A Simplified Liquid Chromatography-Mass Spectrometry Assay for Artesunate and Dihydroartemisinin, Its Metabolite, in Human Plasma

Abstract
:1. Introduction
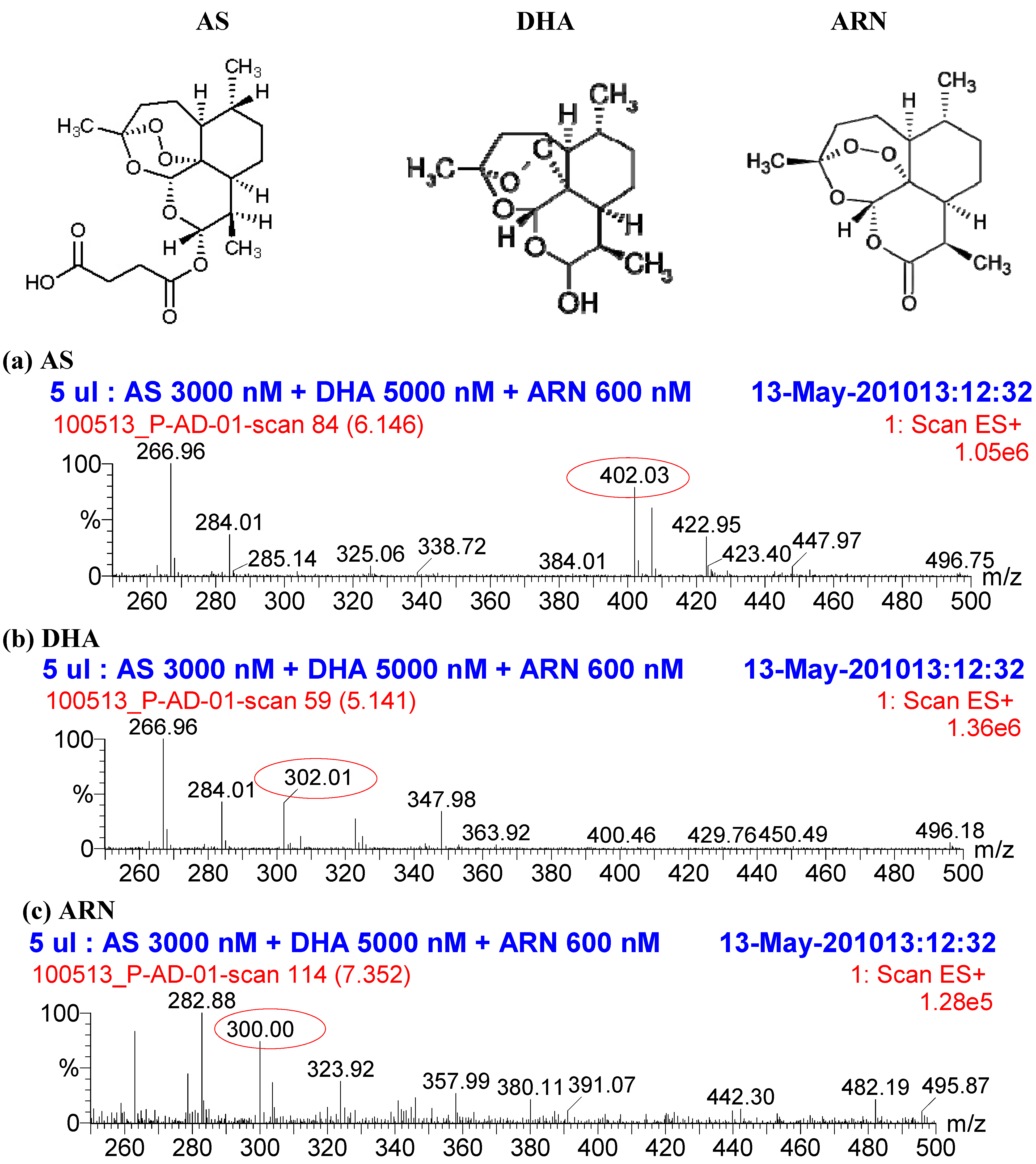
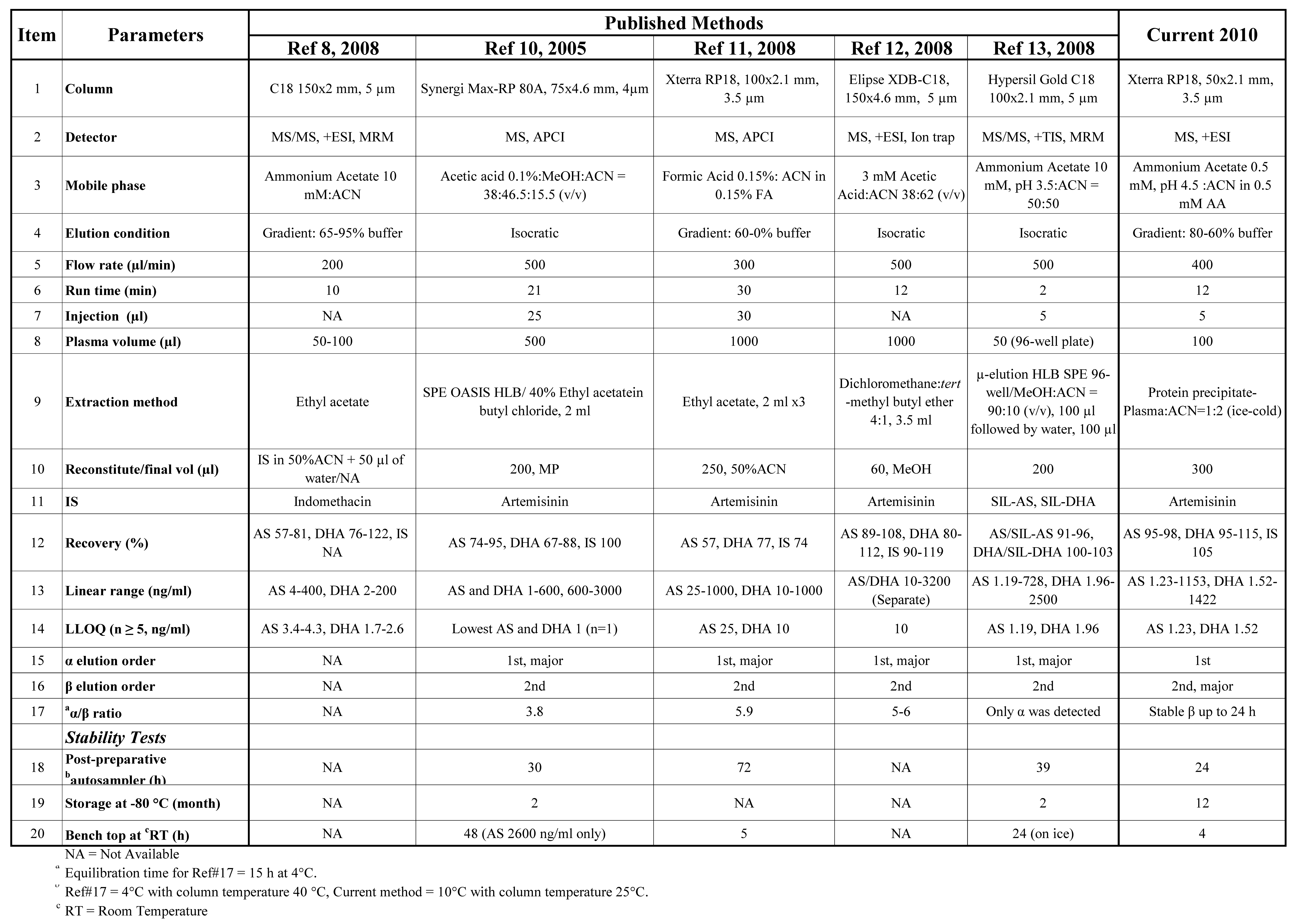
2. Results and Discussion
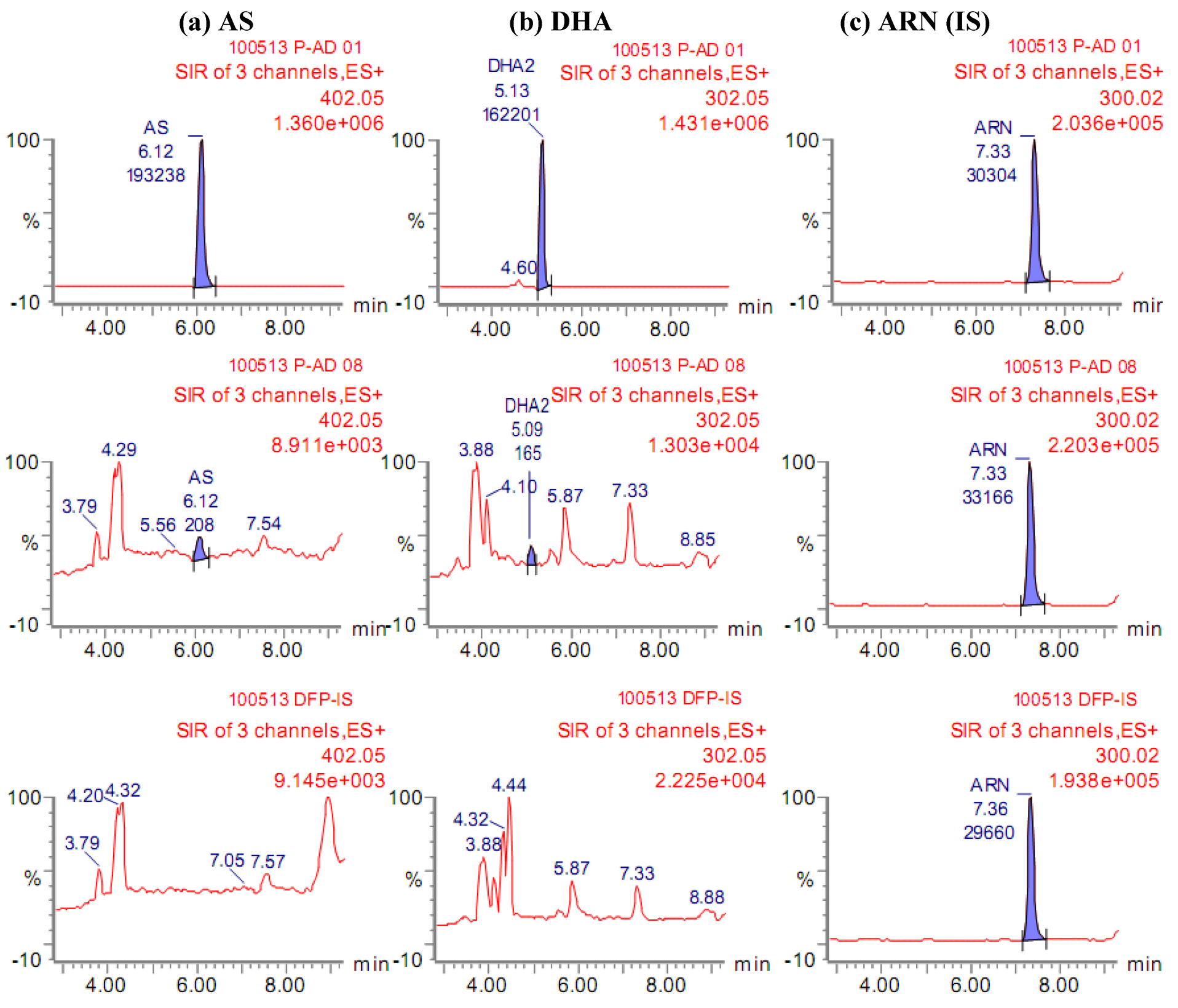
2.1. Quantification of AS and its metabolite DHA

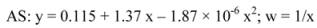
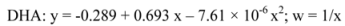
 )/DHA (
)/DHA (  ) at concentrations ranging from 3.20/5.33 to 3,000/5,000 nM. (b) Precision (%CV) and inaccuracy (%) data from back calculated concentrations of AS/DHA. Dotted lines indicate 15% CV and ±15% inaccuracy levels. Data from triplicate analyses on the same day are shown.
)/DHA ( ) at concentrations ranging from 3.20/5.33 to 3,000/5,000 nM. (b) Precision (%CV) and inaccuracy (%) data from back calculated concentrations of AS/DHA. Dotted lines indicate 15% CV and ±15% inaccuracy levels. Data from triplicate analyses on the same day are shown.
) at concentrations ranging from 3.20/5.33 to 3,000/5,000 nM. (b) Precision (%CV) and inaccuracy (%) data from back calculated concentrations of AS/DHA. Dotted lines indicate 15% CV and ±15% inaccuracy levels. Data from triplicate analyses on the same day are shown.
)/DHA ( ) at concentrations ranging from 3.20/5.33 to 3,000/5,000 nM. (b) Precision (%CV) and inaccuracy (%) data from back calculated concentrations of AS/DHA. Dotted lines indicate 15% CV and ±15% inaccuracy levels. Data from triplicate analyses on the same day are shown.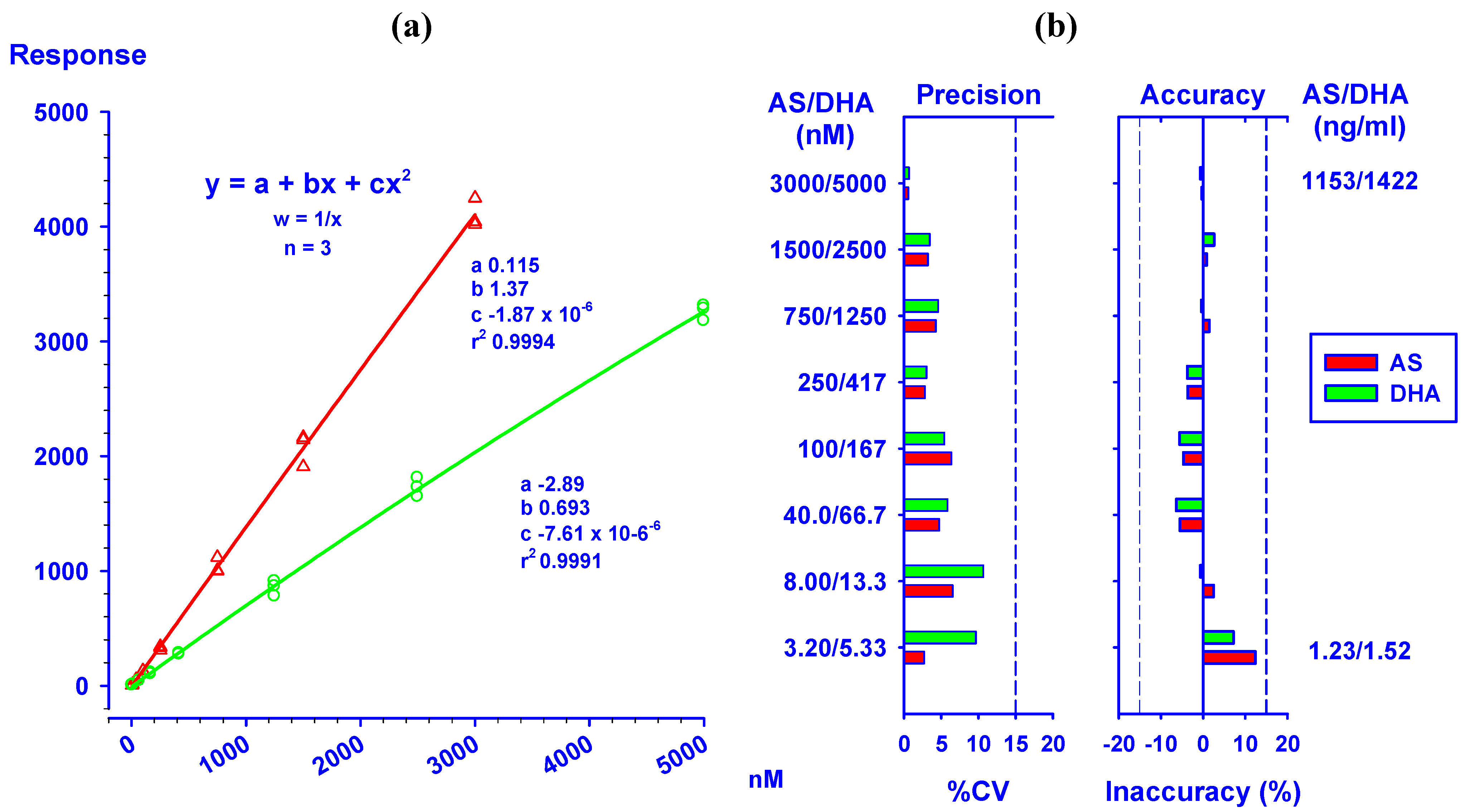

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Theo. Conc . | Found (n = 6) | Inaccuracy (%) | %CV | |||||
|---|---|---|---|---|---|---|---|---|
| AS | DHA | AS | DHA | AS | DHA | AS | DHA | |
| Intra-assay | ||||||||
| 1. LLOQ | 3.20 | 5.33 | 3.02 | 5.53 | -5.73 | 3.66 | 17.6 | 7.15 |
| 2. 3 x LLOQ | 10.0 | 16.0 | 10.7 | 16.3 | 7.12 | 1.71 | 2.76 | 5.29 |
| 3. Medium | 500 | 800 | 533 | 796 | 6.62 | -0.55 | 4.00 | 5.28 |
| 4. 0.8 x ULOQ | 2,500 | 4,000 | 2,481 | 3,810 | -0.77 | -4.74 | 1.68 | 3.11 |
| 5. ULOQ | 3,000 | 5,000 | 2,782 | 4,637 | -7.28 | -7.27 | 3.78 | 4.10 |
| Inter-assay | ||||||||
| 1. LLOQ | 3.20 | 5.33 | 3.28 | 5.65 | 2.36 | 5.93 | 5.44 | 10.2 |
| 2. 3 x LLOQ | 10.0 | 16.0 | 9.93 | 16.7 | -0.69 | 4.25 | 8.88 | 5.19 |
| 3. Medium | 500 | 800 | 496 | 785 | -0.76 | -1.82 | 7.98 | 5.87 |
| 4. 0.8 x ULOQ | 2,500 | 4,000 | 2,445 | 3,872 | -2.21 | -3.20 | 5.48 | 4.89 |
| 5. ULOQ | 3,000 | 5,000 | 2,912 | 4,966 | -2.93 | -0.68 | 5.03 | 5.24 |
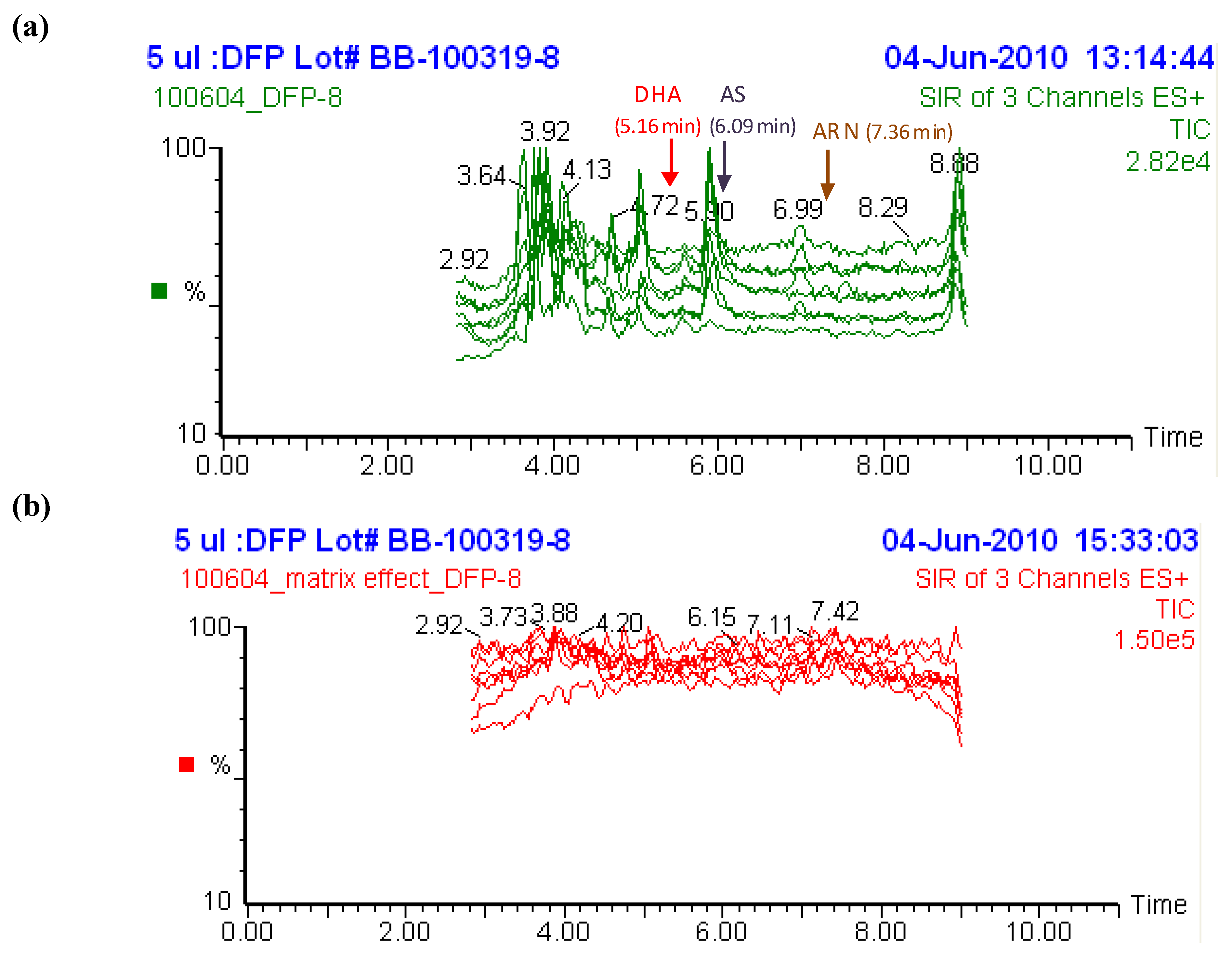
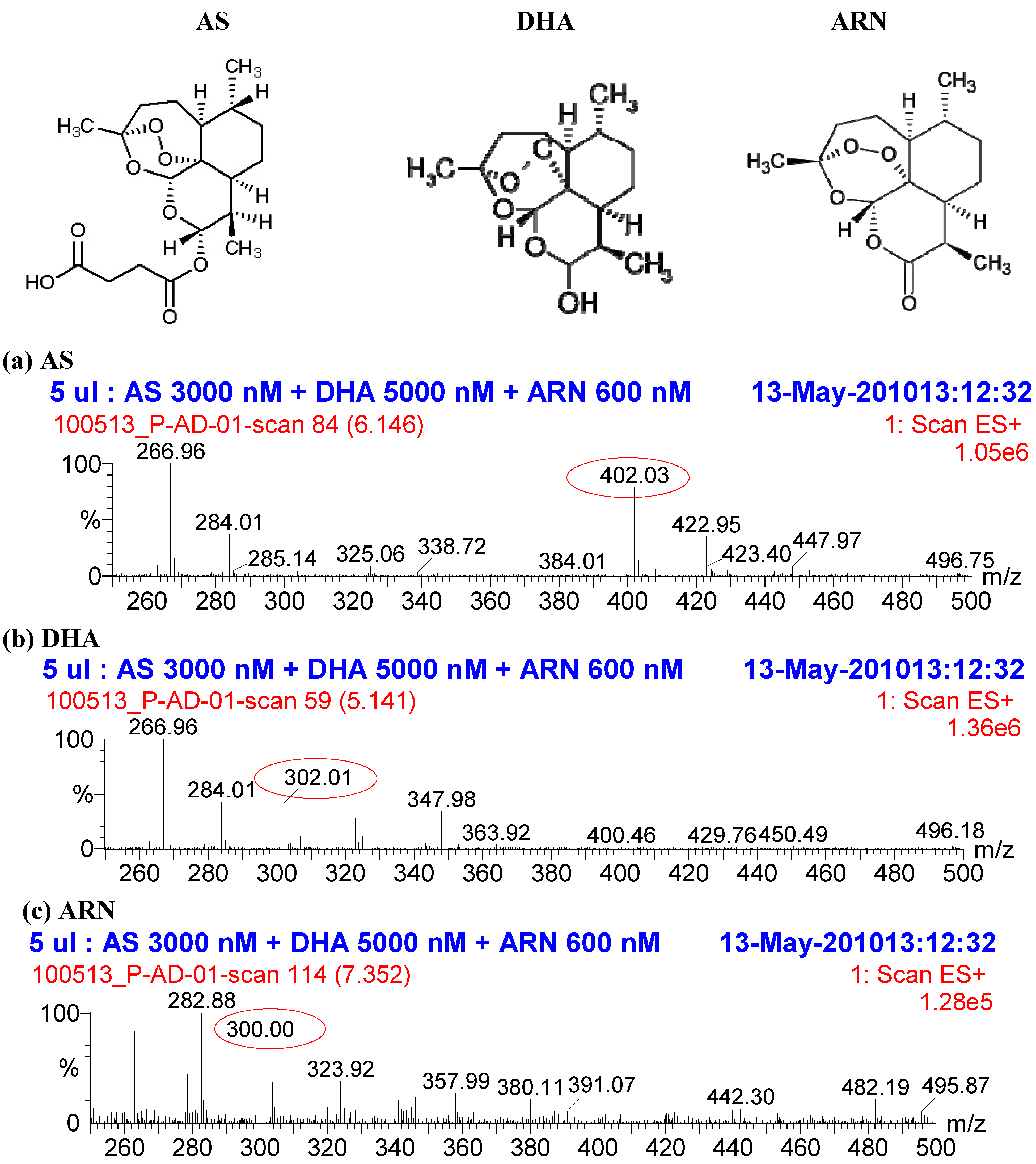
2.2. Selectivity of the assay and interference from other antimalarial drugs
2.3. Post-preparative stability test of DHA and AS
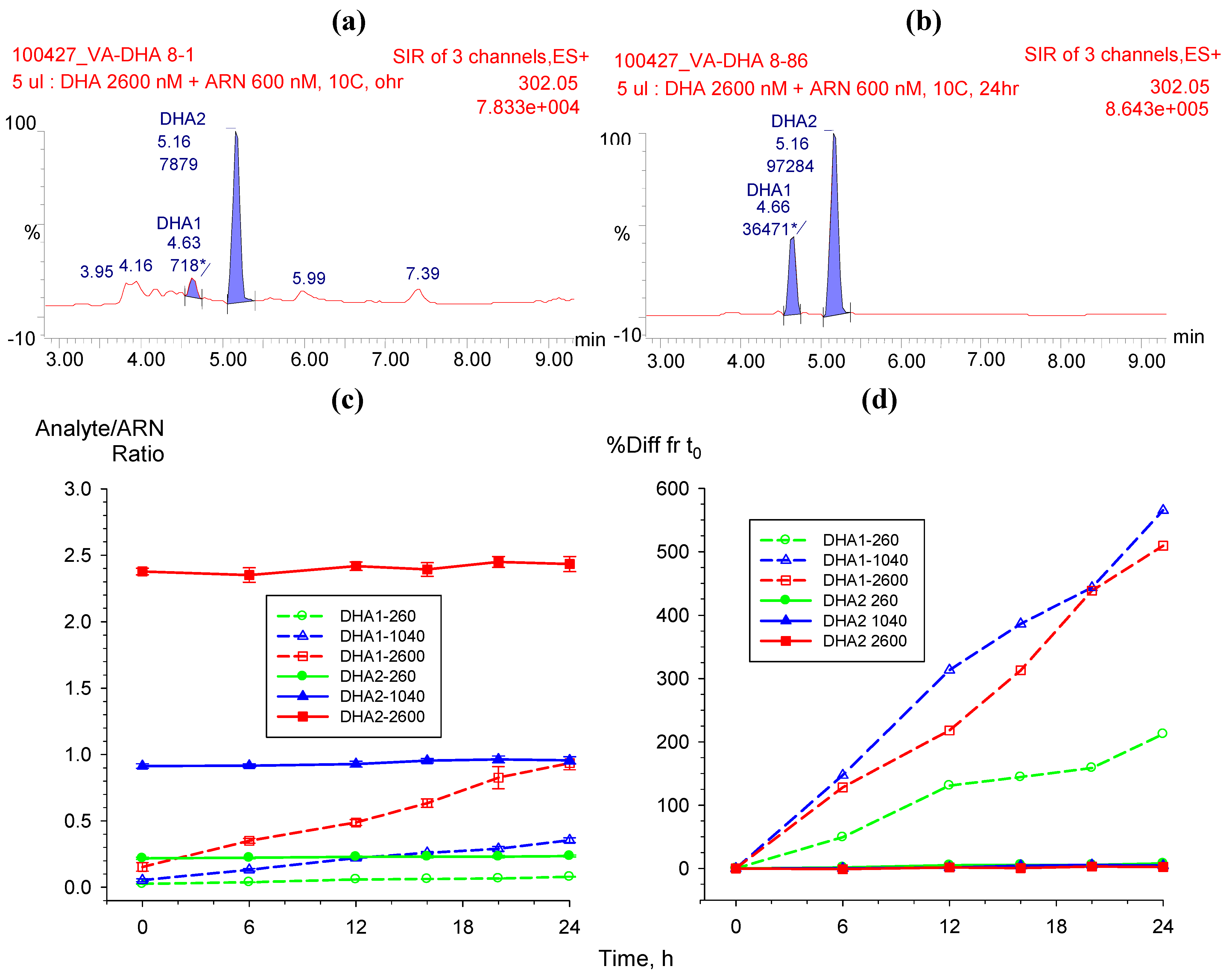
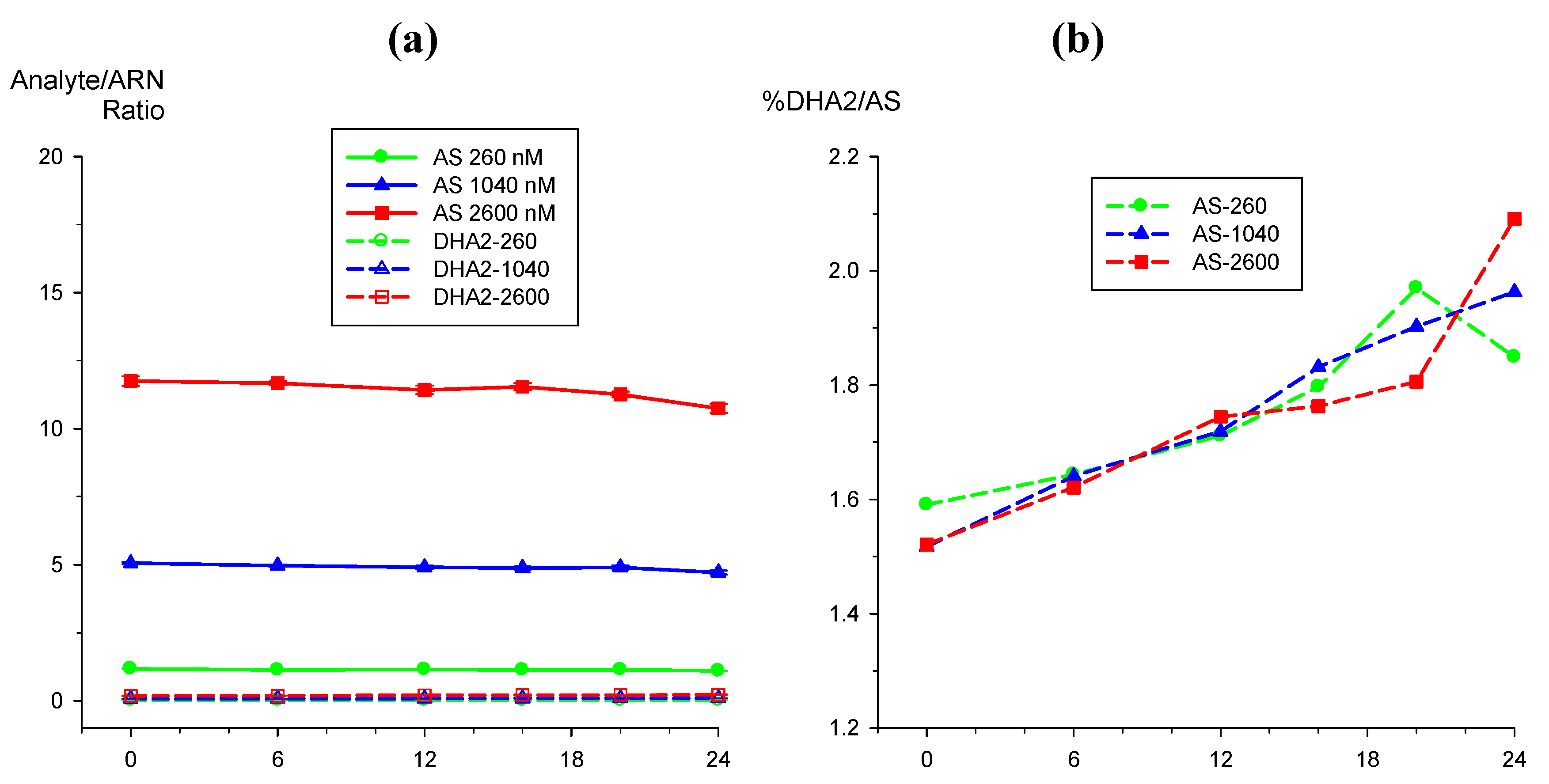
2.3.1. Dihydroartemisinin epimerization
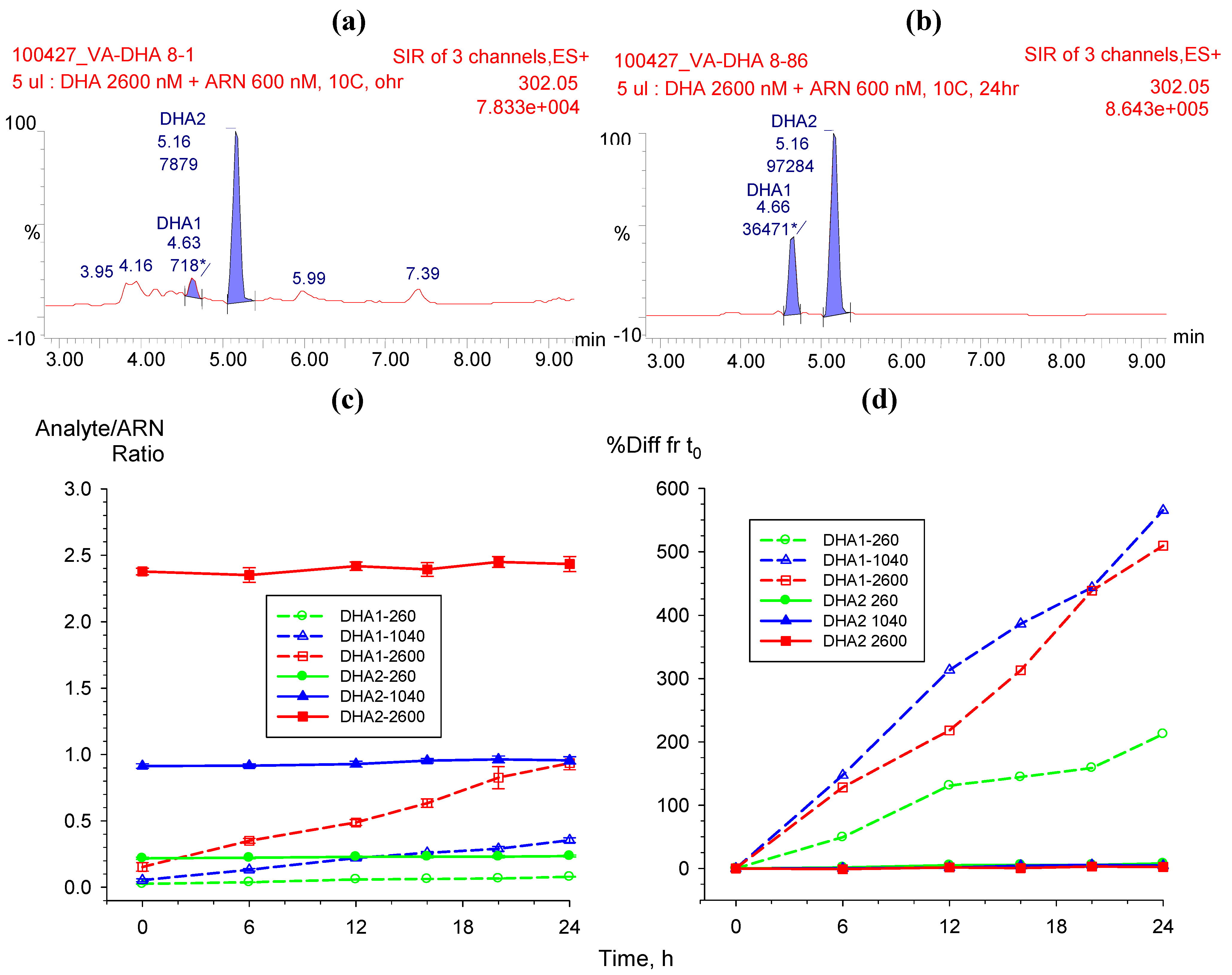
2.3.2. Hydrolysis of AS to DHA
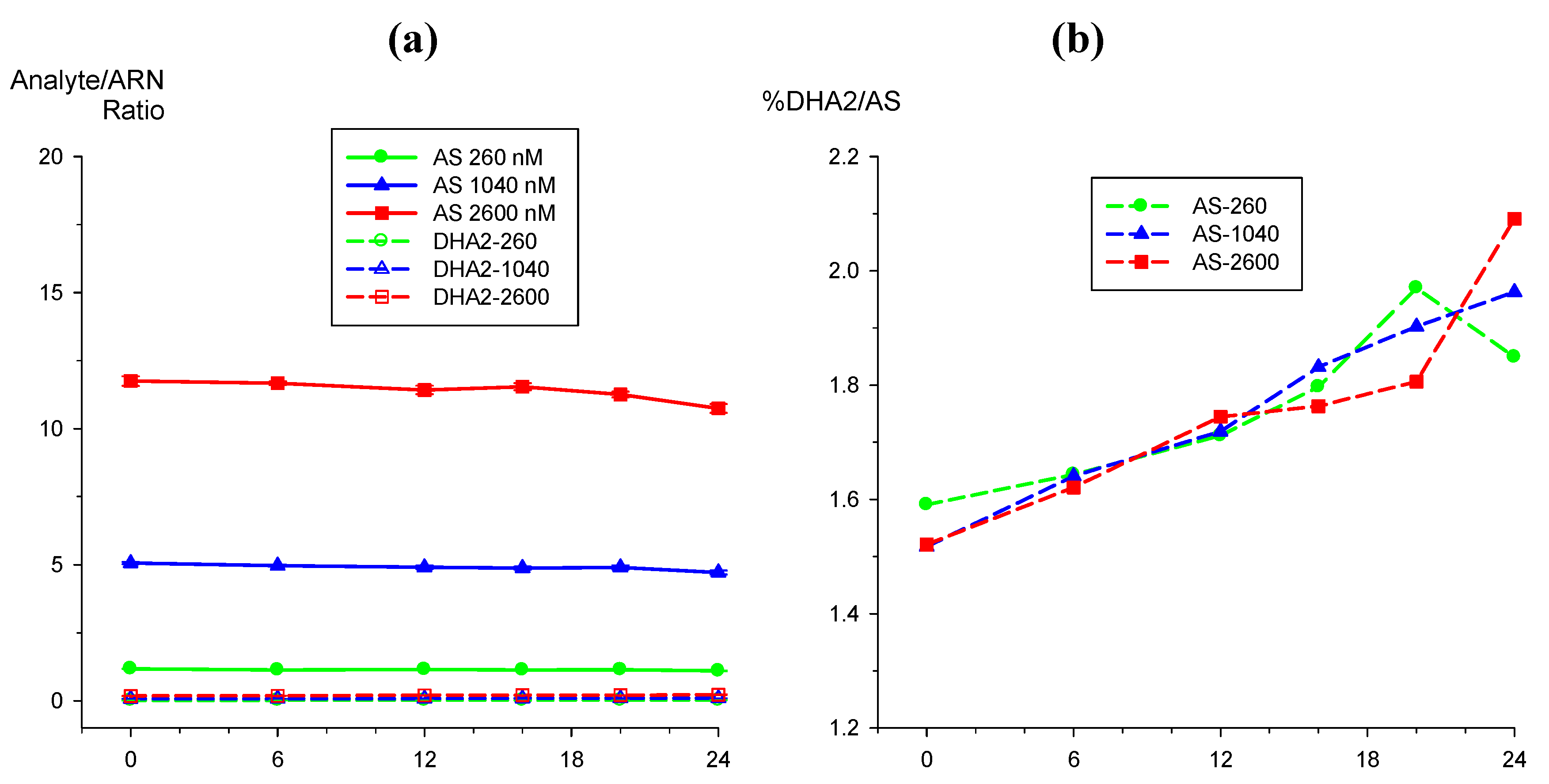
 |
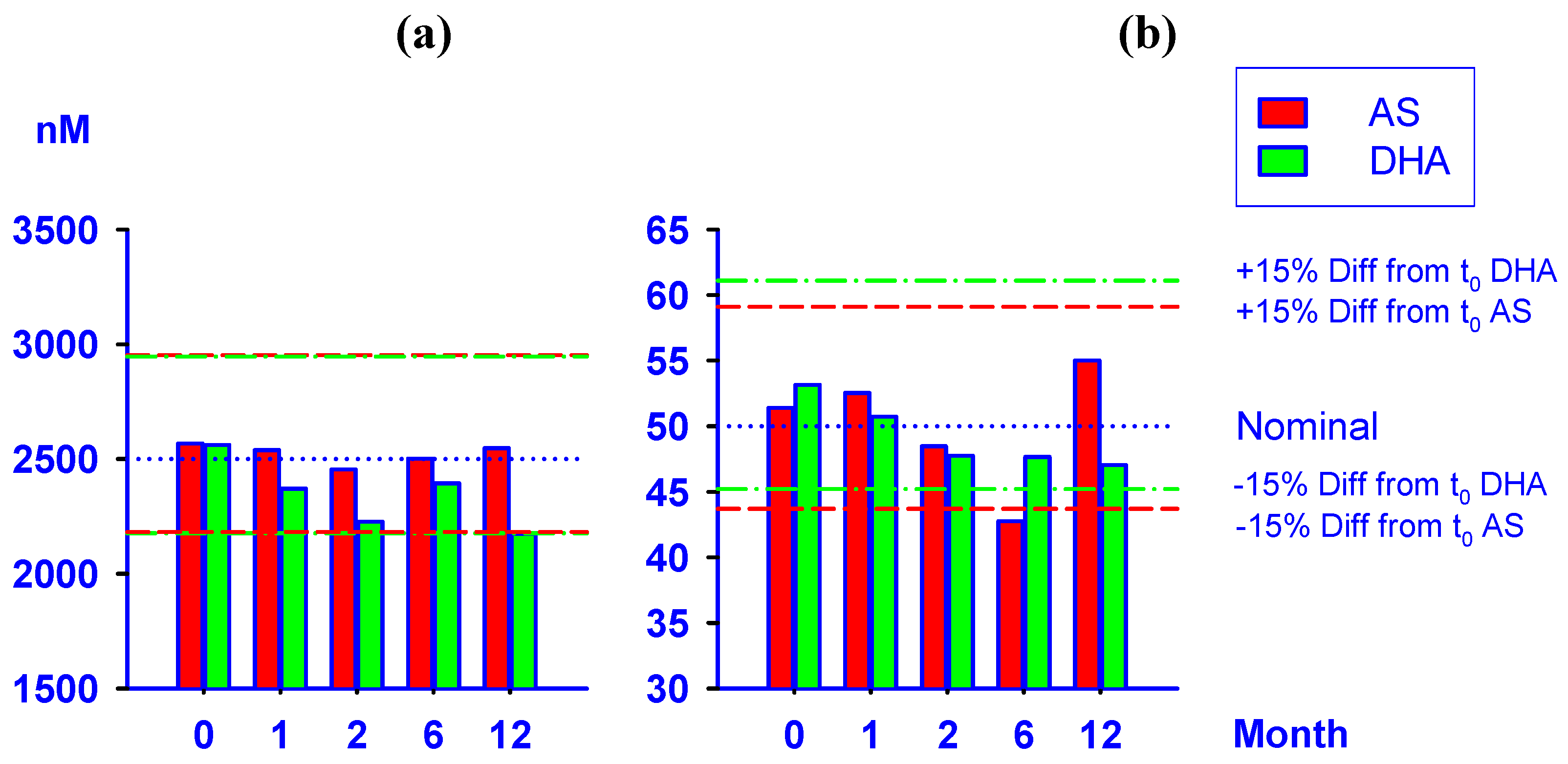
2.4. Stability test of AS and DHA in plasma samples
| 50 nM | 2500 nM | ||||
|---|---|---|---|---|---|
| AS | DHA | AS | DHA | ||
| Room Temperature (Fresh) | 0 h | 52.0 | 54.5 | 2490 | 2547 |
| (0.00, 1.15) | (0.00, 2.11) | (0.00, 13.8) | (0.00, 9.42) | ||
| 2 h | 46.2 | 50.2 | 2256 | 2292 | |
| (-11.2, 0.88) | (-1.67, 3.20) | (-9.40, 10.6) | (-10.0, 7.84) | ||
| 4 h | 44.1 | 46.9 | 2250 | 2243 | |
| (-15.3, 1.84) | (-8.14, 2.69) | (-9.64, 4.23) | (-11.9, 2.27) | ||
| 6 h | 44.5 | 46.4 | 2267 | 2279 | |
| (-14.4, 3.20) | (-9.06, 3.46) | (-8.96, 4.45) | (-10.5, 0.49) | ||
| Freeze/Thaw Cycles | 0 cycle | 47.0 | 50.0 | 2606 | 2467 |
| (0.00, 9.76) | (0.00, 9.96) | (0.00, 1.86) | (0.00, 1.33) | ||
| 3 cycles | 50.3 | 53.2 | 2392 | 2384 | |
| (7.01, 3.75) | (6.39, 0.99) | (-8.23, 0.79) | (-3.35, 1.34) | ||
| Storage Duration | 50 nM | 2500 nM | ||
|---|---|---|---|---|
| (month) | AS | DHA | AS | DHA |
| 0 (Fresh) | 51.4 | 53.2 | 2567 | 2561 |
| (0.00, 2.22) | (0.00, 3.74) | (0.00, 9.15) | (0.00, 6.07) | |
| 1 | 52.5 | 50.7 | 2539 | 2370 |
| (2.22, 11.2) | (-4.61, 10.3) | (-1.10, 8.52) | (-7.46, 7.85) | |
| 2 | 48.5 | 47.7 | 2454 | 2227 |
| (-5.66, 5.98) | (-10.2, 3.17) | (-4.40, 7.95) | (-13.1, 10.7) | |
| 6 | 42.7 | 47.7 | 2501 | 2393 |
| (-16.8, 2.17) | (-10.3, 9.22) * | (-2.56, 9.93) | (-6.58, 2.33) | |
| 12 | 55.0 | 47.0 | 2547 | 2174 |
| (7.04, 10.2) | (-11.5, 15.8) | (-0.80, 15.2) | (-15.1, 13.0) | |
2.5. Application to clinical sample analysis
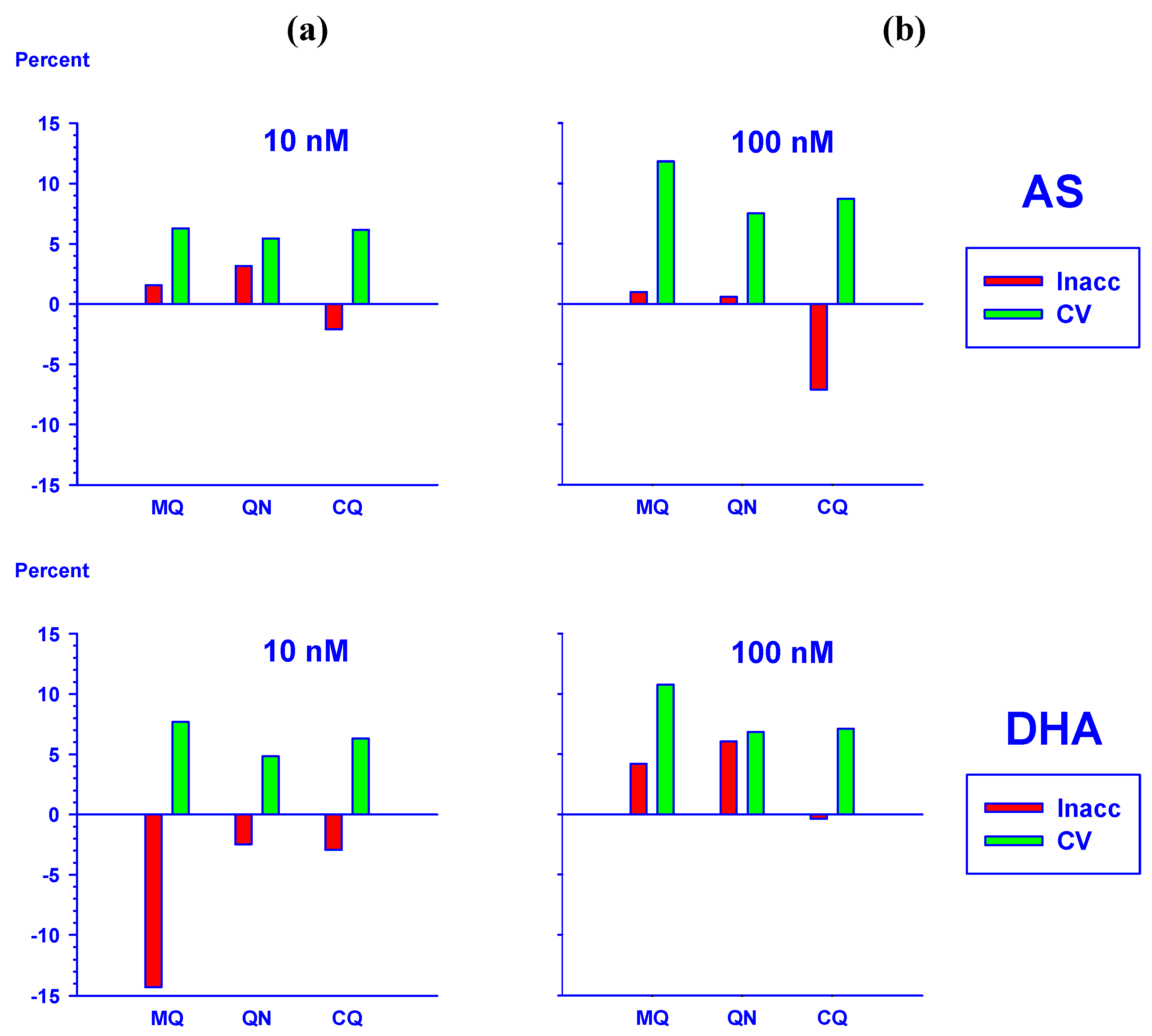
2.5.1. Effect of haemoglobin on the quantification of AS and DHA
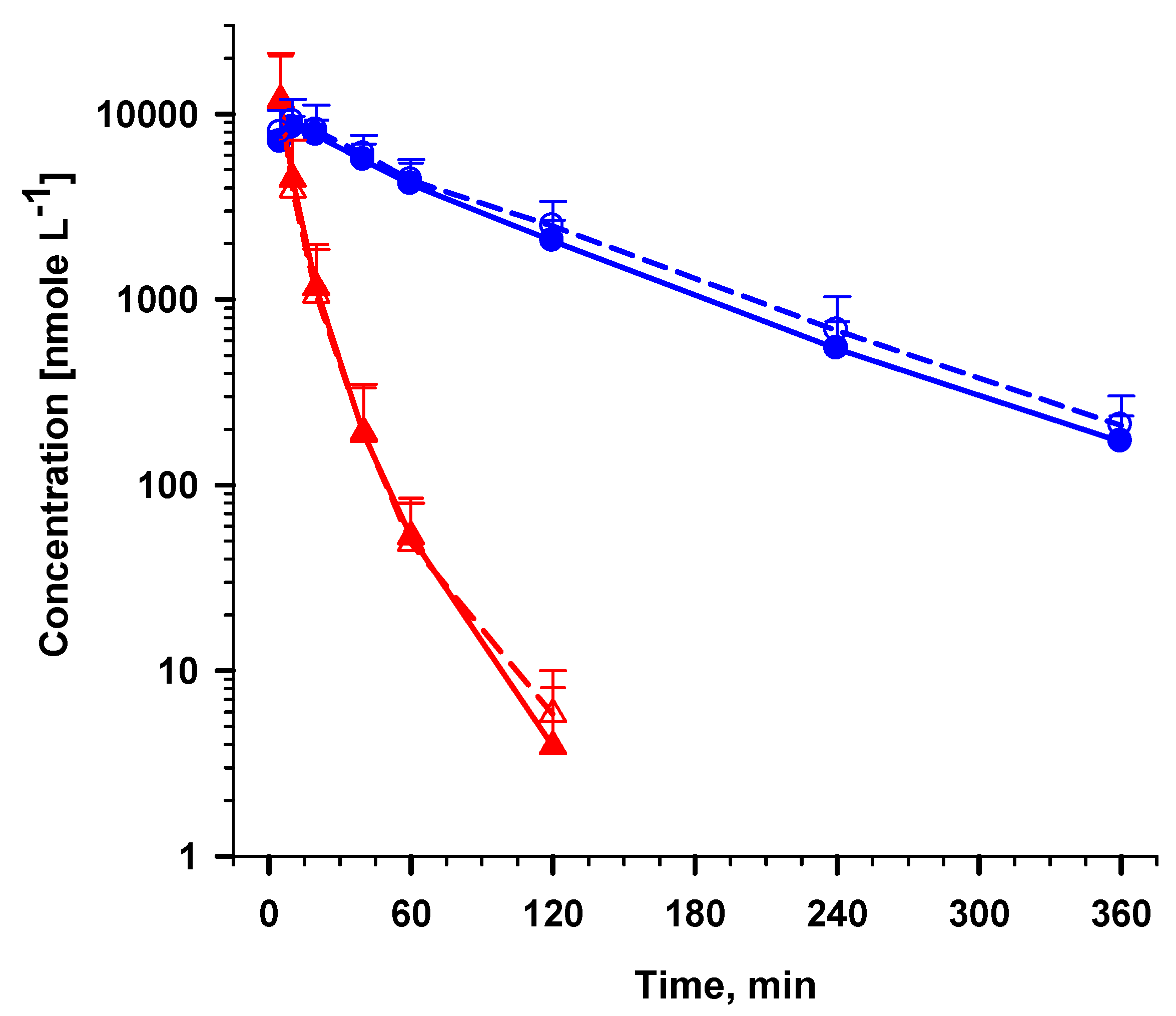
2.5.2. Analysis of AS and DHA in plasma samples from a clinical trial
) and DHA (  ) at (a) 50 nM and (b) 2,500 nM.
) and DHA ( ) at (a) 50 nM and (b) 2,500 nM.
) at (a) 50 nM and (b) 2,500 nM.
) and DHA ( ) at (a) 50 nM and (b) 2,500 nM. ) and DHA ( ) by external standard (solid lines) method compared to internal standard method using ARN as IS (broken lines). Values are mean + SD.
) and DHA ( ) by external standard (solid lines) method compared to internal standard method using ARN as IS (broken lines). Values are mean + SD.
) and DHA ( ) by external standard (solid lines) method compared to internal standard method using ARN as IS (broken lines). Values are mean + SD.
) and DHA ( ) by external standard (solid lines) method compared to internal standard method using ARN as IS (broken lines). Values are mean + SD.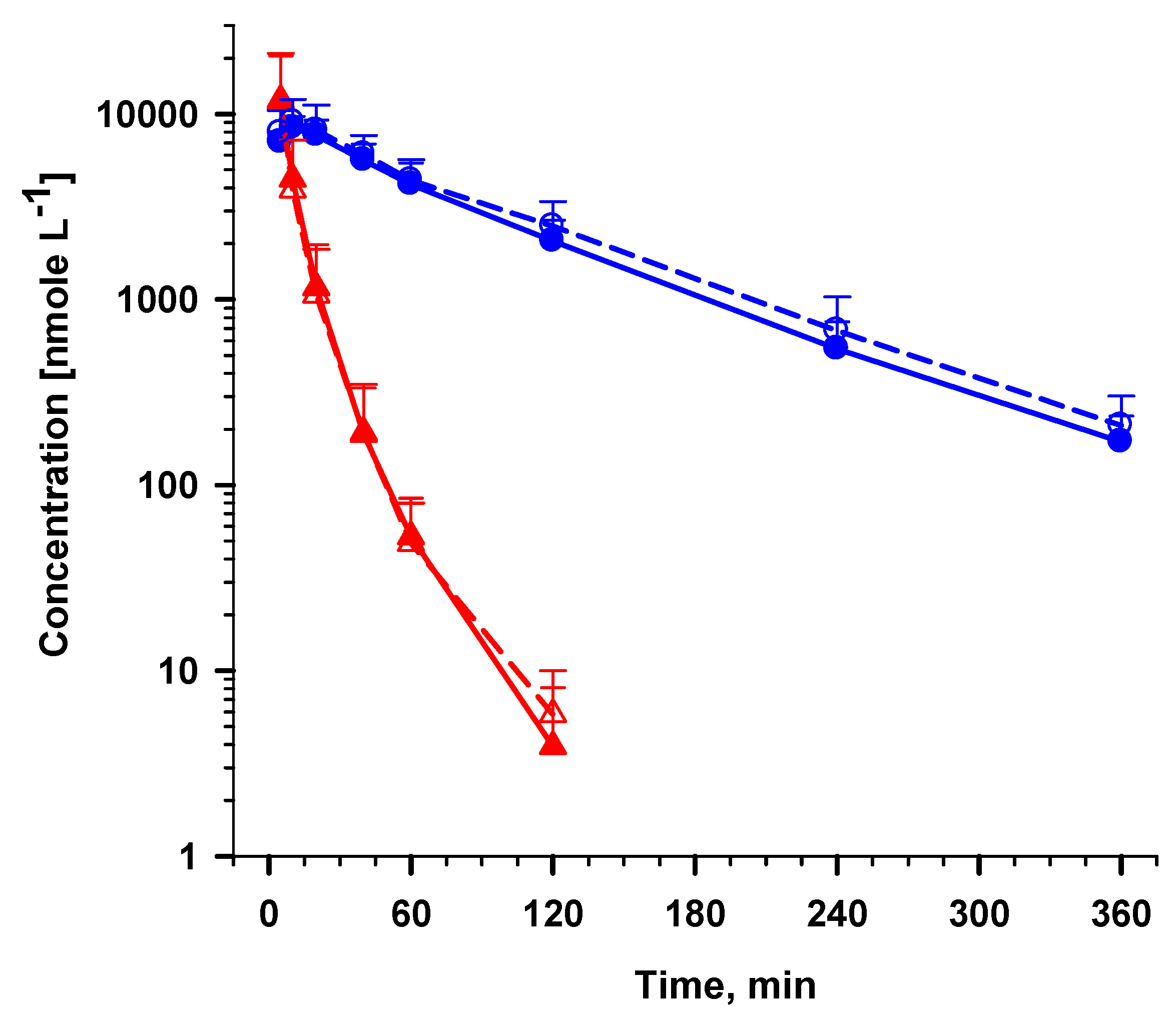
3. Conclusions
4. Experimental
4.1. Reagents
4.2. Instrumentation/Equipments
4.3. Standards
4.3.1. Standard curves
4.3.2. Quality controls
4.3.3. Internal standard
4.4. Sample preparation
4.5. LC-MS conditions
4.6. Quantification method
4.7. Validation
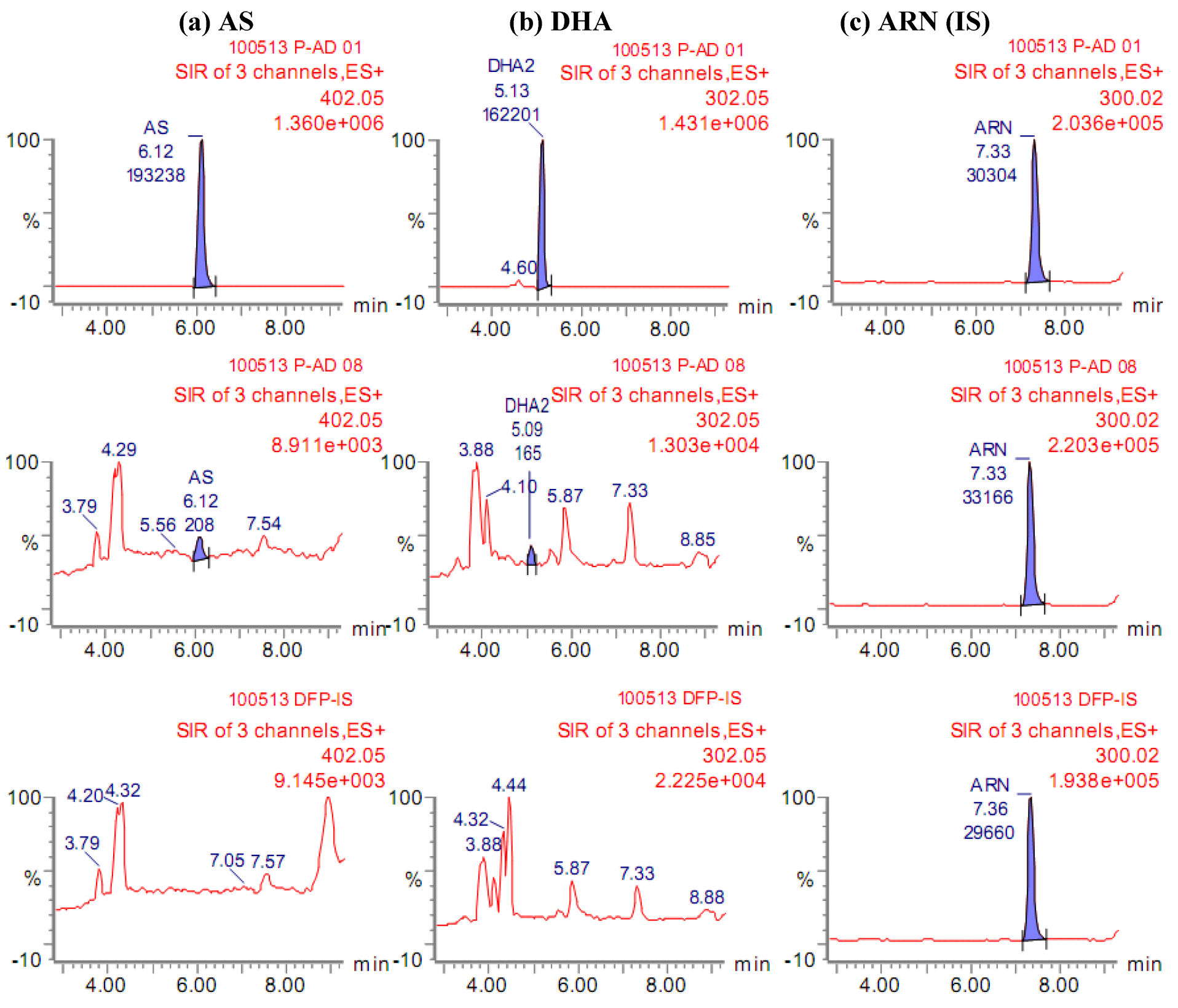
4.7.1. System suitability
4.7.2. Selectivity
4.7.3. Linearity test
4.7.4. Accuracy and precision
4.7.5. Recovery and matrix effect
4.7.6. Stability
4.8. Application to clinical plasma samples
Acknowledgements
Disclaimer
- Sample Availability: Samples of the compounds are available from the authors.
References and Notes
- WHO. Severe falciparum malaria. In Trans. Roy. Soc. Trop. Med. Hyg.; 2000; 94S1, pp. 1–90. [Google Scholar]
- WHO. WHO Guidelines for the treatment of malaria; World Health Organization: Geneva, Switzerland, 2006. Available online: http://www.who.int/malaria/docs/TreatmentGuidelines2006.pdf/ (accessed on 3 December 2009).
- Rosenthal, P.J. Artesunate for the treatment of severe falciparum malaria. New Eng. J. Med. 2008, 358, 1829–1836. [Google Scholar] [CrossRef]
- Kamchonwongpaisan, S.; Meshnick, S. The mode of action of the antimalarialartemisinin and its derivatives. Gen. Pharmacol. 1996, 27, 587–592. [Google Scholar] [CrossRef]
- Eckstein-Ludwig, U.; Webb, R.J.; Van Goethem, I.D.; East, J.M.; Lee, A.G,; Kimura, M.; O'Neill, P.M.; Bray, P.G.; Ward, S.A.; Krishna, S. Artemisinins target the SERCA of Plasmodium falciparum. Nature 2003, 424, 6951–6957. [Google Scholar]
- Day, N.; Dondorp, A.M. The management of patients with severe malaria. Am. J. Trop. Med. Hyg. 2007, 77 (6_Suppl.), 29–35. [Google Scholar]
- Nosten, F.; White, N.J. Artemisinin-based combination treatment of falciparum malaria. Am. J. Trop. Med. Hyg. 2007, 77 (6_Suppl.), 181–192. [Google Scholar]
- Gu, Y.; Li, Q.; Melendez, V.; Weina, P. Comparison of HPLC with electrochemical detection and LC–MS/MS for the separation and validation of artesunate and dihydroartemisinin in animal and human plasma. J. Chromatogr. B 2008, 867, 213–218. [Google Scholar] [CrossRef]
- Lai, C.S.; Nair, N.K.; Mansor, S.M.; Olliaro, P.L.; Navaratnam, V. An analytical method with a single extraction procedure and two separate high performance liquid chromatographic systems for the determination of artesunate, dihydroartemisinin and mefloquine in human plasma for application in clinical pharmacological studies of the drug combination. J. Chromatogr. B 2007, 857, 308–314. [Google Scholar] [CrossRef]
- Naik, H.; Murry, D.J.; Kirsch, L.E.; Fleckenstein, L. Development and validation of a high-performance liquid chromatography-mass spectroscopy assay for determination of artesunate and dihydroartemisinin in human plasma. J. Chromatogr. B 2005, 816, 233–242. [Google Scholar] [CrossRef]
- Van Quekelberghe, S.A.; Soomro, S.A.; Cordonnier, J.A.; Jansen, F.H. Optimization of an LC-MS method for the determination of artesunate and dihydroartemisinin plasma levels using liquid-liquid extraction. J. Anal. Toxicol. 2008, 32, 133–139. [Google Scholar]
- Thuy le, T.D.; Hung le, N.; Danh, P.T.; Na-Bangchang, K. Development and validation of a liquid chromatography-mass spectrometry method for the simultaneous quantification of artesunate and dihydroartemisinin in human plasma. Southeast Asian J. Trop. Med. Public Health 2008, 39, 963–977. [Google Scholar]
- Hanpithakpong, W.; Kamanikom, B.; Dondorp, A.M.; Singhasivanon, P.; White, N.J.; Day, N.P.; Lindegardh, N. A liquid chromatographic-tandem mass spectrometric method for determination of artesunate and its metabolite dihydroartemisinin in human plasma. J. Chromatogr. B 2008, 876, 61–68. [Google Scholar] [CrossRef]
- US Department of Health and Human Services, Food and Drug Administration. Guidance for Industry, Bioanalytical Method Validation; Food and Drug Administration: Rockville, MD, USA, 2001.
- ICH. Guidance for Industry, Q2B Validation of Analytical Procedures: Methodology; Food and Drug Administration: Rockville, MD, USA, 1996.
- Cabri, W.; Ciogli, A.; D'Acquarica, I.; Di Mattia, M.; Galletti, B.; Gasparrini, F.; Giorgi, F.; Lalli, S.; Pierini, M.; Simone, P. On-column epimerization of dihydroartemisinin: An effective analytical approach to overcome the shortcomings of the International Pharmacopoeia monograph. J. Chromatogr. B 2008, 875, 180–191. [Google Scholar] [CrossRef]
- Haynes, R.K. From artemisinin to new artemisinin antimalarials: biosynthesis, extraction, old and new derivatives, stereochemistry and medicinal chemistry requirements. Curr. Top. Med. Chem. 2006, 6, 509–537. [Google Scholar] [CrossRef]
- Batty, K.T.; Thu, L.T.; Davis, T.M.; Ilett, K.F.; Mai, T.X.; Hung, N.C.; Tien, N.P.; Powell, S.M.; Thien, H.V.; Binh, T.Q.; Kim, N.V. A pharmacokinetic and pharmacodynamic study of intravenous vs oral artesunate in uncomplicated falciparum malaria. Br. J. Clin. Pharmacol. 1998, 45, 123–129. [Google Scholar]
- Davis, T.M.; Phuong, H.L.; Ilett, K.F.; Hung, N.C.; Batty, K.T.; Phuong, V.D.; Powell, S.M.; Thien, H.V.; Binh, T.Q. Pharmacokinetics and pharmacodynamics of intravenous artesunate in severe falciparum malaria. Antimicrob. Agents Chemother. 2001, 45, 181–186. [Google Scholar] [CrossRef]
- Li, Q.; Cantilena, L.R.; Leary, K.J.; Saviolakis, G.A.; Miller, R.S.; Melendez, V.; Weina, P.J. Pharmacokinetic profiles of artesunate after single intravenous doses at 0.5, 1, 2, 4, and 8 mg/kg in healthy volunteers: A phase I study. Am. J. Trop. Med. Hyg. 2009, 81, 615–621. [Google Scholar] [CrossRef]
- Lindegardh, N.; Hanpithakpong, W.; Kamanikom, B.; Singhasivanon, P.; Socheat, D.; Yi, P.; Dondorp, A.M.; McGready, R.; Nosten, F.; White, N.J.; Day, N.P. Major pitfalls in the measurement of artemisinin derivatives in plasma in clinical studies. J. Chromatogr. B 2008, 876, 54–60. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Teja-Isavadharm, P.; Siriyanonda, D.; Siripokasupkul, R.; Apinan, R.; Chanarat, N.; Lim, A.; Wannaying, S.; Saunders, D.; Fukuda, M.M.; Miller, R.S.; et al. A Simplified Liquid Chromatography-Mass Spectrometry Assay for Artesunate and Dihydroartemisinin, Its Metabolite, in Human Plasma. Molecules 2010, 15, 8747-8768. https://doi.org/10.3390/molecules15128747
Teja-Isavadharm P, Siriyanonda D, Siripokasupkul R, Apinan R, Chanarat N, Lim A, Wannaying S, Saunders D, Fukuda MM, Miller RS, et al. A Simplified Liquid Chromatography-Mass Spectrometry Assay for Artesunate and Dihydroartemisinin, Its Metabolite, in Human Plasma. Molecules. 2010; 15(12):8747-8768. https://doi.org/10.3390/molecules15128747
Chicago/Turabian StyleTeja-Isavadharm, Paktiya, Duangsuda Siriyanonda, Raveewan Siripokasupkul, Roongnapa Apinan, Nitima Chanarat, Apassorn Lim, Srisombat Wannaying, David Saunders, Mark M. Fukuda, Robert S. Miller, and et al. 2010. "A Simplified Liquid Chromatography-Mass Spectrometry Assay for Artesunate and Dihydroartemisinin, Its Metabolite, in Human Plasma" Molecules 15, no. 12: 8747-8768. https://doi.org/10.3390/molecules15128747
APA StyleTeja-Isavadharm, P., Siriyanonda, D., Siripokasupkul, R., Apinan, R., Chanarat, N., Lim, A., Wannaying, S., Saunders, D., Fukuda, M. M., Miller, R. S., Weina, P. J., & Meléndez, V. (2010). A Simplified Liquid Chromatography-Mass Spectrometry Assay for Artesunate and Dihydroartemisinin, Its Metabolite, in Human Plasma. Molecules, 15(12), 8747-8768. https://doi.org/10.3390/molecules15128747