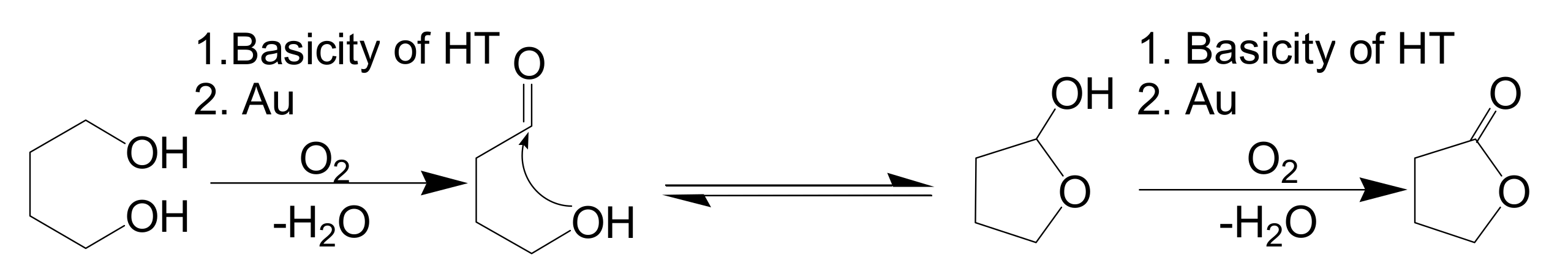
3.1. Deoxygenation of Epoxides Using Au/HT-Alcohols
In the course of our study of Olympic medal metal nanoparticle catalysis for the aerobic oxidation of alcohols, we envisioned that if epoxides could act as hydrogen accepters in place of molecular oxygen, the metal nanoparticles could act as effective catalysts for deoxygenation of epoxides using alcohols as oxygen acceptors (
Scheme 3).
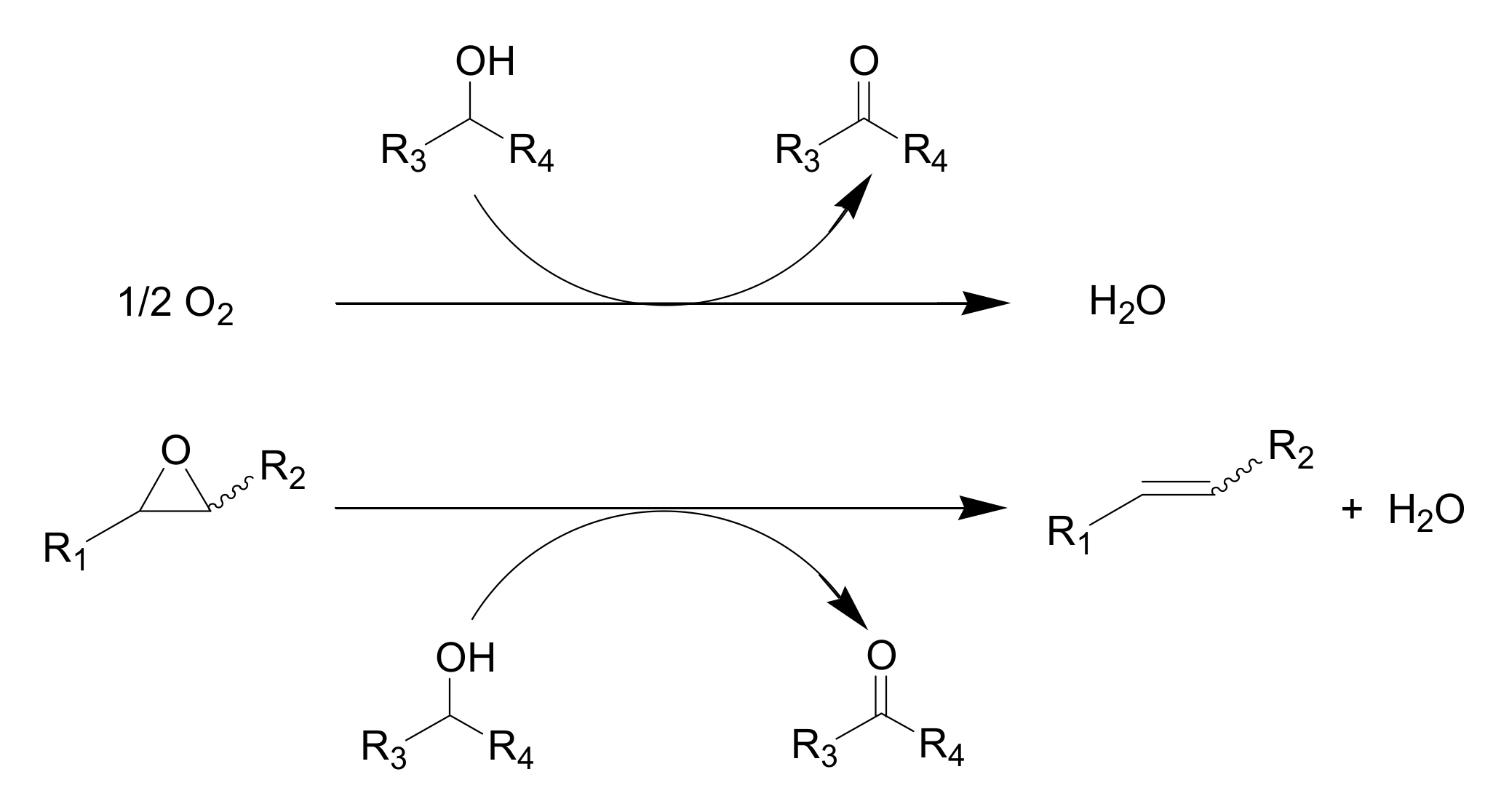
Deoxygenation of epoxides to alkenes is a valuable reaction in organic synthesis because it allows the use of the oxirane ring as a protective group for carbon-carbon double bonds. [
33,
34,
35] The transformation is also of great importance in biological chemistry for the reproduction of vitamin K in the vitamin K cycle [
36,
37]. In addition, it has recently been applied as a quantification method for determination of the epoxide content in graphite epoxide or oxygenated carbon nanotubes [
38,
39]. Stoichiometric deoxygenation of epoxides has been carried out using a variety of reagents [
40]. These reagents, however, are often toxic and/or are employed in large excess, resulting in the production of undesired waste. Several successful catalyst systems have also appeared to date, but these systems suffer from the need for hazardous reductants, are typically air- and moisture-sensitive and often exhibit low catalytic activities and selectivities [
41]. Therefore, the development of an efficient catalytic system for deoxygenation of epoxides remains of great importance.
We discovered the intrinsic ability of gold and silver nanoparticles to catalyze de-epoxidation; gold and silver nanoparticles supported on hydrotalcite (Au/HT and Ag/HT) can promote highly efficient catalytic deoxygenation of epoxides into alkenes using alcohols [
42]. To the best of our knowledge, this is the first report of the catalytic deoxygenation of epoxides using gold and silver nanoparticles.
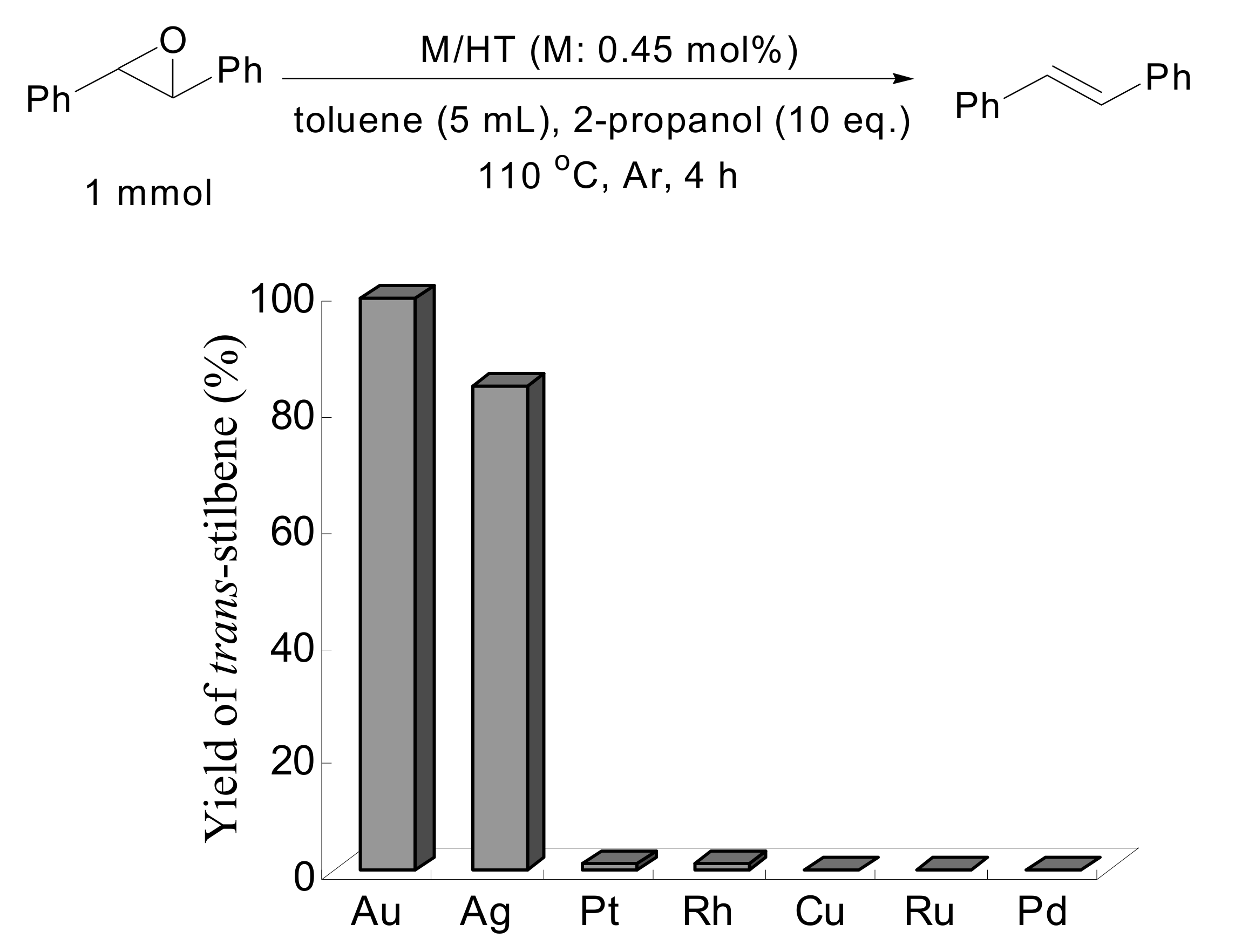
A mixture of solid Au/HT,
trans-stilbene oxide and 2-propanol in toluene was stirred at 110 °C under an Ar atmosphere for 4 h. Selective deoxygenation of
trans-stilbene oxide occurred to afford
trans-stilbene in 99% yield without any byproducts such as 1,2-diphenylethane, 1,2-diphenylethanol or benzyl phenyl ketone, with the reaction occurring through hydrogenation and/or isomerization of
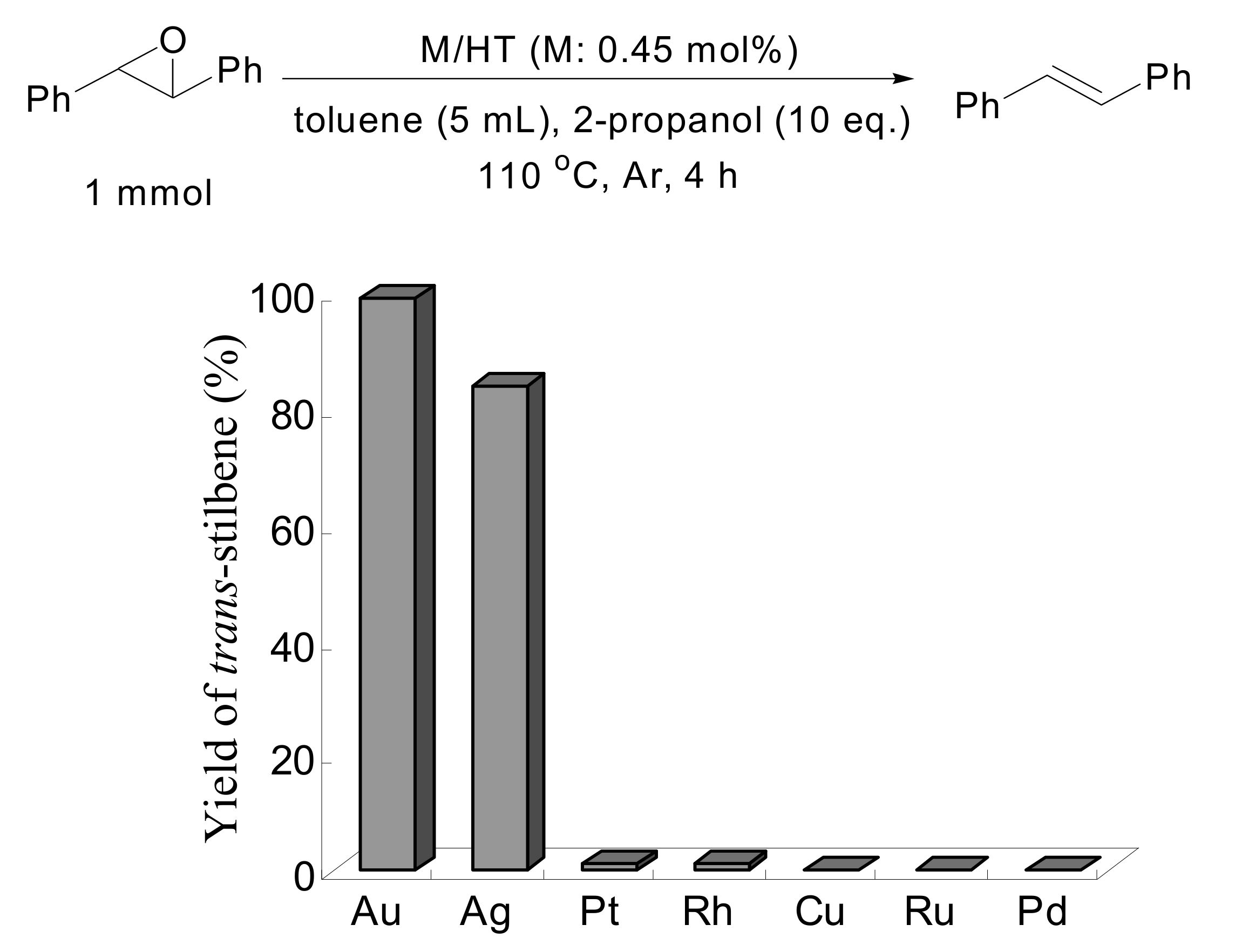
trans-stilbene oxide. Other metal nanoparticles on HT having catalytic potential for aerobic alcohol oxidation such as Pd, Rh, Pt, Ru, Ag and Cu were also examined under similar reaction conditions in the deoxygenation of
trans-stilbene oxide, as shown in
Scheme 4. Notably, HT-supported silver nanoparticles also functioned as an efficient catalyst, giving
trans-stilbene in high yield, while the other metal nanoparticles did not possess any catalytic activity.
The scope of the deoxygenation reaction was then explored with other epoxides, as can be seen in
Table 5. Au/HT deoxygenated a wide range of epoxides, affording the corresponding alkenes in excellent yields. For example, epoxides possessing phenyl (entries 1, 8, 10, 15 and 14), alkyl (entries 20, 22, and 25), ether (entry 16), carbonyl (entry 27), hydroxyl (entry 29) and olefinic (entry 31) groups were successfully employed. Interestingly, Ag/HT showed unique chemoselectivity for the deoxygenation; only styrene oxide derivatives proved to be reactive (entries 5, 9, 11, 13 and 15). Furthermore, these solid catalysts were recoverable by simple filtration following the deoxygenation without any loss in their activities or selectivities, even after several reuse experiments (
Table 5, entries 2, 3, 6 and 7).
The deoxygenation of epoxides using Au/HT was also efficient on a preparative scale. For example,
trans-stilbene oxide (3.9 g; 20 mmol) successfully gave
trans-stilbene (3.4 g; 95% isolated yield) with a turnover frequency (TOF) and a turnover number (TON) of up to 270 h
-1 and 20,000, respectively (
Table 5, entry 4). These values are three orders of magnitude greater than those in previously reported catalyst systems [
43,
44,
45,
46,
47].
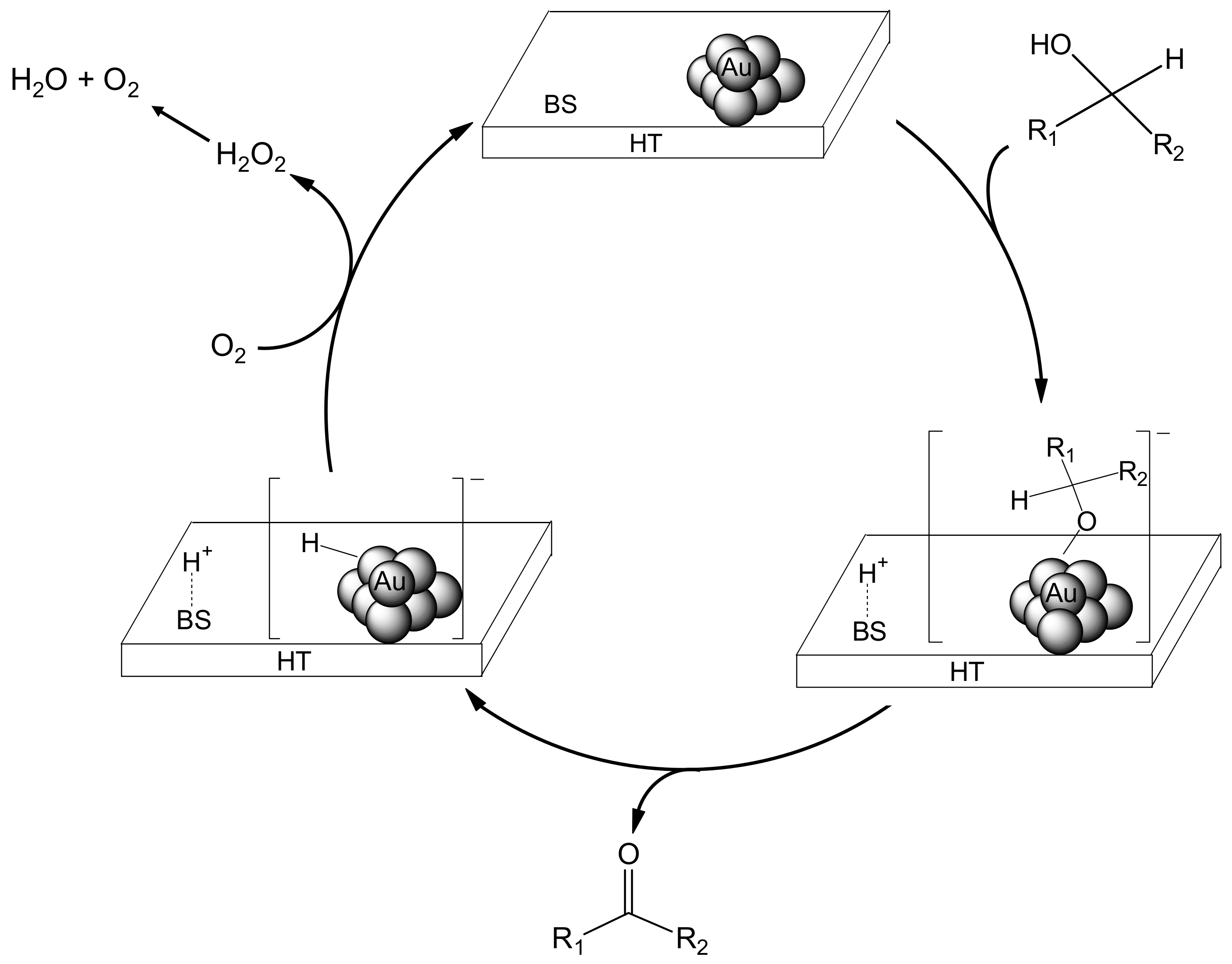
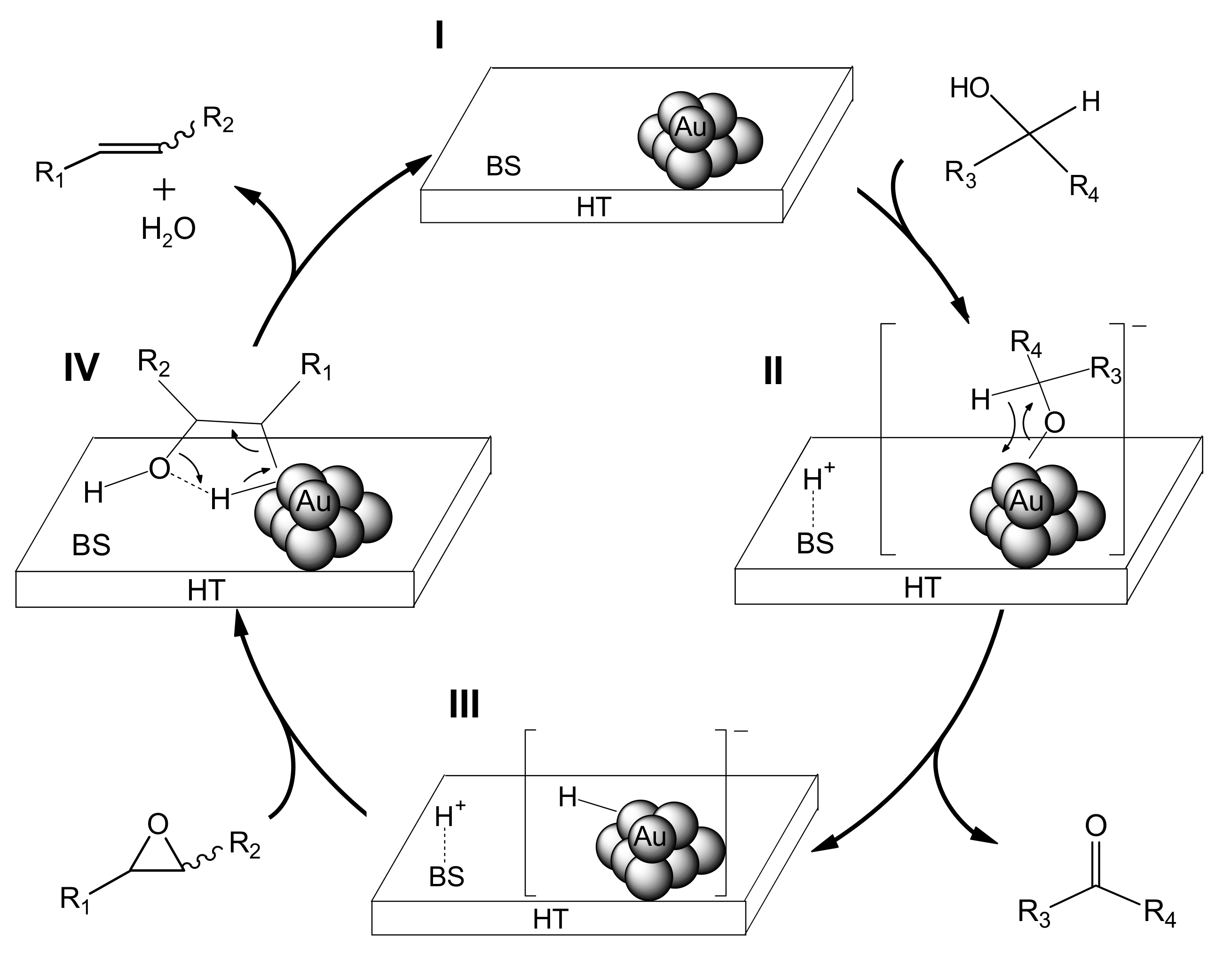
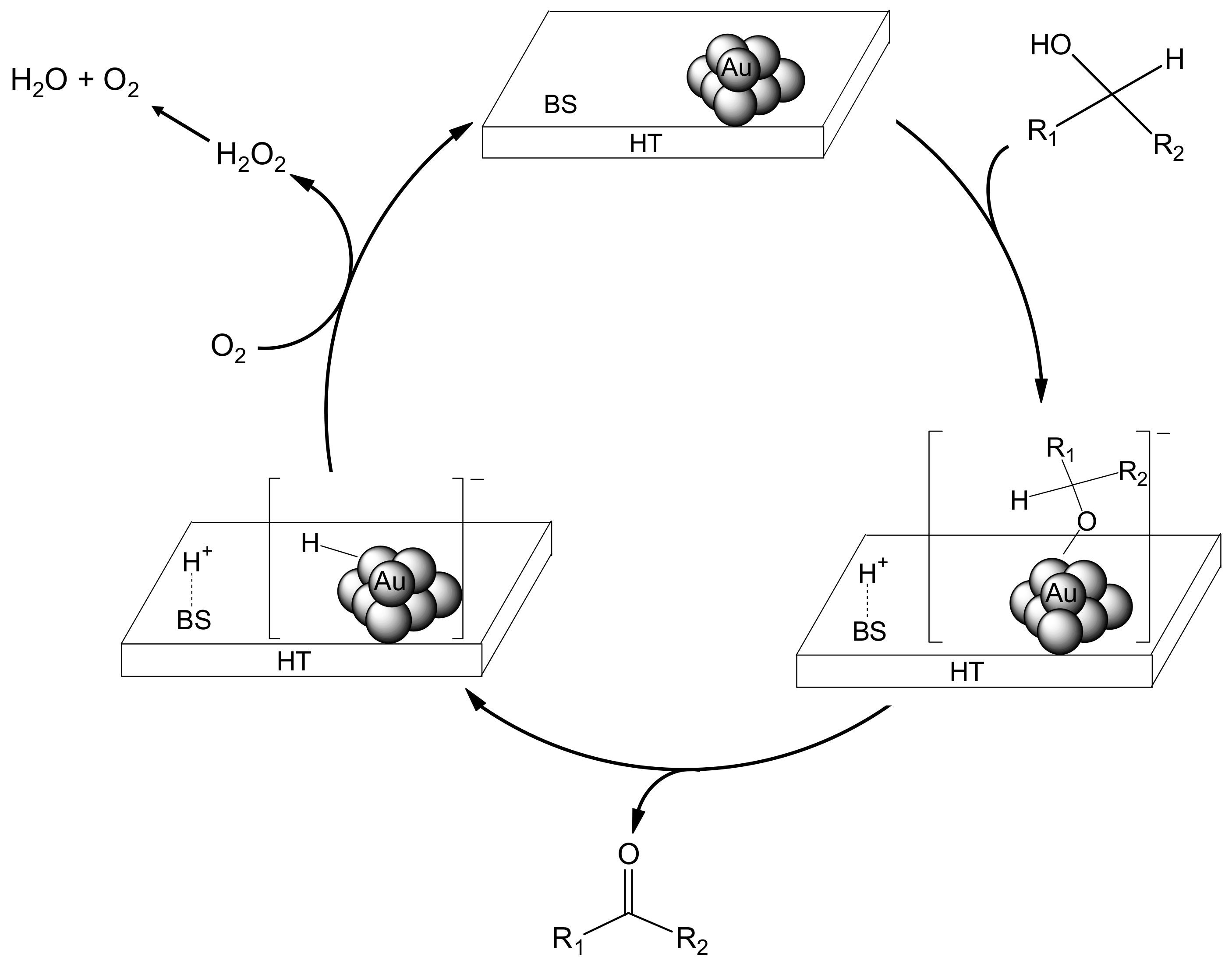
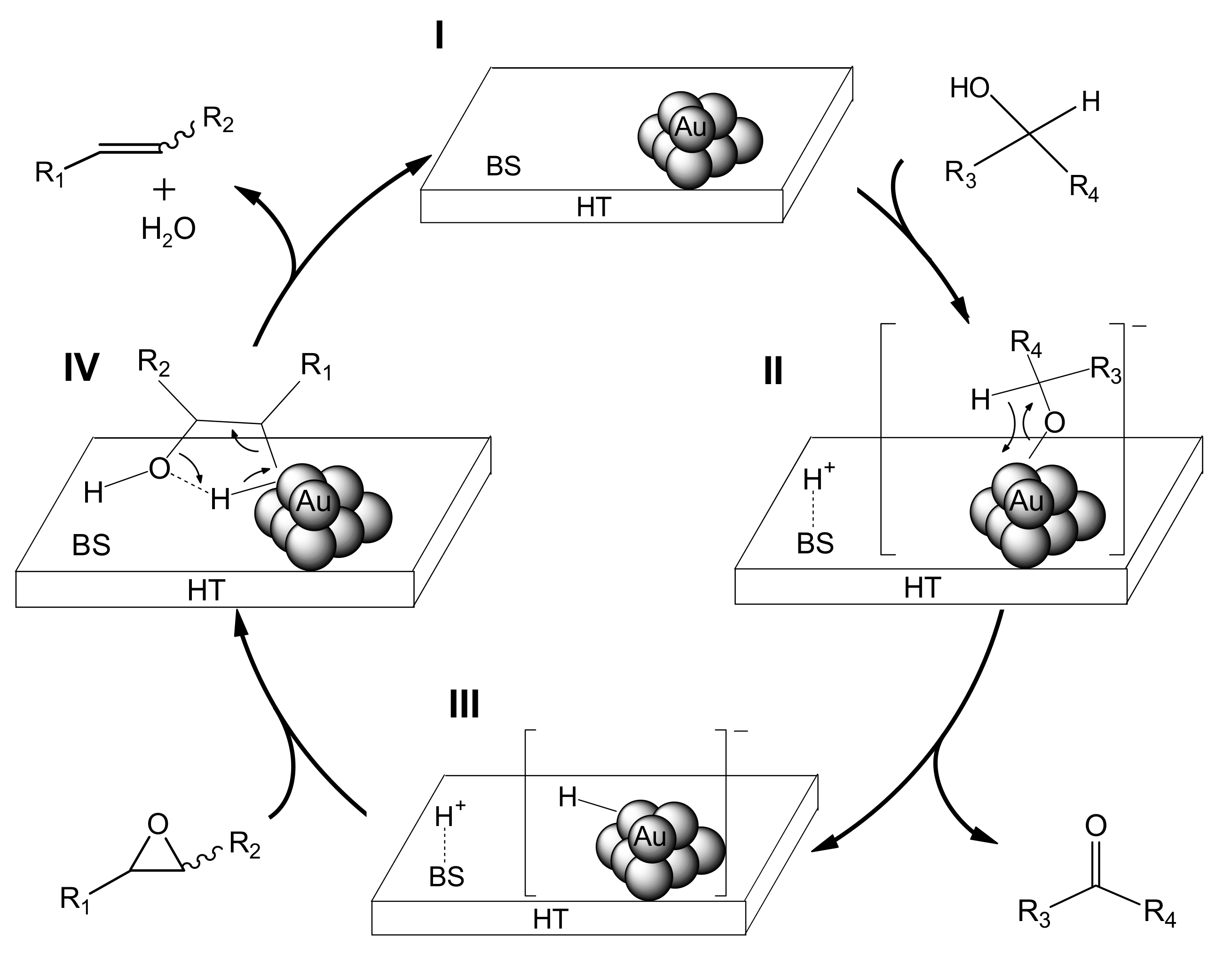
We propose that the deoxygenation proceeds through cooperation between the gold nanoparticles and a basic site of HT (
Scheme 5). The first step involves oxidation of the alcohol on Au/HT (I) via abstraction of the proton of the alcohol by a basic site of HT to generate [H-HT]
+ and a [Au-alcolate] species (II) at the periphery of the AuNP-HT interface. The Au-alcoholate species then undergoes
β-hydride elimination to give an [H-Au]
– species (III) together with the corresponding carbonyl compound. Next, protonation from the [H-HT]
+ species opens the epoxide, which then is attacked by the [H-Au]
– species to give the intermediate (IV). Subsequent dehydration from IV provides the alkene, thereby completing the catalytic cycle.
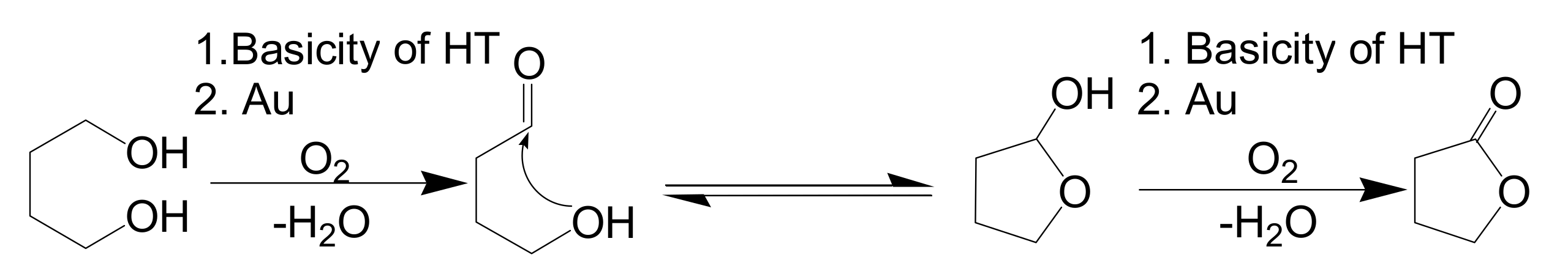
The above reaction pathway is well supported by the following experimental results: (i) almost equimolar amounts of acetophenone and H
2O were formed for every mole of styrene during the deoxygenation of styrene oxide using 1-phenylethanol; (ii) when
trans-2-octenal was used in place of styrene oxide, chemoselective reduction occurred to exclusively afford
trans-2-octenol, preserving the incipient C=C double bond [
48]. It is well-known that all of the metal nanoparticles tested can form metal-hydride species in alcohol oxidation reactions. The distinguished deoxygenation activities of gold and silver nanoparticles from those of other metal NPs can be attributed to their unique reactivity toward an epoxide (Path III→IV→I in
Scheme 3). The solid support of HT can promote the deoxygenation as a base (Path I→II in
Scheme 3) as well as stabilize these metal nanoparticles.
3.2. Deoxygenation of Epoxides Using Au/HT-CO/H2O
From an environmental and practical synthetic point of view, the use of water in organic reactions instead of organic solvents is attractive [
49,
50,
51,
52,
53] because of the low cost, safety (non-explosive, non-flammable and non-toxic), and ease of phase-separation of water-insoluble products in the work-up procedure. For this purpose, our next target focused on the design of a high performance catalyst system for the deoxygenation of epoxides in water.
We found that using CO/H
2O as a reducing agent along with the Au/HT catalyst provides highly efficient deoxygenation of many epoxides into the corresponding alkenes. The reaction takes place under an atmosphere of CO at room temperature in water without any added organic solvents. [
54] Au/HT and styrene oxide were added to water and the heterogeneous mixture was stirred under an atmosphere of CO at room temperature. The reduction of styrene oxide smoothly occurred to give styrene in over 99% yield, accompanied by the formation of an equimolar amount of CO
2 (
Table 6, entry 1). Notably, water was found to be the best solvent to promote efficient deoxygenation, with lower yields being obtained in organic solvents (
Table 6 entries 1 vs. 4-6). When Au/HT was removed from the reaction mixture at 50% styrene conversion, continued stirring of the filtrate under similar conditions did not yield any further products. In addition, no gold ions were detected in the filtrate using ICP-AES analysis, confirming the occurrence of the deoxygenation at the active site of the gold nanoparticles on the Au/HT catalyst.
After completing the deoxygenation of styrene oxide, the reaction mixture was separated into the product phase and the aqueous phase containing the hydrophilic solid Au/HT. Styrene was then easily isolated by extraction using n-hexane. The recovered aqueous phase containing Au/HT was reusable without any loss of activity or selectivity during the recycling experiments (
Table 6, entries 2 and 3). The substrate scope of Au/HT for the deoxygenation of epoxides is exemplified in
Table 6 entries 7-12. Various epoxides were selectively deoxygenated into the corresponding alkenes at room temperature in excellent yields without the use of organic solvents. Notably,
trans-2,3-epoxy-3-phenyl-1-propanol selectively gave cinnamyl alcohol in excellent yield while maintaining the hydroxyl group, and the selectivity for cinnamyl alcohol is much higher in water than in organic solvents (entry 10 vs. entries 11 and 12). These phenomena are in sharp contrast with the results from our previously reported system of gold-catalyzed deoxygenation using alcohols [
42], where the hydroxyl group was not tolerated and cinnamaldehyde was the main product. We believe that a gold-hydride species generated in situ from the reaction of H
2O with CO is the active species for the deoxygenation of epoxides. [
48] The use of D
2O in place of H
2O under identical conditions significantly affected the reaction rate for the deoxygenation of styrene; a
kH/kD value of 3.9 was observed, supporting the theory that not only CO functions as a sole reductant but also water takes part in the deoxygenation.
To verify the above hypothesis, Fourier Transform Infrared (FT-IR) studies of Au/HT were carried out in the presence of CO and H
2O. When Au/HT was treated with CO and H
2O vapor at 298 K, a new band attributed to a gold-hydride species appeared at 1,750 cm
-1 [
55,
56], while no peak was observed at 1,750 cm
-1 when Au/HT was treated with CO. Furthermore, the band attributed to the gold-hydride species gradually disappeared after exposure to styrene oxide vapor.
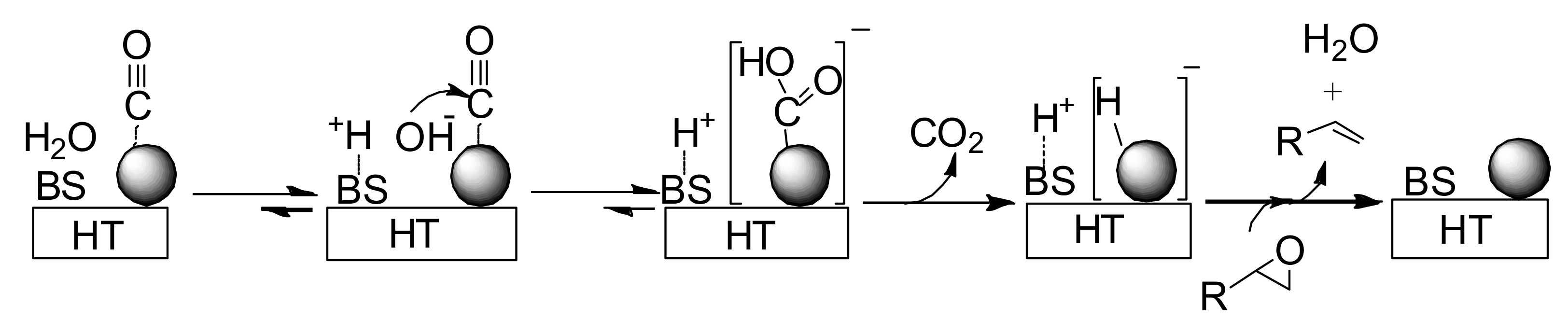
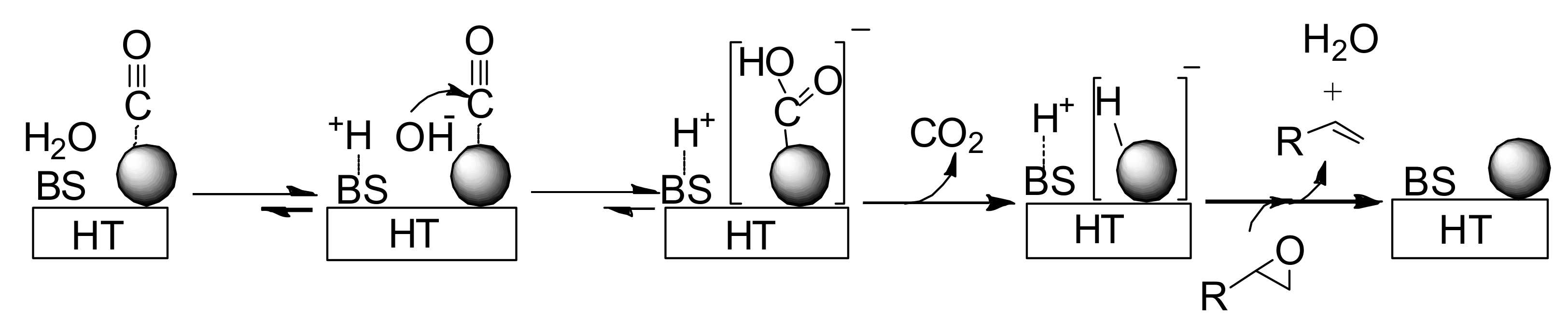
Our proposed reaction pathway can be seen in
Scheme 6. A basic site on HT surface presumably plays an important role in facilitating formation of the gold-hydride species through nucleophilic attack of OH
- on a gold-CO species followed by decarboxylation [
57,
58,
59]. The heterolytic hydrogen species generated on Au/HT then deoxygenates the epoxide to form the corresponding alkene and water.
3.3. Deoxygenation of Nitro Compounds Catalyzed by Ag/HT Using CO/H2O
We next succeeded in extending our deoxygenation methodology using Olympic medal metal NP catalysts with CO/H
2O as a reductant to the selective deoxygenation of nitro compounds. Namely, Ag/HT functioned as an effective heterogeneous catalyst for the quantitative chemoselective reduction of various nitroaromatics to the corresponding anilines with >99% selectivity, even in the presence of a C=C double bond. This methodology completely suppresses the reduction of C=C double bonds during the reduction of nitro functionalities [
60].
Aniline derivatives are valuable intermediates in the production of agrochemicals, pharmaceuticals and dyes [
61]. Although the reduction of aromatic nitro compounds is the most straightforward method for the synthesis of the corresponding anilines [
62], it is difficult to reduce only the nitro functionality in the presence of other reducible substituents [
63]. The chemoselective reduction of nitro compounds bearing C=C bonds to the corresponding anilines has been achieved using a large excess of stoichiometric reducing agents such as Fe, Sn, Zn and NaS
2O
4 [
64]. These reaction systems, however, have typically suffered from the production of harmful wastes, the need for neutralization of acid additives used as a hydrogen source and low atom efficiencies. To date, TiO
2-supported gold NPs (Au/TiO
2) may be the best heterogeneous catalyst for the chemoselective reduction of the nitro functionality under an H
2 atmosphere [
65].
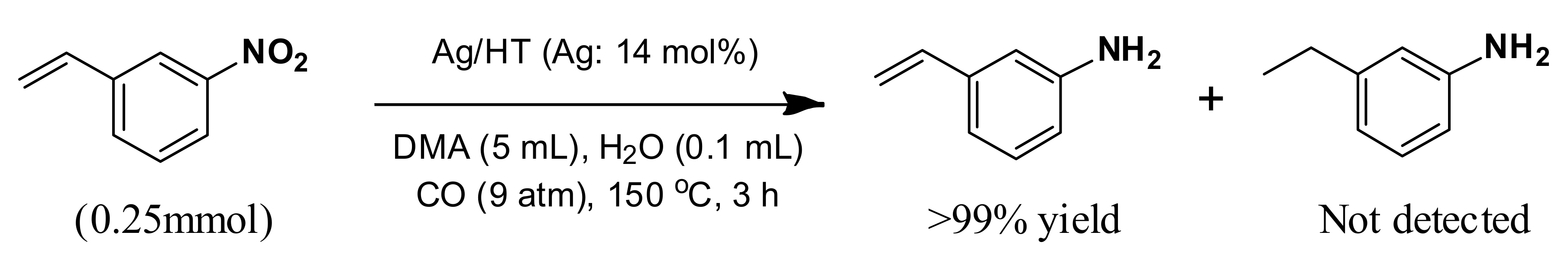
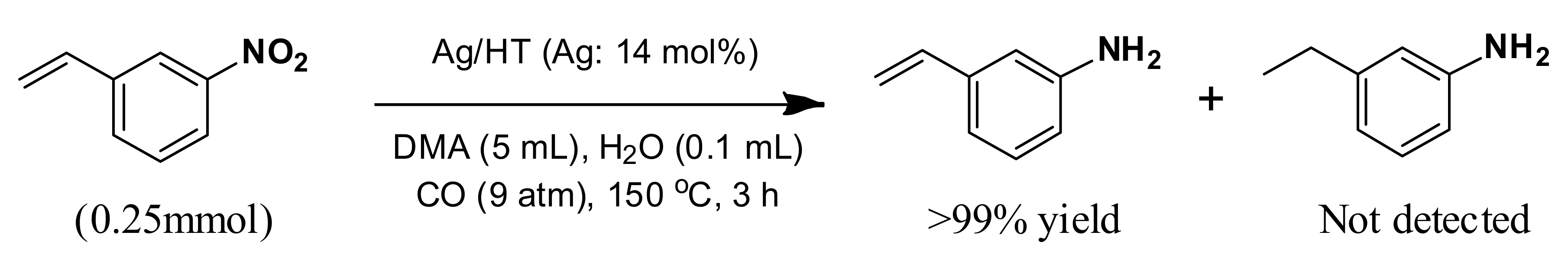
With our system, 3-nitrostyrene and Ag/HT were mixed in DMA solvent under 9 atm of CO at 150 °C in the presence of water. 3-Aminostyrene was formed in over 99% yield in 3 h without production of any reduction products of the C=C double bond such as 3-ethylaniline or 3-ethylnitrobenzene (
Scheme 7).
The time courses for the reduction of 3-nitrostyrene using Ag/HT and other HT-supported metal nanoparticles (Au/HT, Pd/HT, Pt/HT and Rh/HT) are shown in
Figure 1.
Although the activity of the Au/HT catalyst was higher than that of Ag/HT, an over reduction to 3-ethylaniline was observed, resulting in lower selectivity toward 3-aminostyrene. Pt/HT and Rh/HT functioned as good catalysts, giving 3-aminostyrene in moderate yields with high selectivity, while Pd/HT did not exhibit high activity or chemoselectivity. These results indicate that Ag/HT gives the highest yield of 3-aminostyrene. Notably, the C=C bond of the product 3-aminostyrene was maintained intact even after the complete conversion of 3-nitrostyrene. This chemoselectivity of Ag/HT towards 3-aminostyrene in the reduction of 3-nitrostyrene is greater than that of previously reported catalyst systems [
66,
67,
68,
69,
70,
71,
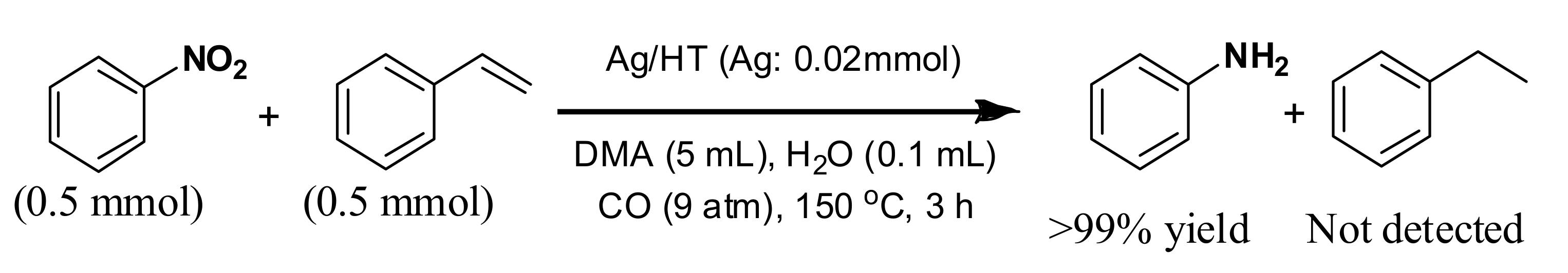
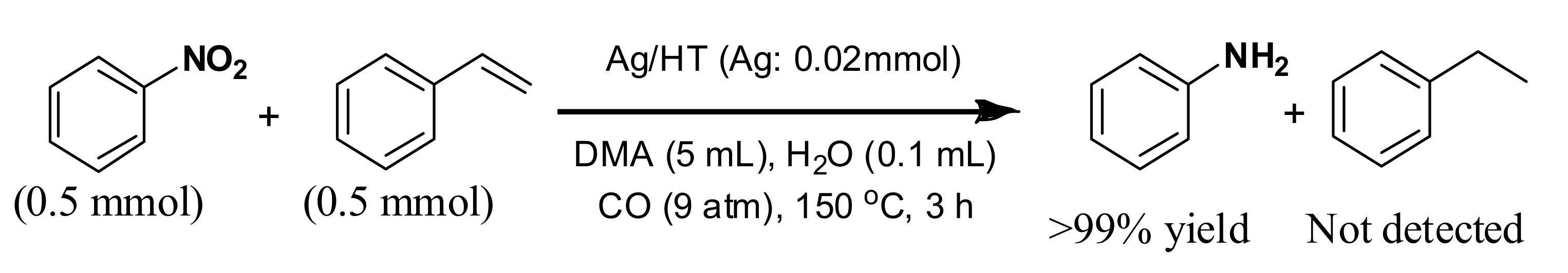
72]. The high chemoselectivity of Ag/HT for nitro functionalities was further investigated in the intermolecular competitive reaction of nitrobenzene and styrene (
Scheme 8).
Interestingly, nitrobenzene was reduced to give aniline in over 99% yield, but styrene was not reduced at all. These results clearly demonstrate that the Ag/HT catalyst system using CO/H2O shows complete chemoselectivity for nitro functionalities in the presence of inter- and intra-molecular olefinic functionalities.
This result is in sharp contrast with that obtained for a previously reported Au/TiO
2 catalyst system in which reduction of styrene did occur in the competitive reduction of nitrobenzene and styrene [
69]. Furthermore, the Ag/HT catalyst was applicable to the reduction of various nitro compounds bearing C=C double bonds, such as 4-nitrostyrene, 4-nitrostilbene, 1-nitro-4-propenyl benzene and 5-nitro-indole; the corresponding anilines were obtained in high yields with over 99% selectivity (
Table 7).
In separate experiments under identical conditions without 3-nitrostyrene, H
2 was not generated from the water-gas shift reaction. Moreover, when H
2 was used as a reducing agent instead of CO/H
2O, selective reduction of 3-nitrostyrene to 3-aminostyrene did not occur. These results rule out the participation of H
2 in the Ag/HT-catalyzed reduction reaction described above. [
73,
74] In-situ silver hydride species generated from the reaction of H
2O with CO at the surface of the Ag nanoparticles may be the active species that leads to highly chemoselective reduction of nitroaromatics with suppression of the reduction of the olefinic bond.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









