Role of Oxidant Scavengers in the Prevention of Ca2+ Homeostasis Disorders


Abstract
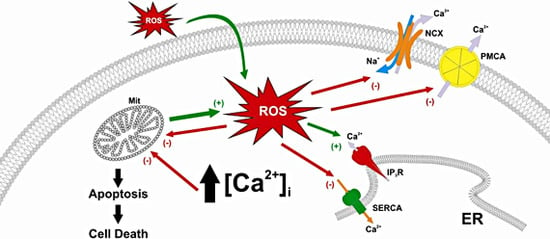
:
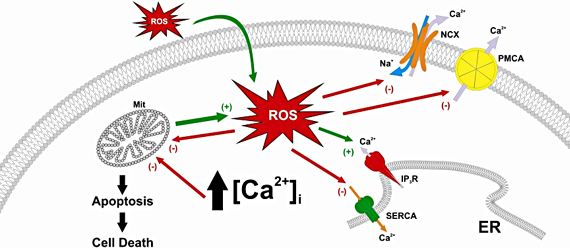{kind=link}
1. Calcium Homeostasis
2. Calcium Homeostasis Abnormalities Induced by Reactive Oxygen Species
3. Disorders Caused by Reactive Oxygen Species and Therapeutic Strategies Based on the Use of Antioxidants
Neurodegenerative diseases
4. Conclusions
Acknowledgments
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E.; Santella, L.; Branca, D.; Brini, M. Generation, control, and processing of cellular calcium signals. Crit. Rev. Biochem. Molec. Biol. 2001, 36, 107–260. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Salido, G.M.; Sage, S.O.; Rosado, J.A. Biochemical and functional properties of the store-operated Ca2+ channels. Cell. Signal. 2009, 21, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Strehler, E.E.; Zacharias, D.A. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol. Rev. 2001, 81, 21–50. [Google Scholar] [CrossRef] [PubMed]
- Exton, J.H. New developments in phospholipase D. J. Biol. Chem. 1997, 272, 15579–15582. [Google Scholar] [CrossRef] [PubMed]
- Cavallini, L.; Coassin, M.; Alexandre, A. Two classes of agonist-sensitive Ca2+ stores in platelets, as identified by their differential sensitivity to 2,5-di-(tert-butyl)-1,4-benzohydroquinone and thapsigargin. Biochem. J. 1995, 310, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E. Calcium signaling: a tale for all seasons. Proc. Natl. Acad. Sci. USA 2002, 99, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Guerini, D.; Coletto, L.; Carafoli, E. Exporting calcium from cells. Cell Calcium 2005, 38, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Choe, C.U.; Ehrlich, B.E. The inositol 1,4,5-trisphosphate receptor (IP3R) and its regulators: sometimes good and sometimes bad teamwork. Science’s STKE 2006, 2006, re15. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Churamani, D.; Cai, X.; Schrlau, M.G.; Brailoiu, G.C.; Gao, X.; Hooper, R.; Boulware, M.J.; Dun, N.J.; Marchant, J.S.; Patel, S. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J. Cell Biol. 2009, 186, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Redondo, P.C.; Harper, M.T.; Rosado, J.A.; Sage, S.O. A role for cofilin in the activation of store-operated calcium entry by de novo conformational coupling in human platelets. Blood 2006, 107, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Montell, C.; Birnbaumer, L.; Flockerzi, V. The TRP channels, a remarkably functional family. Cell 2002, 108, 595–598. [Google Scholar] [CrossRef]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Hogan, P.G.; Rao, A. Dissecting ICRAC, a store-operated calcium current. Trends Biochem. Sci. 2007, 32, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Zweifach, A.; Lewis, R.S. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc. Natl. Acad. Sci. USA 1993, 90, 6295–6299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; McCloskey, M.A. Immunoglobulin E receptor-activated calcium conductance in rat mast cells. J. Physiol. 1995, 483, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; Beck, A.; Billingsley, J.M.; Lis, A.; Parvez, S.; Peinelt, C.; Koomoa, D.L.; Soboloff, J.; Gill, D.L.; Fleig, A.; Kinet, J.P.; Penner, R. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 2006, 16, 2073–2079. [Google Scholar] [CrossRef] [PubMed]
- Mignen, O.; Thompson, J.L.; Shuttleworth, T.J. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J. Physiol. 2008, 586, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Ambudkar, I.S. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J. Biol. Chem. 2008, 283, 12935–12940. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Gomez, L.J.; Salido, G.M.; Rosado, J.A. Dynamic interaction of hTRPC6 with the Orai1-STIM1 complex or hTRPC3 mediates its role in capacitative or non-capacitative Ca2+ entry pathways. Biochem. J. 2009, 420, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J. Biol. Chem. 2008, 283, 25296–25304. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.L.; Cheng, K.T.; Liu, X.; Bandyopadhyay, B.C.; Paria, B.C.; Soboloff, J.; Pani, B.; Gwack, Y.; Srikanth, S.; Singh, B.B.; Gill, D.L.; Ambudkar, I.S. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J. Biol. Chem. 2007, 282, 9105–9116. [Google Scholar]
- Ambudkar, I.S.; Ong, H.L.; Liu, X.; Bandyopadhyay, B.; Cheng, K.T. TRPC1: the link between functionally distinct store-operated calcium channels. Cell Calcium 2007, 42, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Functional relevance of the de novo coupling between hTRPC1 and type II IP3 receptor in store-operated Ca2+ entry in human platelets. Cell. Signal. 2008, 20, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; Velicelebi, G.; Stauderman, K.A. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.J.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J. Biol. Chem. 2006, 281, 28254–28264. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Salido, G.M.; Rosado, J.A. Role of lipid rafts in the interaction between hTRPC1, Orai1 and STIM1. Channels (Austin) 2008, 2, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.J.; Jardin, I.; Bobe, R.; Pariente, J.A.; Enouf, J.; Salido, G.M.; Rosado, J.A. STIM1 regulates acidic Ca2+ store refilling by interaction with SERCA3 in human platelets. Biochem. Pharmacol. 2008, 75, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Rosado, J.A. Store-operated Ca2+ entry is sensitive to the extracellular Ca2+ concentration through plasma membrane STIM1. Biochim. Biophys. Acta 2009, 1793, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Luik, R.M.; Wu, M.M.; Buchanan, J.; Lewis, R.S. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006, 174, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Swain, J.H.; Alekel, D.L.; Dent, S.B.; Peterson, C.T.; Reddy, M.B. Iron indexes and total antioxidant status in response to soy protein intake in perimenopausal women. Amer. J. Clin. Nutr. 2002, 76, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Biological reactivity and biomarkers of the neutrophil oxidant, hypochlorous acid. Toxicology 2002, 181–182, 223–227. [Google Scholar]
- Yasui, H.; Hayashi, S.; Sakurai, H. Possible involvement of singlet oxygen species as multiple oxidants in p450 catalytic reactions. Drug Metab. Pharmacokinet. 2005, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K. Cellular thiols and redox-regulated signal transduction. Curr. Topics Cell Regul. 2000, 36, 1–30. [Google Scholar]
- Lopez, J.J.; Salido, G.M.; Gomez-Arteta, E.; Rosado, J.A.; Pariente, J.A. Thrombin induces apoptotic events through the generation of reactive oxygen species in human platelets. J. Thromb Haemost. 2007, 5, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Rosado, J.A.; Lopez, J.J.; Gomez-Arteta, E.; Redondo, P.C.; Salido, G.M.; Pariente, J.A. Early caspase-3 activation independent of apoptosis is required for cellular function. J. Cell Physiol. 2006, 209, 142–152. [Google Scholar]
- Rosado, J.A.; Gonzalez, A.; Salido, G.M.; Pariente, J.A. Effects of reactive oxygen species on actin filament polymerisation and amylase secretion in mouse pancreatic acinar cells. Cell. Signal. 2002, 14, 547–556. [Google Scholar] [CrossRef]
- Pariente, J.A.; Camello, C.; Camello, P.J.; Salido, G.M. Release of calcium from mitochondrial and nonmitochondrial intracellular stores in mouse pancreatic acinar cells by hydrogen peroxide. J. Membr. Biol. 2001, 179, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Redondo, P.C.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. Dual effect of hydrogen peroxide on store-mediated calcium entry in human platelets. Biochem. Pharmacol. 2004, 67, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Redondo, P.C.; Salido, G.M.; Rosado, J.A.; Pariente, J.A. Effect of hydrogen peroxide on Ca2+ mobilisation in human platelets through sulphydryl oxidation dependent and independent mechanisms. Biochem. Pharmacol. 2004, 67, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.; Barron, L.; Sharov, V.S.; Schoneich, C.; Michaelis, E.K.; Michaelis, M.L. Oxidative inactivation of purified plasma membrane Ca2+-ATPase by hydrogen peroxide and protection by calmodulin. Biochemistry 2003, 42, 12001–12010. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.; Michaelis, M.L. Effects of reactive oxygen species on brain synaptic plasma membrane Ca2+-ATPase. Free Radical Biol. Med. 1999, 27, 810–821. [Google Scholar] [CrossRef]
- Hidalgo, C.; Donoso, P. Crosstalk between calcium and redox signaling: from molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 1275–1312. [Google Scholar] [CrossRef] [PubMed]
- Lushington, G.H.; Zaidi, A.; Michaelis, M.L. Theoretically predicted structures of plasma membrane Ca2+-ATPase and their susceptibilities to oxidation. J. Mol. Graph. Model. 2005, 24, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Osborn, K.D.; Zaidi, A.; Urbauer, R.J.; Michaelis, M.L.; Johnson, C.K. Single-molecule characterization of the dynamics of calmodulin bound to oxidatively modified plasma-membrane Ca2+-ATPase. Biochemistry 2005, 44, 11074–11081. [Google Scholar] [CrossRef] [PubMed]
- Goldhaber, J.I. Free radicals enhance Na+/Ca2+ exchange in ventricular myocytes. Amer. J. Physiol. 1996, 271, H823–833. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, W.A.; Ichikawa, H.; Hearse, D.J. Oxidant stress inhibits Na-Ca-exchange current in cardiac myocytes: mediation by sulfhydryl groups? Amer. J. Physiol. 1994, 266, H909–919. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Kako, K.J. Na+/Ca2+ exchange of isolated sarcolemmal membrane: effects of insulin, oxidants and insulin deficiency. Mol. Cell. Biochem. 1988, 83, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Barnes, K.A.; Samson, S.E.; Grover, A.K. Sarco/endoplasmic reticulum Ca2+-pump isoform SERCA3a is more resistant to superoxide damage than SERCA2b. Mol. Cell. Biochem. 2000, 203, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.K.; Samson, S.E. Peroxide resistance of ER Ca2+ pump in endothelium: implications to coronary artery function. Amer. J. Physiol. 1997, 273, C1250–C1258. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.Y.; Zweier, J.L.; Becker, L.C. Hydroxyl radical inhibits sarcoplasmic reticulum Ca2+-ATPase function by direct attack on the ATP binding site. Circ. Res. 1997, 80, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Burk, S.E.; Lytton, J.; MacLennan, D.H.; Shull, G.E. cDNA cloning, functional expression, and mRNA tissue distribution of a third organellar Ca2+ pump. J. Biol. Chem. 1989, 264, 18561–18568. [Google Scholar] [PubMed]
- Trebak, M.; Ginnan, R.; Singer, H.A.; Jourd’heuil, D. Interplay between calcium and reactive oxygen/nitrogen species: an essential paradigm for vascular smooth muscle signaling. Antioxid. Redox Signal. 2010, 12, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Kourie, J.I. Interaction of reactive oxygen species with ion transport mechanisms. Amer. J. Physiol. 1998, 275, C1–24. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Taylor, C.W.; Berridge, M.J. The thiol reagent, thimerosal, evokes Ca2+ spikes in HeLa cells by sensitizing the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 1992, 267, 25113–25119. [Google Scholar] [PubMed]
- Missiaen, L.; Taylor, C.W.; Berridge, M.J. Spontaneous calcium release from inositol trisphosphate-sensitive calcium stores. Nature 1991, 352, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Oba, T.; Kurono, C.; Nakajima, R.; Takaishi, T.; Ishida, K.; Fuller, G.A.; Klomkleaw, W.; Yamaguchi, M. H2O2 activates ryanodine receptor but has little effect on recovery of releasable Ca2+ content after fatigue. J. Appl. Physiol. 2002, 93, 1999–2008. [Google Scholar] [CrossRef] [PubMed]
- Favero, T.G.; Zable, A.C.; Abramson, J.J. Hydrogen peroxide stimulates the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1995, 270, 25557–25563. [Google Scholar] [CrossRef] [PubMed]
- Zissimopoulos, S.; Docrat, N.; Lai, F.A. Redox sensitivity of the ryanodine receptor interaction with FK506-binding protein. J. Biol. Chem. 2007, 282, 6976–6983. [Google Scholar] [CrossRef] [PubMed]
- Balshaw, D.M.; Xu, L.; Yamaguchi, N.; Pasek, D.A.; Meissner, G. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J. Biol. Chem. 2001, 276, 20144–20153. [Google Scholar] [CrossRef] [PubMed]
- Hool, L.C.; Corry, B. Redox control of calcium channels: from mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2007, 9, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Lacampagne, A.; Duittoz, A.; Bolanos, P.; Peineau, N.; Argibay, J.A. Effect of sulfhydryl oxidation on ionic and gating currents associated with L-type calcium channels in isolated guinea-pig ventricular myocytes. Cardiovasc. Res. 1995, 30, 799–806. [Google Scholar] [CrossRef]
- Viola, H.M.; Arthur, P.G.; Hool, L.C. Transient exposure to hydrogen peroxide causes an increase in mitochondria-derived superoxide as a result of sustained alteration in L-type Ca2+ channel function in the absence of apoptosis in ventricular myocytes. Circ. Res. 2007, 100, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Hudasek, K.; Brown, S.T.; Fearon, I.M. H2O2 regulates recombinant Ca2+ channel alpha1C subunits but does not mediate their sensitivity to acute hypoxia. Biochem. Biophys. Res. Commun. 2004, 318, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Segui, J.; Heinemann, S.H.; Hoshi, T. Oxidation regulates cloned neuronal voltage-dependent Ca2+ channels expressed in Xenopus oocytes. J. Neurosci. 1998, 18, 6740–6747. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Bernardi, P.; Pozzan, T. Mitochondria as all-round players of the calcium game. J. Physiol. 2000, 529 Pt 1, 37–47. [Google Scholar] [CrossRef]
- Tornquist, K.; Vainio, P.J.; Bjorklund, S.; Titievsky, A.; Dugue, B.; Tuominen, R.K. Hydrogen peroxide attenuates store-operated calcium entry and enhances calcium extrusion in thyroid FRTL-5 cells. Biochem. J. 2000, 351, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Rosado, J.A.; Redondo, P.C.; Salido, G.M.; Gomez-Arteta, E.; Sage, S.O.; Pariente, J.A. Hydrogen peroxide generation induces pp60src activation in human platelets: evidence for the involvement of this pathway in store-mediated calcium entry. J. Biol. Chem. 2004, 279, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Groschner, K.; Rosker, C.; Lukas, M. Role of TRP channels in oxidative stress. Novart. Fdn. Symp. 2004, 258, 222–230. [Google Scholar]
- Aarts, M.M.; Tymianski, M. TRPMs and neuronal cell death. Pflugers Arch. 2005, 451, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.M.; Tymianski, M. TRPM7 and ischemic CNS injury. Neuroscientist 2005, 11, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Montalbetti, N.; Cantero, M.R.; Dalghi, M.G.; Cantiello, H.F. Reactive oxygen species inhibit polycystin-2 (TRPP2) cation channel activity in term human syncytiotrophoblast. Placenta 2008, 29, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Bogeski, I.; Kummerow, C.; Al-Ansary, D.; Schwarz, E.C.; Koehler, R.; Kozai, D.; Takahashi, N.; Peinelt, C.; Griesemer, D.; Bozem, M.; Mori, Y.; Hoth, M.; Niemeyer, B.A. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci. Signal. 2010, 3, ra24. [Google Scholar] [CrossRef] [PubMed]
- Trebak, M.; Ginnan, R.; Singer, H.A.; Jourd’heuil, D. Interplay between calcium and reactive oxygen/nitrogen species: an essential paradigm for vascular smooth muscle signaling. Antioxid. Redox Signal. 2010, 12, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wei, C.L.; Zhang, W.R.; Cheng, H.P.; Liu, J. Cross-talk between calcium and reactive oxygen species signaling. Acta Pharmacol. Sin. 2006, 27, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Waring, P. Redox active calcium ion channels and cell death. Arch. Biochem. Biophys. 2005, 434, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Alexandru, N.; Jardin, I.; Popov, D.; Simionescu, M.; Garcia-Estan, J.; Salido, G.M.; Rosado, J.A. Effect of homocysteine on calcium mobilization and platelet function in type 2 diabetes mellitus. J. Cell. Mol. Med. 2008, 12, 2586–2597. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Damrauer, S.M.; Lee, M.; Sellke, F.W.; Ferran, C.; Abid, M.R. Endothelium-dependent coronary vasodilatation requires NADPH oxidase-derived reactive oxygen species. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Schiffrin, E.L. Reactive oxygen species in vascular biology: implications in hypertension. Histochemistry Cell Biol. 2004, 122, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Alexander, R.W. Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase. Mol. Cell. Biochem. 2004, 264, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yi, J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Redondo, P.C.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. Endogenously generated reactive oxygen species reduce PMCA activity in platelets from patients with non-insulin-dependent diabetes mellitus. Platelets 2006, 17, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part II: animal and human studies. Circulation 2003, 108, 2034–2040. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Altered Lipid Metabolism in Brain Injury and Disorders. Subcell. Biochem. 2008, 48, nihpa41041. [Google Scholar]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Munzel, T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 2001, 104, 2673–2678. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Monticone, R.E.; Lakatta, E.G. Arterial aging: a journey into subclinical arterial disease. Curr. Opin. Nephrol. Hypertens. 2010, 19, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Stumvoll, M.; Goldstein, B.J.; van Haeften, T.W. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 2005, 365, 1333–1346. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Bosca, L.; Zeini, M.; Traves, P.G.; Hortelano, S. Nitric oxide and cell viability in inflammatory cells: a role for NO in macrophage function and fate. Toxicology 2005, 208, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Flamand, L.; Tremblay, M.J.; Borgeat, P. Leukotriene B4 triggers the in vitro and in vivo release of potent antimicrobial agents. J. Immunol. 2007, 178, 8036–8045. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Heininger, Y.M.; Aesif, S.W.; van der Velden, J.; Guala, A.S.; Reiss, J.N.; Roberson, E.C.; Budd, R.C.; Reynaert, N.L.; Anathy, V. Regulation of apoptosis through cysteine oxidation: implications for fibrotic lung disease. Ann. N. Y. Acad. Sci. 2010, 1203, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Cardaci, S.; Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Reactive oxygen species mediate p53 activation and apoptosis induced by sodium nitroprusside in SH-SY5Y cells. Mol. Pharmacol. 2008, 74, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, A.; Ichijo, H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 2008, 1780, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- D’Archivio, M.; Santangelo, C.; Scazzocchio, B.; Vari, R.; Filesi, C.; Masella, R.; Giovannini, C. Modulatory effects of polyphenols on apoptosis induction: relevance for cancer prevention. Int. J. Mol. Sci. 2008, 9, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K. Anti-oxidants for therapeutic use: why are only a few drugs in clinical use? Advan. Drug Delivery Rev. 2009, 61, 287–289. [Google Scholar] [CrossRef] [PubMed]
- Cimminiello, C.; Milani, M. Diabetes mellitus and peripheral vascular disease: is aspirin effective in preventing vascular events? Diabetologia 1996, 39, 1402–1404. [Google Scholar] [CrossRef] [PubMed]
- Sobol, A.B.; Watala, C. The role of platelets in diabetes-related vascular complications. Diabetes Res. Clin. Pract. 2000, 50, 1–16. [Google Scholar] [CrossRef]
- Vericel, E.; Januel, C.; Carreras, M.; Moulin, P.; Lagarde, M. Diabetic patients without vascular complications display enhanced basal platelet activation and decreased antioxidant status. Diabetes 2004, 53, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Alexandru, N.; Constantin, A.; Popov, D. Carbonylation of platelet proteins occurs as consequence of oxidative stress and thrombin activation, and is stimulated by ageing and type 2 diabetes. Clin. Chem. Lab. Med. 2008, 46, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Rosado, J.A.; Saavedra, F.R.; Redondo, P.C.; Hernandez-Cruz, J.M.; Salido, G.M.; Pariente, J.A. Reduced plasma membrane Ca2+-ATPase function in platelets from patients with non-insulin-dependent diabetes mellitus. Haematologica 2004, 89, 1142–1144. [Google Scholar] [PubMed]
- Belia, S.; Santilli, F.; Beccafico, S.; De Feudis, L.; Morabito, C.; Davi, G.; Fano, G.; Mariggio, M.A. Oxidative-induced membrane damage in diabetes lymphocytes: effects on intracellular Ca(2 +) homeostasis. Free Radical Res. 2009, 43, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Redondo, P.C.; Jardin, I.; Hernandez-Cruz, J.M.; Pariente, J.A.; Salido, G.M.; Rosado, J.A. Hydrogen peroxide and peroxynitrite enhance Ca2+ mobilization and aggregation in platelets from type 2 diabetic patients. Biochem. Biophys. Res. Commun. 2005, 333, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.A. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, F.R.; Redondo, P.C.; Hernandez-Cruz, J.M.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. Store-operated Ca2+ entry and tyrosine kinase pp60src hyperactivity are modulated by hyperglycemia in platelets from patients with non insulin-dependent diabetes mellitus. Arch. Biochem. Biophys. 2004, 432, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Hers, I. Insulin-like growth factor-1 potentiates platelet activation via the IRS/PI3Kalpha pathway. Blood 2007, 110, 4243–4252. [Google Scholar] [CrossRef] [PubMed]
- Matrougui, K. Diabetes and microvascular pathophysiology: role of epidermal growth factor receptor tyrosine kinase. Diabetes Metab. Res. Rev. 2010, 26, 13–16. [Google Scholar] [CrossRef] [PubMed]
- van Guldener, C.; Stehouwer, C.D. Diabetes mellitus and hyperhomocysteinemia. Semin. Vasc. Med. 2002, 2, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Signorello, M.G.; Viviani, G.L.; Armani, U.; Cerone, R.; Minniti, G.; Piana, A.; Leoncini, G. Homocysteine, reactive oxygen species and nitric oxide in type 2 diabetes mellitus. Thromb Res. 2007, 120, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Kadono, K.; Tetsutani, T.; Yasunaga, K. Platelet free Ca2+ concentration in non-insulin-dependent diabetes mellitus. Diabetes Res. 1991, 18, 89–94. [Google Scholar] [PubMed]
- Vangheluwe, P.; Raeymaekers, L.; Dode, L.; Wuytack, F. Modulating sarco(endo)plasmic reticulum Ca2+ ATPase 2 (SERCA2) activity: cell biological implications. Cell Calcium 2005, 38, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Dean, W.L.; Chen, D.; Brandt, P.C.; Vanaman, T.C. Regulation of platelet plasma membrane Ca2+-ATPase by cAMP-dependent and tyrosine phosphorylation. J. Biol. Chem. 1997, 272, 15113–15119. [Google Scholar] [CrossRef] [PubMed]
- Begonja, A.J.; Teichmann, L.; Geiger, J.; Gambaryan, S.; Walter, U. Platelet regulation by NO/cGMP signaling and NAD(P)H oxidase-generated ROS. Blood Cells Molecules Dis. 2006, 36, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Redondo, P.C.; Ben-Amor, N.; Salido, G.M.; Bartegi, A.; Pariente, J.A.; Rosado, J.A. Ca2+-independent activation of Bruton’s tyrosine kinase is required for store-mediated Ca2+ entry in human platelets. Cell. Signal. 2005, 17, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Chapado, L.; Linares-Palomino, P.J.; Salido, S.; Altarejos, J.; Rosado, J.A.; Salido, G.M. Synthesis and evaluation of the platelet antiaggregant properties of phenolic antioxidants structurally related to rosmarinic acid. Bioorg. Chem. 2010, 38, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Bouaziz, A.; Salido, S.; Linares-Palomino, P.J.; Sanchez, A.; Altarejos, J.; Bartegi, A.; Salido, G.M.; Rosado, J.A. Cinnamtannin B-1 from bay wood reduces abnormal intracellular Ca2+ homeostasis and platelet hyperaggregability in type 2 diabetes mellitus patients. Arch. Biochem. Biophys. 2007, 457, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Maiorino, M.I.; Ceriello, A.; Giugliano, D. Prevention and control of type 2 diabetes by Mediterranean diet: a systematic review. Diabetes Res. Clin. Pract. 2010, 89, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.Z.; O’Dea, K.; Gomez, M.; Girgis, S.; Colagiuri, R. Diet and exercise in the prevention of diabetes. J. Hum. Nutr. Diet. 2010, 23, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Fenercioglu, A.K.; Saler, T.; Genc, E.; Sabuncu, H.; Altuntas, Y. The effects of polyphenol-containing antioxidants on oxidative stress and lipid peroxidation in Type 2 diabetes mellitus without complications. J. Endocrinol. Invest. 2010, 33, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.A. Mitochondrial oxidative stress and inflammation: an slalom to obesity and insulin resistance. J. Physiol. Biochem. 2006, 62, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Schulz, E.; Oelze, M.; Muller, J.; Schuhmacher, S.; Alhamdani, M.S.; Debrezion, J.; Hortmann, M.; Reifenberg, K.; Fleming, I.; Munzel, T.; Daiber, A. AT1-receptor blockade by telmisartan upregulates GTP-cyclohydrolase I and protects eNOS in diabetic rats. Free Radical Biol. Med. 2008, 45, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Mechlovich, D.; Amit, T.; Mandel, S.A.; Bar-Am, O.; Bloch, K.; Vardi, P.; Youdim, M.B. The novel multifunctional, iron-chelating drugs M30 and HLA20 protect pancreatic beta-cell lines from oxidative stress damage. J. Pharmacol. Exp. Ther. 2010, 333, 874–882. [Google Scholar] [CrossRef] [PubMed]
- San Jose, G.; Fortuno, A.; Beloqui, O.; Diez, J.; Zalba, G. NADPH oxidase CYBA polymorphisms, oxidative stress and cardiovascular diseases. Clin. Sci. (Lond). 2008, 114, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Judge, E.P.; Phelan, D.; O’Shea, D. Beyond statin therapy: a review of the management of residual risk in diabetes mellitus. J. Roy. Soc. Med. 2010, 103, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Selemidis, S. Suppressing NADPH oxidase-dependent oxidative stress in the vasculature with nitric oxide donors. Clin. Exp. Pharmacol. Physiol. 2008, 35, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Marzotti, S.; Guglielmini, G.; Momi, S.; Giannini, S.; Minuz, P.; Lucidi, P.; Bolli, G.B. Hyperglycemia-induced platelet activation in type 2 diabetes is resistant to aspirin but not to a nitric oxide-donating agent. Diabetes Care 2010, 33, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Missiaen, L.; Robberecht, W.; van den Bosch, L.; Callewaert, G.; Parys, J.B.; Wuytack, F.; Raeymaekers, L.; Nilius, B.; Eggermont, J.; De Smedt, H. Abnormal intracellular Ca2+ homeostasis and disease. Cell Calcium 2000, 28, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W., Jr. Capacitative calcium entry in the nervous system. Cell Calcium 2003, 34, 339–344. [Google Scholar] [CrossRef]
- Yamamoto, S.; Wajima, T.; Hara, Y.; Nishida, M.; Mori, Y. Transient receptor potential channels in Alzheimer’s disease. Biochim. Biophys. Acta 2007, 1772, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Sun, Y.; Singh, B.B. TRPC channels and their implication in neurological diseases. CNS Neurol. Disord. Drug Target. 2010, 9, 94–104. [Google Scholar] [CrossRef]
- Rao, M.V.; Mohan, P.S.; Peterhoff, C.M.; Yang, D.S.; Schmidt, S.D.; Stavrides, P.H.; Campbell, J.; Chen, Y.; Jiang, Y.; Paskevich, P.A.; Cataldo, A.M.; Haroutunian, V.; Nixon, R.A. Marked calpastatin (CAST) depletion in Alzheimer’s disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by CAST overexpression. J. Neurosci. 2008, 28, 12241–12254. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, F.; Marcilhac, A. Implication of calpain in neuronal apoptosis. A possible regulation of Alzheimer’s disease. FEBS J. 2006, 273, 3437–3443. [Google Scholar]
- Fifre, A.; Sponne, I.; Koziel, V.; Kriem, B.; Yen Potin, F.T.; Bihain, B.E.; Olivier, J.L.; Oster, T.; Pillot, T. Microtubule-associated protein MAP1A, MAP1B, and MAP2 proteolysis during soluble amyloid beta-peptide-induced neuronal apoptosis. Synergistic involvement of calpain and caspase-3. J. Biol. Chem. 2006, 281, 229–240. [Google Scholar]
- Lipton, S.A.; Choi, Y.B.; Pan, Z.H.; Lei, S.Z.; Chen, H.S.; Sucher, N.J.; Loscalzo, J.; Singel, D.J.; Stamler, J.S. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 1993, 364, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Dargusch, R.; Piasecki, D.; Tan, S.; Liu, Y.; Schubert, D. The role of Bax in glutamate-induced nerve cell death. J. Neurochem. 2001, 76, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Culmsee, C.; Zhu, C.; Landshamer, S.; Becattini, B.; Wagner, E.; Pellecchia, M.; Blomgren, K.; Plesnila, N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J. Neurosci. 2005, 25, 10262–10272. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.; Closhen, D.; Croxford, A.; White, R.; Kulig, P.; Pietrowski, E.; Bechmann, I.; Becher, B.; Luhmann, H.J.; Waisman, A.; Kuhlmann, C.R. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010, 24, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Rattan, V.; Sultana, C.; Shen, Y.; Kalra, V.K. Oxidant stress-induced transendothelial migration of monocytes is linked to phosphorylation of PECAM-1. Amer. J. Physiol. 1997, 273, E453–461. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hao, W.; Letiembre, M.; Walter, S.; Kulanga, M.; Neumann, H.; Fassbender, K. Suppression of microglial inflammatory activity by myelin phagocytosis: role of p47-PHOX-mediated generation of reactive oxygen species. J. Neurosci. 2006, 26, 12904–12913. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Phospholipase A(2), reactive oxygen species, and lipid peroxidation in CNS pathologies. BMB Rep. 2008, 41, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Koppal, T.; Howard, B.; Subramaniam, R.; Hall, N.; Hensley, K.; Yatin, S.; Allen, K.; Aksenov, M.; Aksenova, M.; Carney, J. Structural and functional changes in proteins induced by free radical-mediated oxidative stress and protective action of the antioxidants N-tert-butyl-alpha-phenylnitrone and vitamin E. Ann. N. Y. Acad. Sci. 1998, 854, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Yoshida, M.; Yamato, M.; Ide, T.; Wu, Z.; Ochi-Shindou, M.; Kanki, T.; Kang, D.; Sunagawa, K.; Tsutsui, H.; Nakanishi, H. Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. J. Neurosci. 2008, 28, 8624–8634. [Google Scholar] [CrossRef] [PubMed]
- Bettens, K.; Sleegers, K.; Van Broeckhoven, C. Current status on Alzheimer disease molecular genetics: from past, to present, to future. Hum. Mol Genet. 2010, 19, R4–R11. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Thinakaran, G. Amyloidogenic processing of beta-amyloid precursor protein in intracellular compartments. Neurology 2006, 66, S69–S73. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Zhang, Y.W.; Xu, H.; Thinakaran, G. Pathological and physiological functions of presenilins. Mol. Neurodegeneration 2006, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Robinson, N.; Mattson, M.P. Secreted beta-amyloid precursor protein counteracts the proapoptotic action of mutant presenilin-1 by activation of NF-kappaB and stabilization of calcium homeostasis. J. Biol. Chem. 1998, 273, 12341–12351. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Calcium and neurodegeneration. Aging Cell 2007, 6, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Cecchi, C.; Pensalfini, A.; Bonini, S.A.; Ferrari-Toninelli, G.; Liguri, G.; Memo, M.; Uberti, D. Generation of reactive oxygen species by beta amyloid fibrils and oligomers involves different intra/extracellular pathways. Amino Acids 2010, 38, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Surh, Y.J. Beta-amyloid-induced apoptosis is associated with cyclooxygenase-2 up-regulation via the mitogen-activated protein kinase-NF-kappaB signaling pathway. Free Radical Biol. Med. 2005, 38, 1604–1613. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992, 12, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Kuroda, Y. Molecular mechanism of neurodegeneration induced by Alzheimer’s beta-amyloid protein: channel formation and disruption of calcium homeostasis. Brain Res. Bull. 2000, 53, 389–397. [Google Scholar] [CrossRef]
- Yagami, T.; Ueda, K.; Sakaeda, T.; Itoh, N.; Sakaguchi, G.; Okamura, N.; Hori, Y.; Fujimoto, M. Protective effects of a selective L-type voltage-sensitive calcium channel blocker, S-312-d, on neuronal cell death. Biochem. Pharmacol. 2004, 67, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Kasturirangan, S.; Boddapati, S.; Sierks, M.R. Engineered proteolytic nanobodies reduce Abeta burden and ameliorate Abeta-induced cytotoxicity. Biochemistry 2010, 49, 4501–4508. [Google Scholar] [CrossRef] [PubMed]
- Biscaro, B.; Lindvall, O.; Hock, C.; Ekdahl, C.T.; Nitsch, R.M. Abeta immunotherapy protects morphology and survival of adult-born neurons in doubly transgenic APP/PS1 mice. J. Neurosci. 2009, 29, 14108–14119. [Google Scholar] [CrossRef] [PubMed]
- Bacher, M.; Depboylu, C.; Du, Y.; Noelker, C.; Oertel, W.H.; Behr, T.; Henriksen, G.; Behe, M.; Dodel, R. Peripheral and central biodistribution of (111)In-labeled anti-beta-amyloid autoantibodies in a transgenic mouse model of Alzheimer’s disease. Neurosci. Lett. 2009, 449, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L. NADPH oxidase as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008, 9, S8. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Gardoni, F.; Di Luca, M. Molecular rationale for the pharmacological treatment of Alzheimer’s disease. Drugs Aging 2005, 22, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Castegna, A.; Lauderback, C.M.; Drake, J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol. Aging 2002, 23, 655–664. [Google Scholar] [CrossRef]
- Butterfield, D.A. Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radical Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef]
- Nelson, C.; Wengreen, H.J.; Munger, R.G.; Corcoran, C.D. Dietary folate, vitamin B-12, vitamin B-6 and incident Alzheimer’s disease: the cache county memory, health and aging study. J. Nutr. Health Aging 2009, 13, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer’s disease. CNS Spectr. 2009, 14, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Schubert, D.R. The specificity of neuroprotection by antioxidants. J. Biomed. Sci. 2009, 16, 98. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.J.; Jardin, I.; Salido, G.M.; Rosado, J.A. Cinnamtannin B-1 as an antioxidant and platelet aggregation inhibitor. Life Sci. 2008, 82, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, S.H.; Ko, W.C.; Ko, F.N.; Teng, C.M. Inhibition of platelet aggregation by some flavonoids. Thromb Res. 1991, 64, 91–100. [Google Scholar] [CrossRef]
- Bucki, R.; Pastore, J.J.; Giraud, F.; Sulpice, J.C.; Janmey, P.A. Flavonoid inhibition of platelet procoagulant activity and phosphoinositide synthesis. J. Thromb Haemost. 2003, 1, 1820–1828. [Google Scholar] [CrossRef] [PubMed]
- Beretz, A.; Cazenave, J.P.; Anton, R. Inhibition of aggregation and secretion of human platelets by quercetin and other flavonoids: structure-activity relationships. Agent. Action 1982, 12, 382–387. [Google Scholar]
- Polette, A.; Lemaitre, D.; Lagarde, M.; Vericel, E. N-3 fatty acid-induced lipid peroxidation in human platelets is prevented by catechins. Thromb Haemost. 1996, 75, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Pastore, J.J.; Funaki, M.; Janmey, P.A.; Bucki, R. Flavonoid-mediated inhibition of actin polymerization in cold-activated platelets. Platelets 2005, 16, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, J.A.; Navarro-Nunez, L.; Lozano, M.L.; Martinez, C.; Vicente, V.; Gibbins, J.M.; Rivera, J. Flavonoids inhibit the platelet TxA(2) signalling pathway and antagonize TxA(2) receptors (TP) in platelets and smooth muscle cells. Br. J. Clin. Pharmacol. 2007, 64, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, J.A.; Lozano, M.L.; Castillo, J.; Benavente-Garcia, O.; Vicente, V.; Rivera, J. Flavonoids inhibit platelet function through binding to the thromboxane A2 receptor. J. Thromb Haemost. 2005, 3, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Bouaziz, A.; Romera-Castillo, C.; Salido, S.; Linares-Palomino, P.J.; Altarejos, J.; Bartegi, A.; Rosado, J.A.; Salido, G.M. Cinnamtannin B-1 from bay wood exhibits antiapoptotic effects in human platelets. Apoptosis 2007, 12, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Ben Amor, N.; Bouaziz, A.; Romera-Castillo, C.; Salido, S.; Linares-Palomino, P.J.; Bartegi, A.; Salido, G.M.; Rosado, J.A. Characterization of the intracellular mechanisms involved in the antiaggregant properties of cinnamtannin B-1 from bay wood in human platelets. J. Med. Chem. 2007, 50, 3937–3944. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.; Perez Vizcaino, F.; Utrilla, P.; Jimenez, J.; Tamargo, J.; Zarzuelo, A. Vasodilatory effects of flavonoids in rat aortic smooth muscle. Structure-activity relationships. Gen. Pharmacol. 1993, 24, 857–862. [Google Scholar]
- Middleton, E., Jr. Effect of plant flavonoids on immune and inflammatory cell function. Adv. Exp. Med. Biol. 1998, 439, 175–182. [Google Scholar] [PubMed]
- Peluso, M.R. Flavonoids attenuate cardiovascular disease, inhibit phosphodiesterase, and modulate lipid homeostasis in adipose tissue and liver. Exp. Biol. Med. (Maywood). 2006, 231, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Galan, C.; Jardín, I.; Dionisio, N.; Salido, G.; Rosado, J.A. Role of Oxidant Scavengers in the Prevention of Ca2+ Homeostasis Disorders. Molecules 2010, 15, 7167-7187. https://doi.org/10.3390/molecules15107167
Galan C, Jardín I, Dionisio N, Salido G, Rosado JA. Role of Oxidant Scavengers in the Prevention of Ca2+ Homeostasis Disorders. Molecules. 2010; 15(10):7167-7187. https://doi.org/10.3390/molecules15107167
Chicago/Turabian StyleGalan, Carmen, Isaac Jardín, Natalia Dionisio, Ginés Salido, and Juan A. Rosado. 2010. "Role of Oxidant Scavengers in the Prevention of Ca2+ Homeostasis Disorders" Molecules 15, no. 10: 7167-7187. https://doi.org/10.3390/molecules15107167
APA StyleGalan, C., Jardín, I., Dionisio, N., Salido, G., & Rosado, J. A. (2010). Role of Oxidant Scavengers in the Prevention of Ca2+ Homeostasis Disorders. Molecules, 15(10), 7167-7187. https://doi.org/10.3390/molecules15107167