Salen-type” ferrocenyl ligands. Tetrahedron Asymmetry 2007, 18, 2377–2380. [Google Scholar] [CrossRef]
- Dalla Cort, A.; Gasparrini, F.; Lunazzi, L.; Mandolini, L.; Mozzanti, A.; Pasquini, C.; Pierini, M.; Rompietti, R.; Schiaffino, L. Stereomutations of atropisomers of sterically hindered salophen ligands. J. Org. Chem. 2005, 70, 8877–8883. [Google Scholar]
- Dalla Cort, A.; Mandolini, L.; Calmieri, G.; Pasquini, C.; Schiaffino, L. Unprecedented detection of inherent chirality in uranyl–salophen complexes. Chem. Commun. 2003, 2178–2179. [Google Scholar]
- Kureshy, R.I.; Khan, N.H.; Abdi, S.H.; Patel, S.T.; Iyer, P.K.; Jasra, R.V. Enantioselective epoxidation of chromenes using chiral Mn(III) salen catalysts with built-in phase-transfer capability. Tetrahedron Lett. 2002, 43, 2665–2668. [Google Scholar]
- Xia, Q.H.; Ge, H.Q.; Ye, C.P.; Liu, Z.M.; Su, K.X. Advances in homogeneous and heterogeneous catalytic asymmetric epoxidation. Chem. Rev. 2005, 105, 1603–1662. [Google Scholar] [CrossRef]
- Shitama, H.; Katsuki, T. Synthesis of metal-(pentadentate-salen) complexes: asymmetric epoxidation with aqueous hydrogen peroxide and asymmetric cyclopropanation (salenH2: N,N-bis(salicylidene)ethylene-1,2-diamine). Chem. Eur. J. 2007, 13, 4849–4858. [Google Scholar] [CrossRef]
- Scheurer, A.; Mosset, P.; Spiegel, M.; Saalfrank, R.W. Reverse asymmetric catalytic epoxidation of unfunctionalized alkenes. Tetrahedron 1999, 55, 1063–1078. [Google Scholar]
- Bergmann, R.; Gericke, R. Synthesis and antihypertensive activity of 4-(1,2-dihydro-2-oxo-1-pyridyl)-2H-1-benzopyrans and related compounds, new potassium channel activators. J. Med. Chem. 1990, 33, 492–504. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Amato, M.E.; Ballistreri, F.P.; Pappalardo, A.; Tomaselli, G.A.; Toscano, R.M. Synthesis and Conformational Study of a Novel Macrocyclic Chiral(Salen) ligand and its Uranyl and Mn Complexes. Molecules 2010, 15, 1442-1452. https://doi.org/10.3390/molecules15031442
Amato ME, Ballistreri FP, Pappalardo A, Tomaselli GA, Toscano RM. Synthesis and Conformational Study of a Novel Macrocyclic Chiral(Salen) ligand and its Uranyl and Mn Complexes. Molecules. 2010; 15(3):1442-1452. https://doi.org/10.3390/molecules15031442
Chicago/Turabian StyleAmato, Maria E., Francesco P. Ballistreri, Andrea Pappalardo, Gaetano A. Tomaselli, and Rosa M. Toscano. 2010. "Synthesis and Conformational Study of a Novel Macrocyclic Chiral(Salen) ligand and its Uranyl and Mn Complexes" Molecules 15, no. 3: 1442-1452. https://doi.org/10.3390/molecules15031442
APA StyleAmato, M. E., Ballistreri, F. P., Pappalardo, A., Tomaselli, G. A., & Toscano, R. M. (2010). Synthesis and Conformational Study of a Novel Macrocyclic Chiral(Salen) ligand and its Uranyl and Mn Complexes. Molecules, 15(3), 1442-1452. https://doi.org/10.3390/molecules15031442