Synthesis and In Vitro Evaluation of N-(Bromobut-3-en-2-yl)-7-methoxy-1,2,3,4-tetrahydroacridin-9-amine as a Cholinesterase Inhibitor with Regard to Alzheimer's Disease Treatment

 ,
, 
Abstract
:1. Introduction
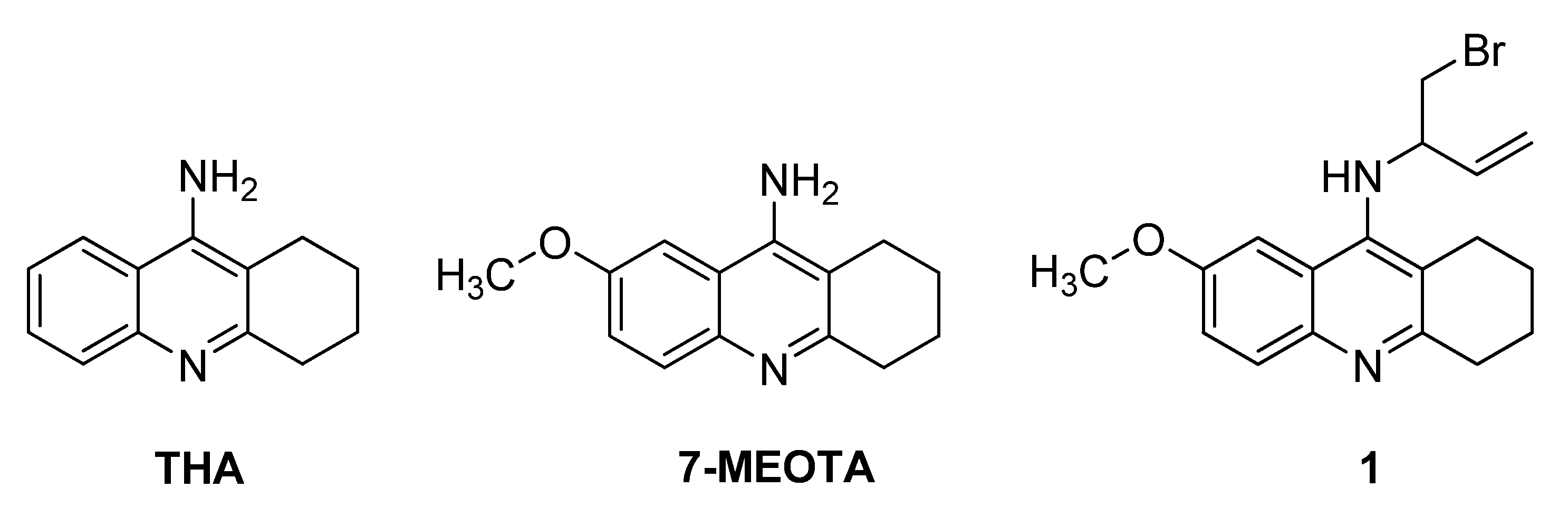
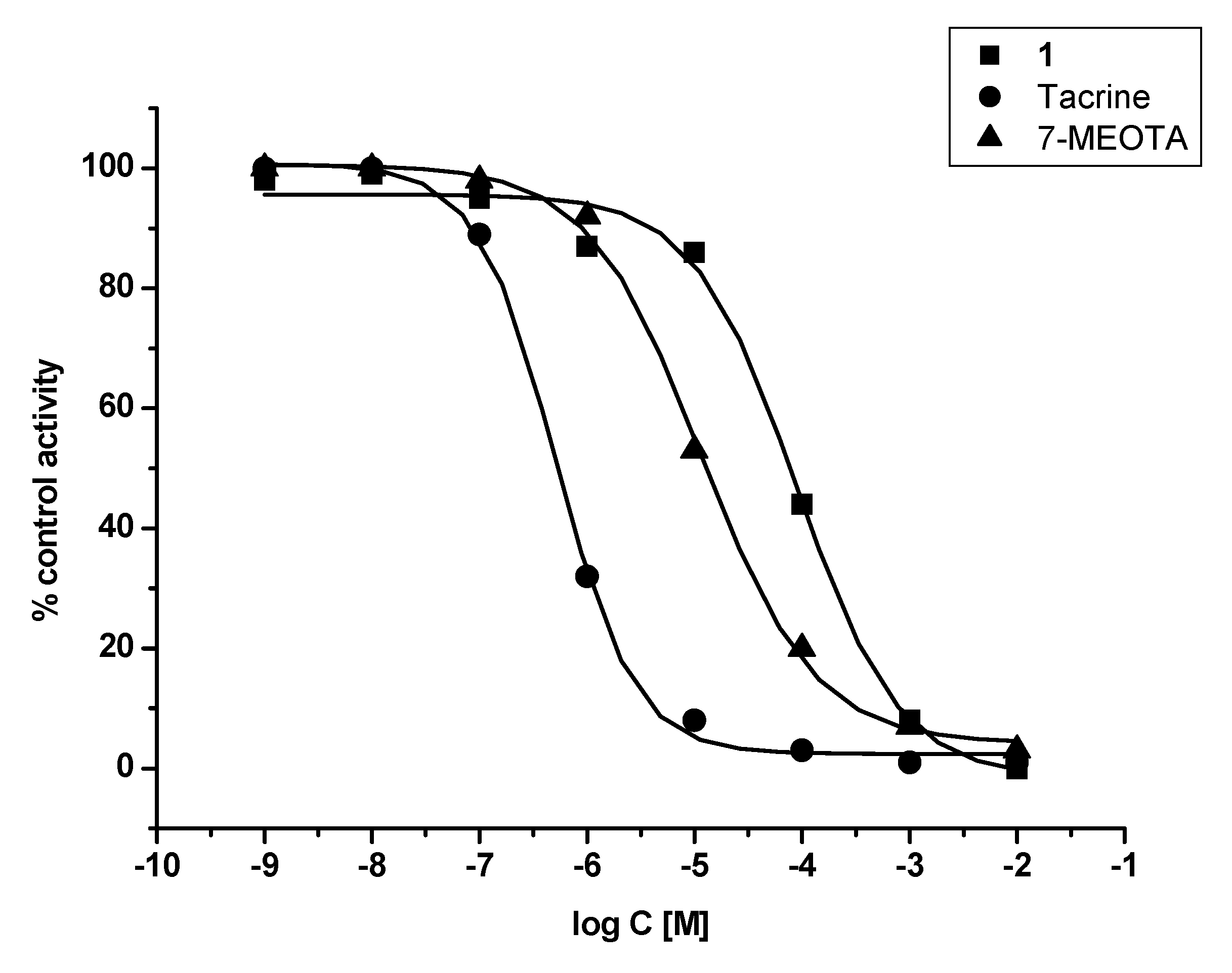
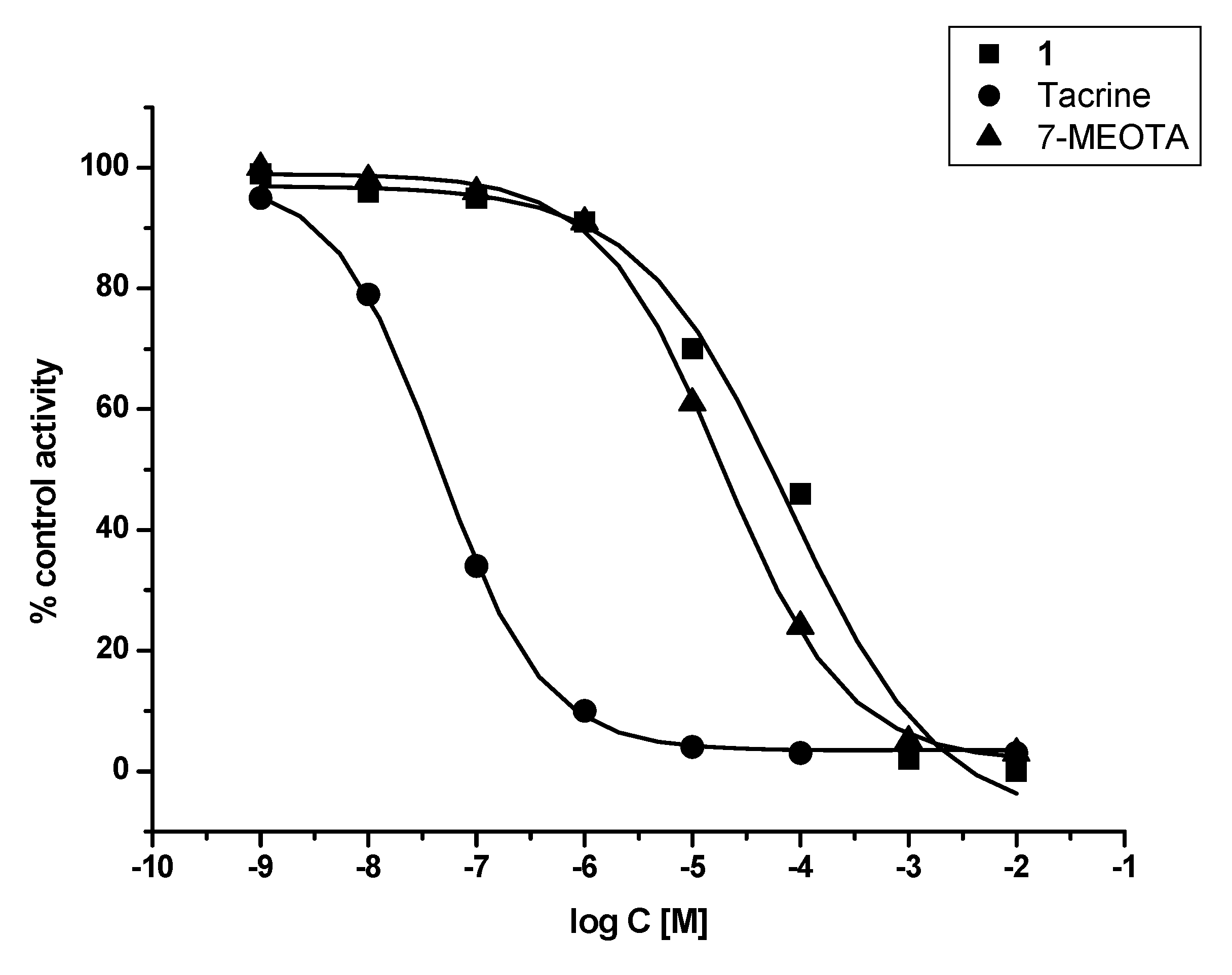
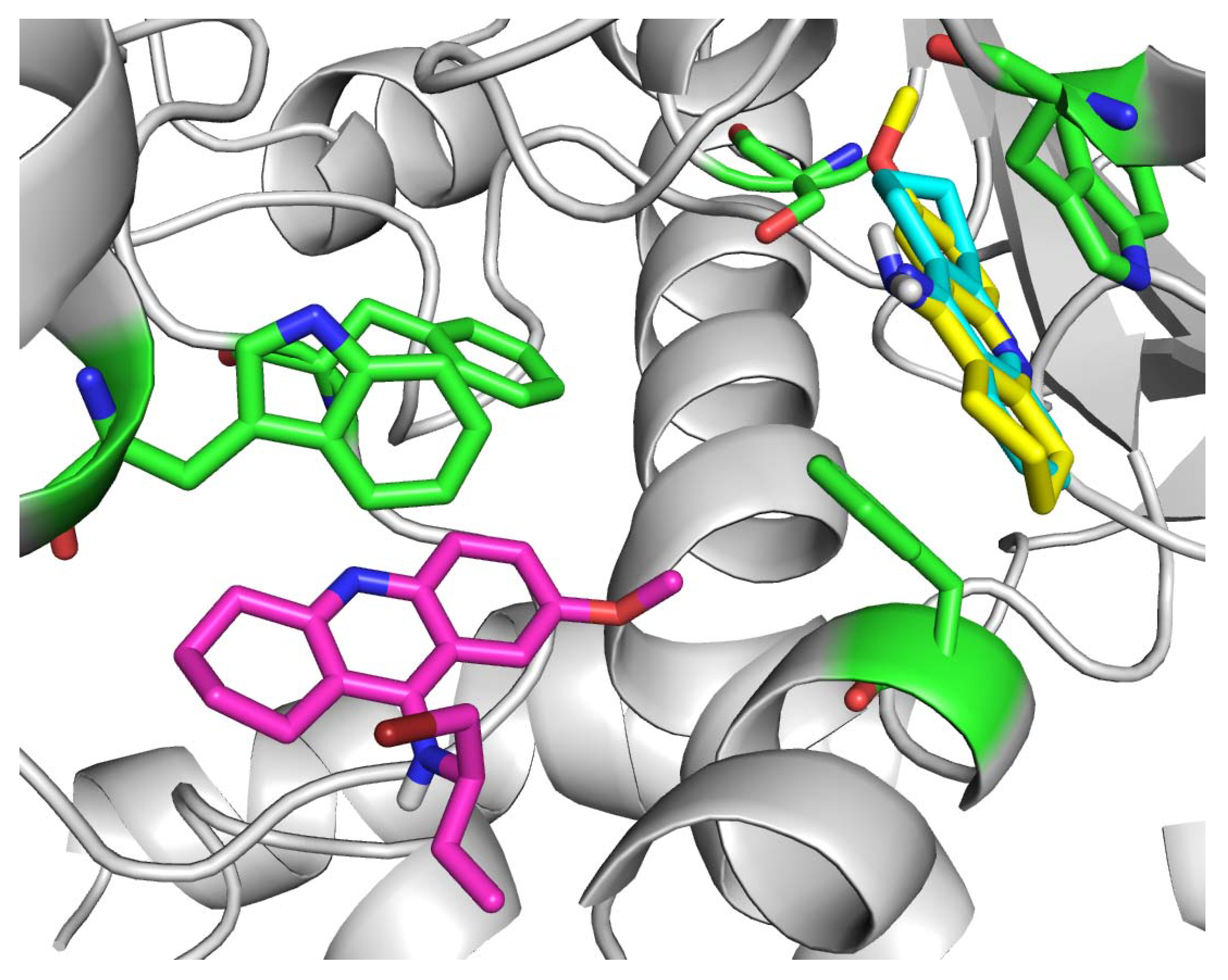
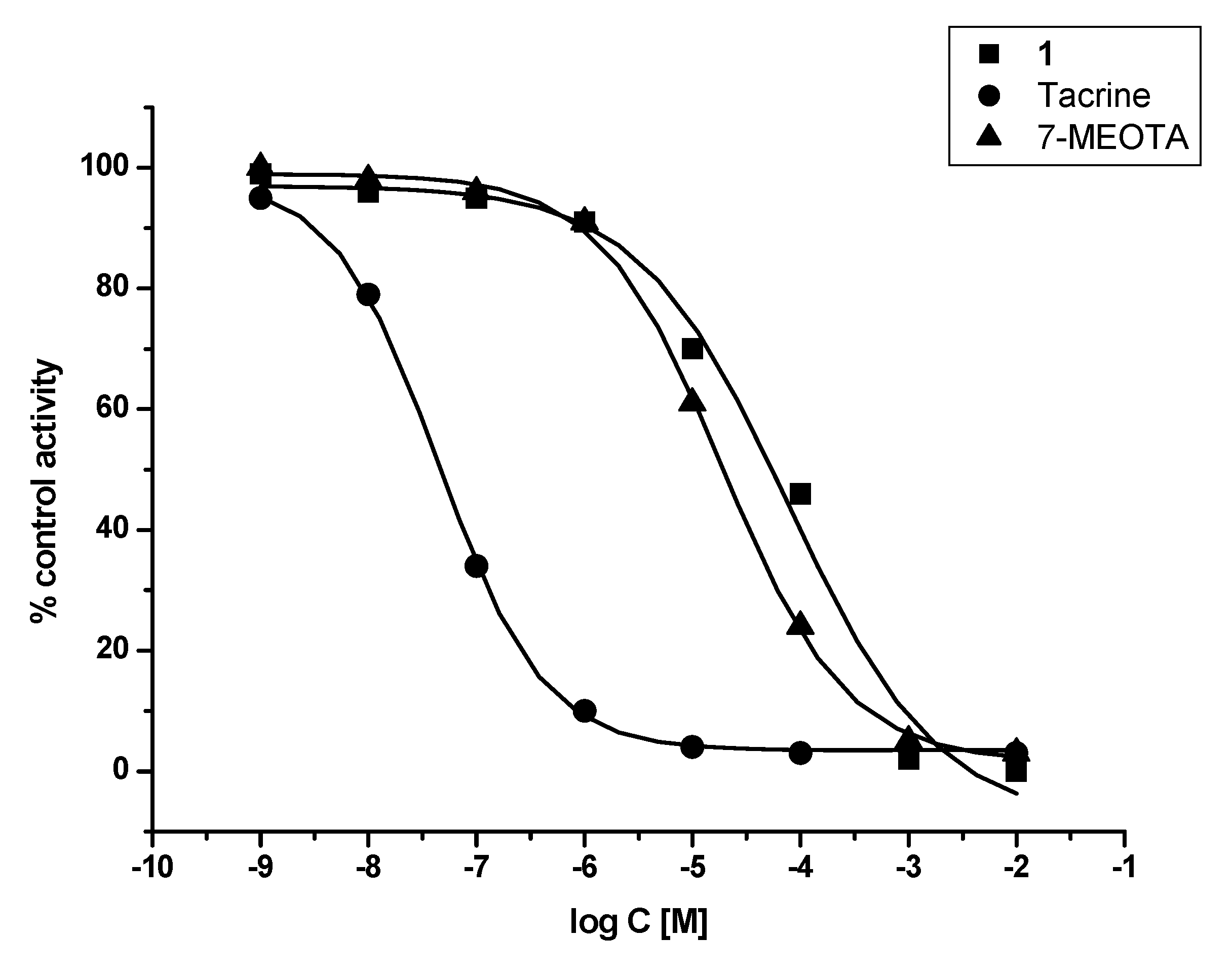
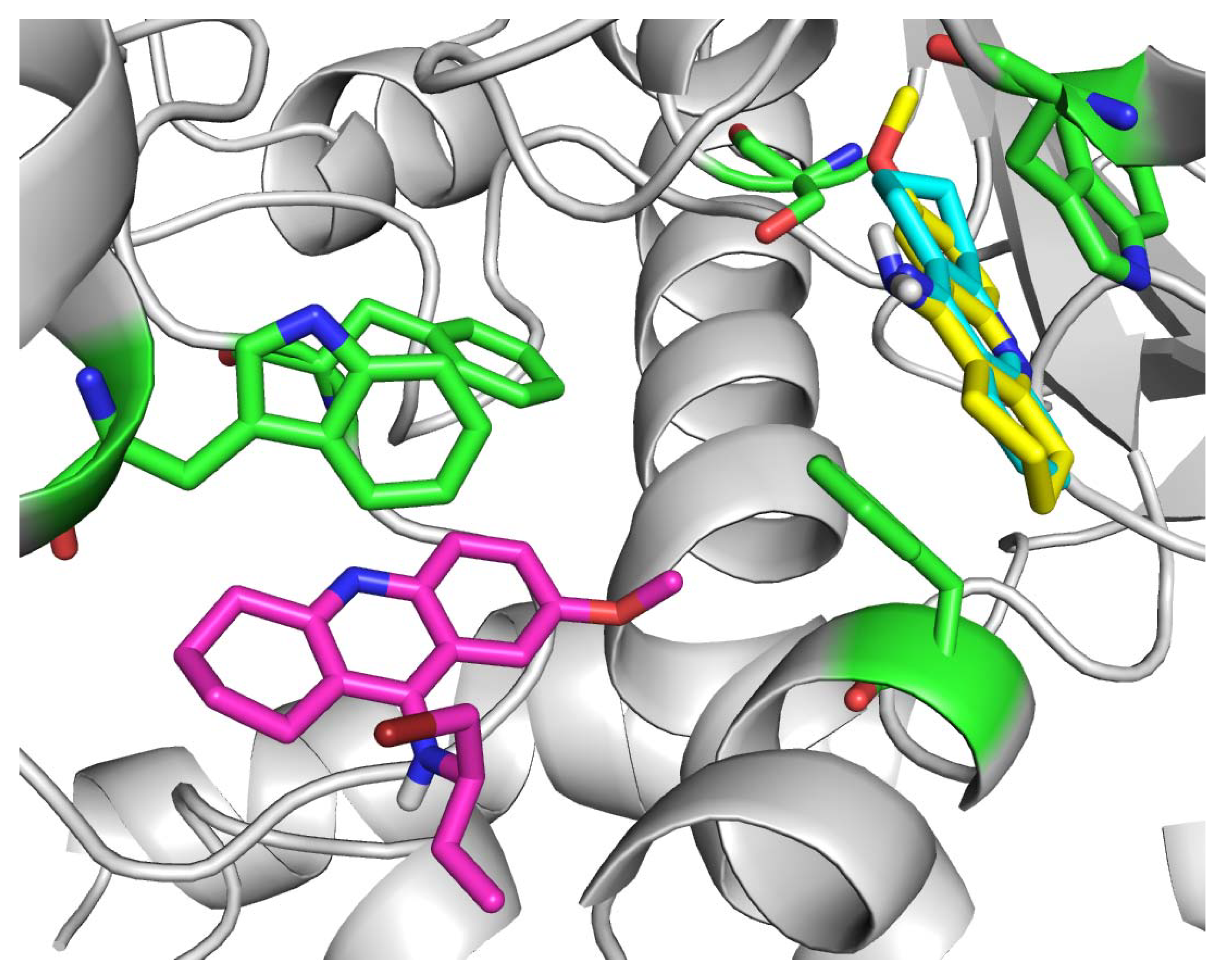
2. Results and Discussion
3. Experimental
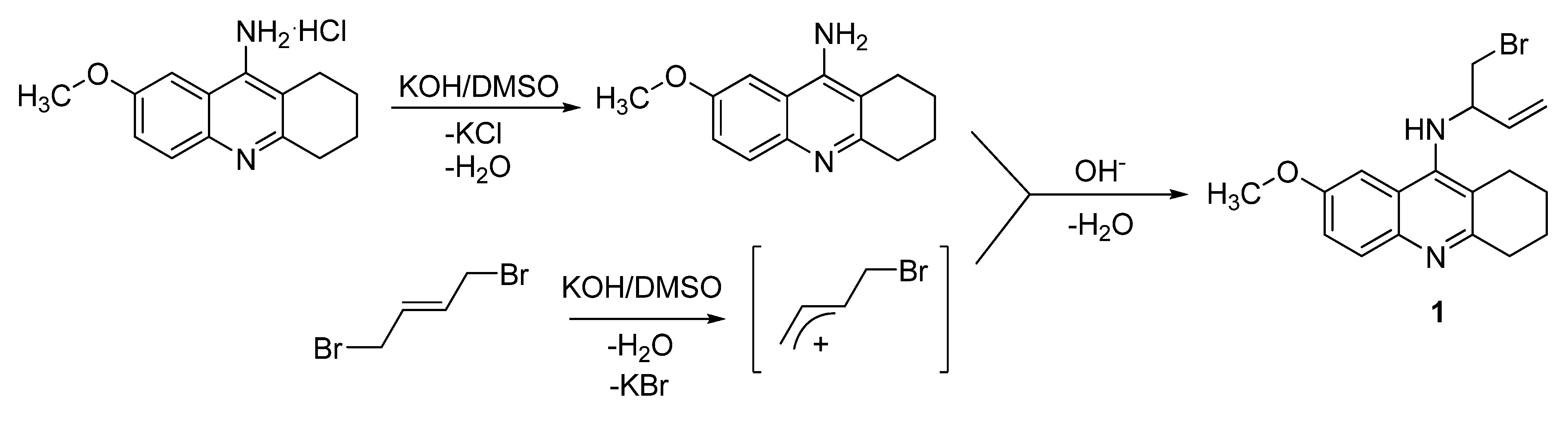
3.1. Chemistry
3.2. In vitro evaluation
3.3. Synthesis of N-(bromobut-3-en-2-yl)-7-methoxy-1,2,3,4-tetrahydroacridin-9-amine (1)
3.4. Molecular docking
4. Conclusion
Acknowledgements
References
- Selkoe, D.J. Alzheimer's disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 2004, 44, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J.F. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Perry, E.K.; Perry, R.H.; Blessed, G.; Tomlinson, B.E. Necropsy evidence of central cholinergic deficits in senile dementia. Lancet 1997, 1, 189. [Google Scholar] [CrossRef]
- Summers, W.K.; Majowski, L.V.; Marsh, G.M.; Tachiki, K.; Kling, A. Oral tetrahydroacridineamine in long term treatment of senile dementia, Alzheimer type. N. Engl. J. Med. 1986, 315, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.B.; Zimmerman, H.J.; Knapp, M.J.; Gracon, S.I.; Lewis, K.W. Hepatotoxic effects of tacrine administration in patients with Alzheimer-disease. Jama-J. Am. Med. Assoc. 1994, 271, 992–998. [Google Scholar] [CrossRef]
- Marx, J.L. Alzheimer's drug trial put on hold. Science 1987, 238, 1041–1042. [Google Scholar] [CrossRef] [PubMed]
- Ames, D.J.; Bhathal, P.S.; Davies, B.M.; Fraser, J.R.E. Hepatotoxicity of tetrahydroacridine. Lancet 1988, 1, 887. [Google Scholar] [CrossRef]
- Patocka, J.; Jun, D.; Kuca, K. Possible role of hydroxylated metabolites of tacrine in drug toxicity and therapy of Alzheimer's disease. Curr. Drug Met. 2008, 9, 332–335. [Google Scholar] [CrossRef]
- Filip, V.; Vachek, J.; Albrecht, V.; Dvorak, I.; Dvorakova, J.; Fusek, J.; Havluj, J. Pharmacokinetics and tolerance of 7-methoxytacrine following the single dose administration in healthy-volunteers. Int. J. Clin. Pharm. Ther. Toxicol. 1991, 29, 431–436. [Google Scholar]
- Korabecny, J.; Holas, O.; Musilek, K.; Pohanka, M.; Opletalova, V.; Dohnal, V.; Kuca, K. Synthesis and In Vitro Evaluation of New Tacrine Derivates-Bis-Alkylene Linked 7-MEOTA. Lett. Org. Chem. 2010, 7, 327–331. [Google Scholar] [CrossRef]
- Korabecny, J.; Musilek, K.; Holas, O.; Binder, J.; Zemek, F.; Marek, J.; Pohanka, M.; Opletalova, V.; Dohnal, V.; Kuca, K. Synthesis and in vitro evaluation of N-alkyl-7-methoxytacrine hydrochlorides as potential cholinesterase inhibitors in Alzheimer disease. Bioorg. Med. Chem. Lett. 2010, 20, 6093–6095. [Google Scholar] [CrossRef] [PubMed]
- McLennan, D.J. A revised transition state spectrum for concerted bimolecular β-eliminations. Tetrahedron 1975, 31, 2999. [Google Scholar] [CrossRef]
- Harel, M.; Schalk, I.; Ehret-Sabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef] [PubMed]
- Patocka, J. Anticholinesterase activity of 9-amino-7-methoxy-1,2,3,4-tetrahydroacridine and some derivatives and analogues. Sbornik Ved. Prac. VLVDU Hradec Kralove 1986, 102, 123–140. [Google Scholar]
- Pohanka, M.; Jun, D.; Kuca, K. Improvement of acetylcholinesterase-based assay for organophosphates in way of identification by reactivators. Talanta 2008, 77, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.-P.; Hong, F.; Quiram, P.; Jelacic, T.; Brimijon, S. Synthesis of alkylene linked bis-THA and alkylene linked benzyl- THA as highly potent and selective inhibitors and molecular probes of acetylcholinesterase. J. Chem. Soc., Perkin Trans. 1 1997, 171–176. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Potein crystal structures are available online: http://www.pdb.org Accessed on 1st October 2010.
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera – A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System (2002). Available online: http://www.pymol.org/ Accessed on 1 October 2010.
Sample Availability: Samples of the compounds THA, 7-MEOTA and 1 are available from authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | hAChE IC50 ± SD (µM) | hBChE IC50 ± SD (µM) | SI hBChE/hAChE |
|---|---|---|---|
| THA | 0.5 ± 0.1 | 0.02 ± 0.003 | 0.05 |
| 7-MEOTA | 15.0 ± 2.4 | 21.0 ± 3.4 | 1.4 |
| 1 | 89.1 ± 17.3 | 78.8 ± 12.8 | 0.88 |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Korabecny, J.; Musilek, K.; Holas, O.; Nepovimova, E.; Jun, D.; Zemek, F.; Opletalova, V.; Patocka, J.; Dohnal, V.; Nachon, F.; et al. Synthesis and In Vitro Evaluation of N-(Bromobut-3-en-2-yl)-7-methoxy-1,2,3,4-tetrahydroacridin-9-amine as a Cholinesterase Inhibitor with Regard to Alzheimer's Disease Treatment. Molecules 2010, 15, 8804-8812. https://doi.org/10.3390/molecules15128804
Korabecny J, Musilek K, Holas O, Nepovimova E, Jun D, Zemek F, Opletalova V, Patocka J, Dohnal V, Nachon F, et al. Synthesis and In Vitro Evaluation of N-(Bromobut-3-en-2-yl)-7-methoxy-1,2,3,4-tetrahydroacridin-9-amine as a Cholinesterase Inhibitor with Regard to Alzheimer's Disease Treatment. Molecules. 2010; 15(12):8804-8812. https://doi.org/10.3390/molecules15128804
Chicago/Turabian StyleKorabecny, Jan, Kamil Musilek, Ondrej Holas, Eugenie Nepovimova, Daniel Jun, Filip Zemek, Veronika Opletalova, Jiri Patocka, Vlastimil Dohnal, Florian Nachon, and et al. 2010. "Synthesis and In Vitro Evaluation of N-(Bromobut-3-en-2-yl)-7-methoxy-1,2,3,4-tetrahydroacridin-9-amine as a Cholinesterase Inhibitor with Regard to Alzheimer's Disease Treatment" Molecules 15, no. 12: 8804-8812. https://doi.org/10.3390/molecules15128804
APA StyleKorabecny, J., Musilek, K., Holas, O., Nepovimova, E., Jun, D., Zemek, F., Opletalova, V., Patocka, J., Dohnal, V., Nachon, F., Hroudova, J., Fisar, Z., & Kuca, K. (2010). Synthesis and In Vitro Evaluation of N-(Bromobut-3-en-2-yl)-7-methoxy-1,2,3,4-tetrahydroacridin-9-amine as a Cholinesterase Inhibitor with Regard to Alzheimer's Disease Treatment. Molecules, 15(12), 8804-8812. https://doi.org/10.3390/molecules15128804