Synthesis, ex Vivo and in Vitro Hydrolysis Study of an Indoline Derivative Designed as an Anti-Inflammatory with Reduced Gastric Ulceration Properties

Abstract
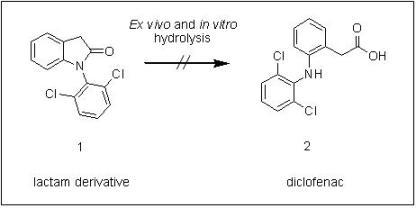
:
1. Introduction
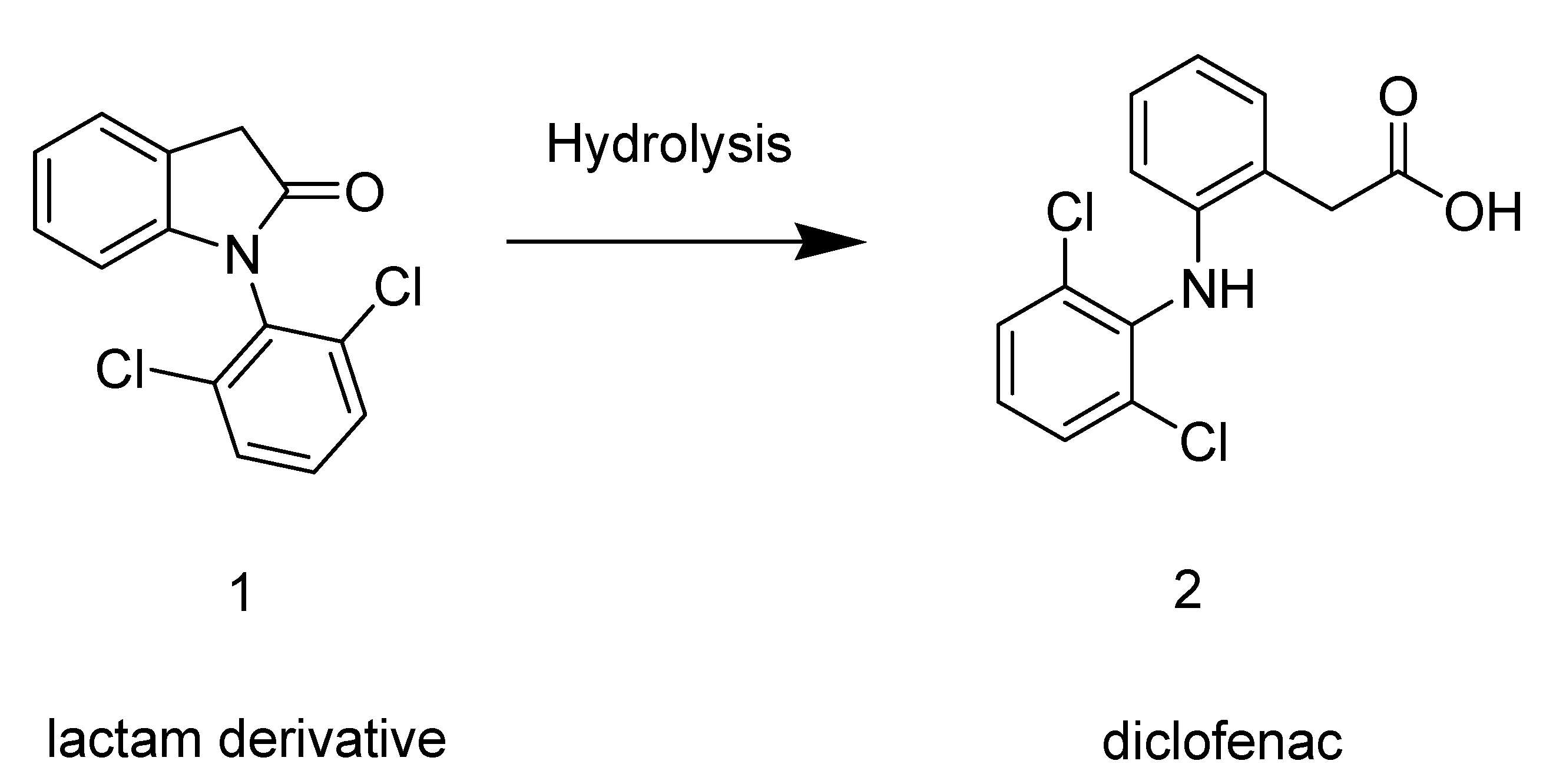
2. Results and Discussion
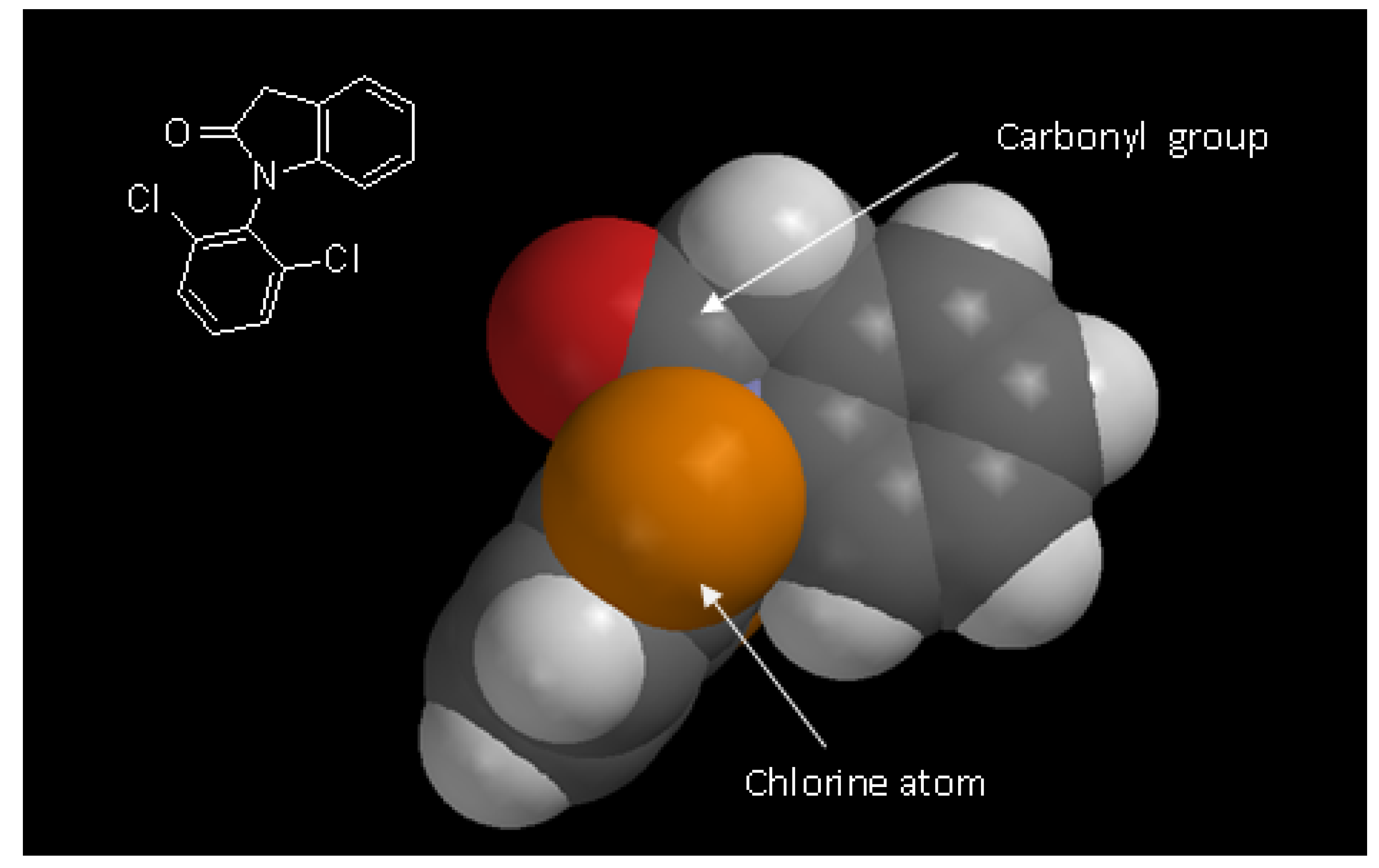
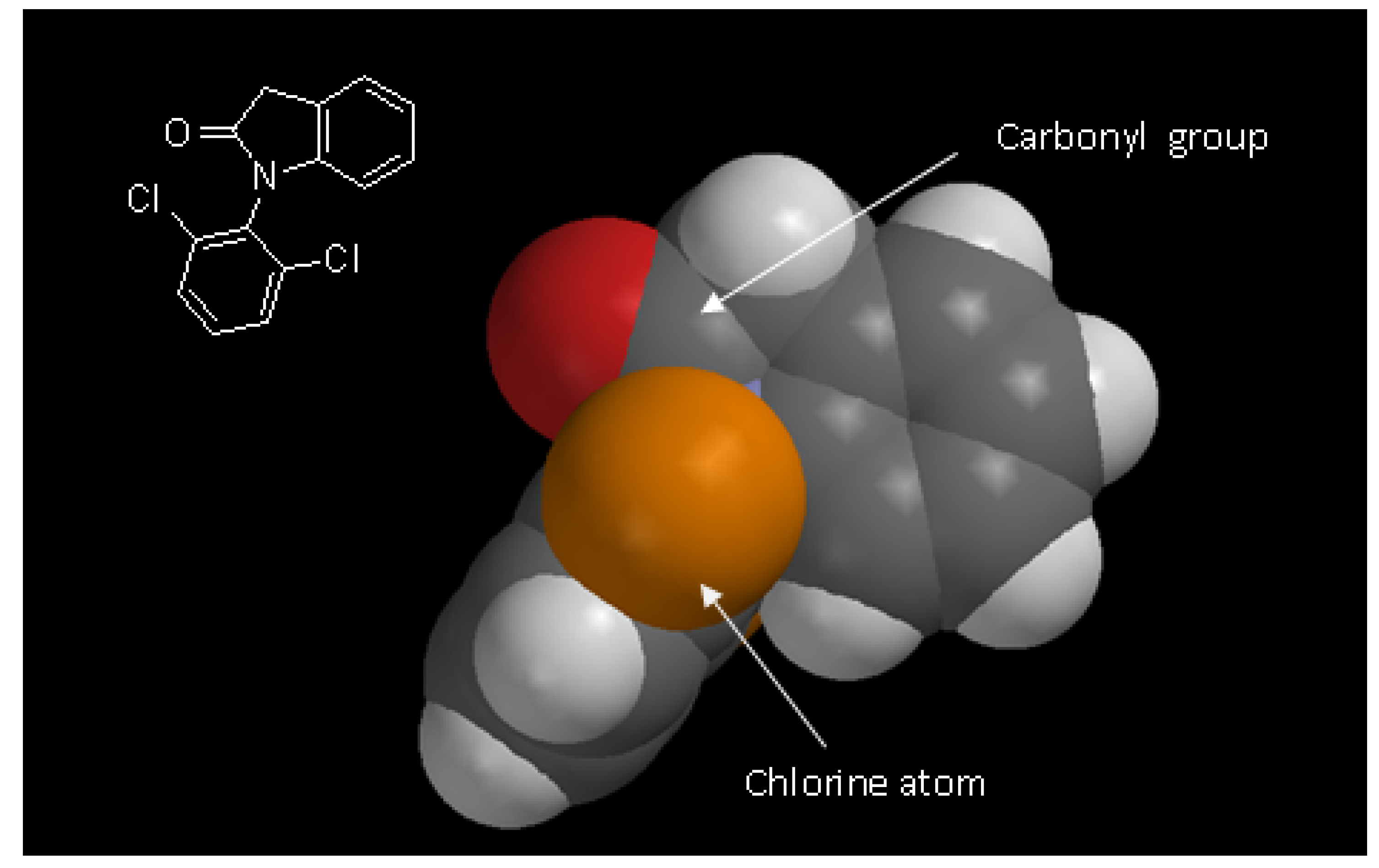
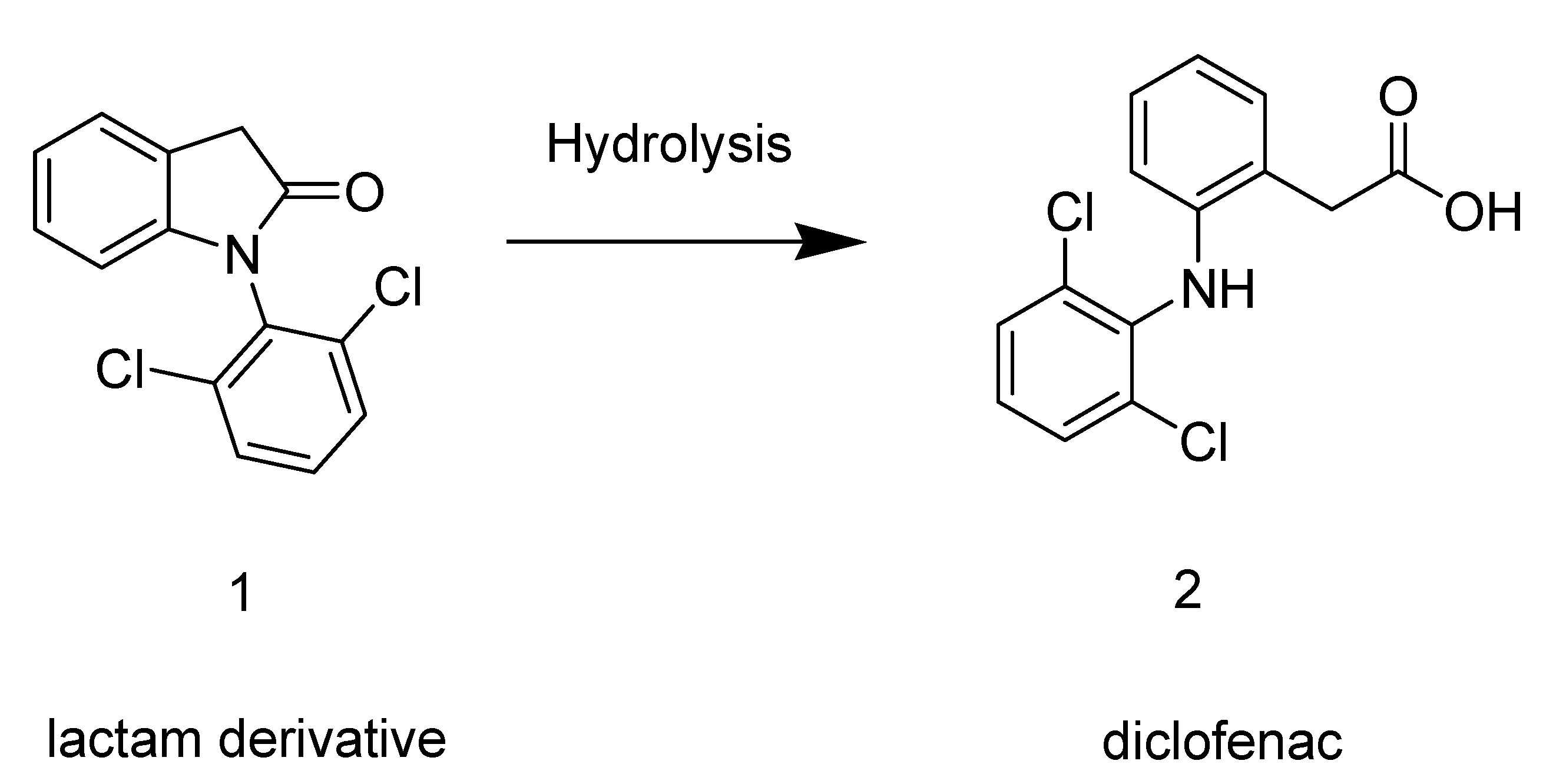
2.1. Synthesis
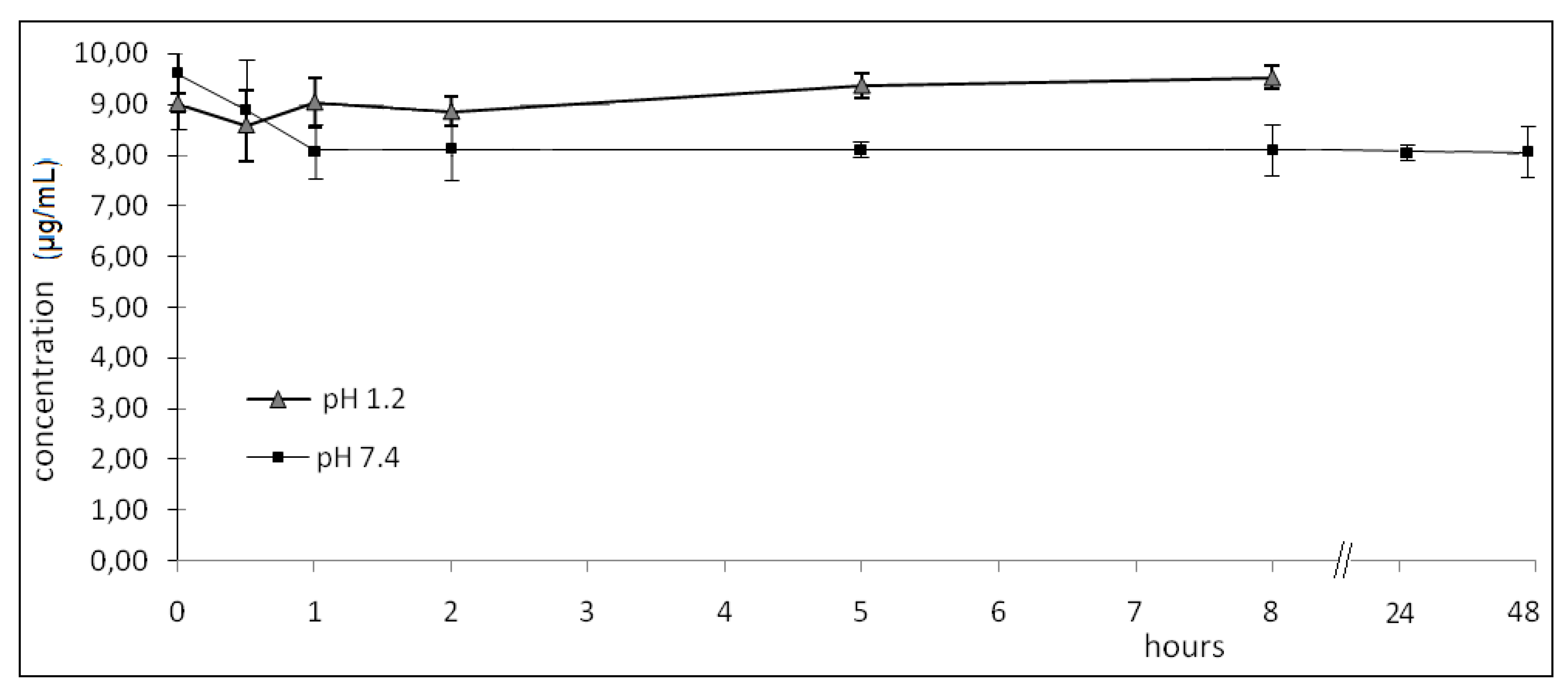
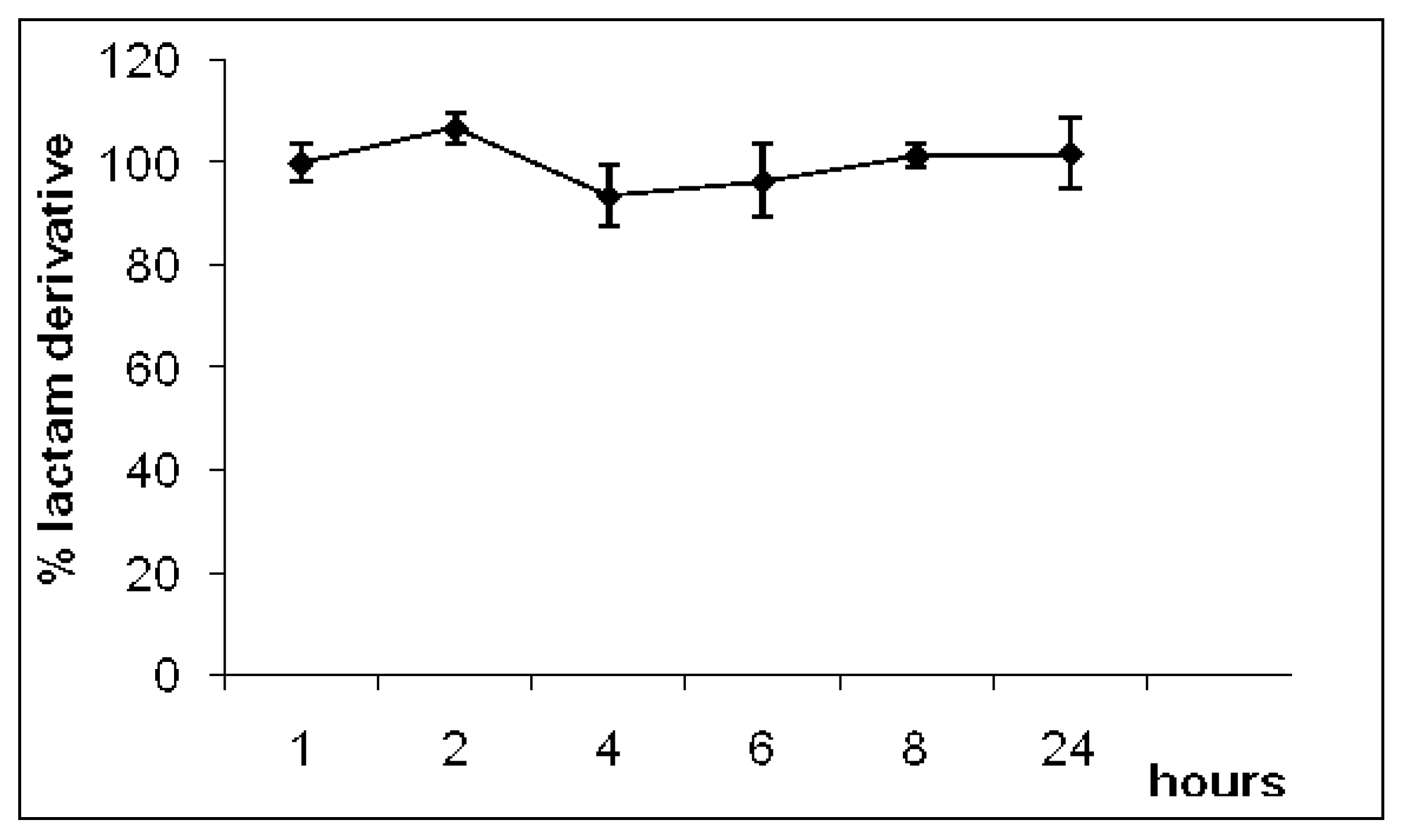
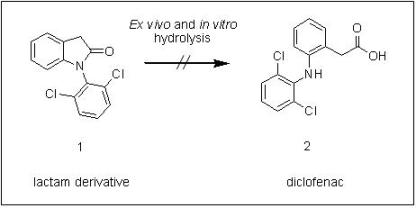
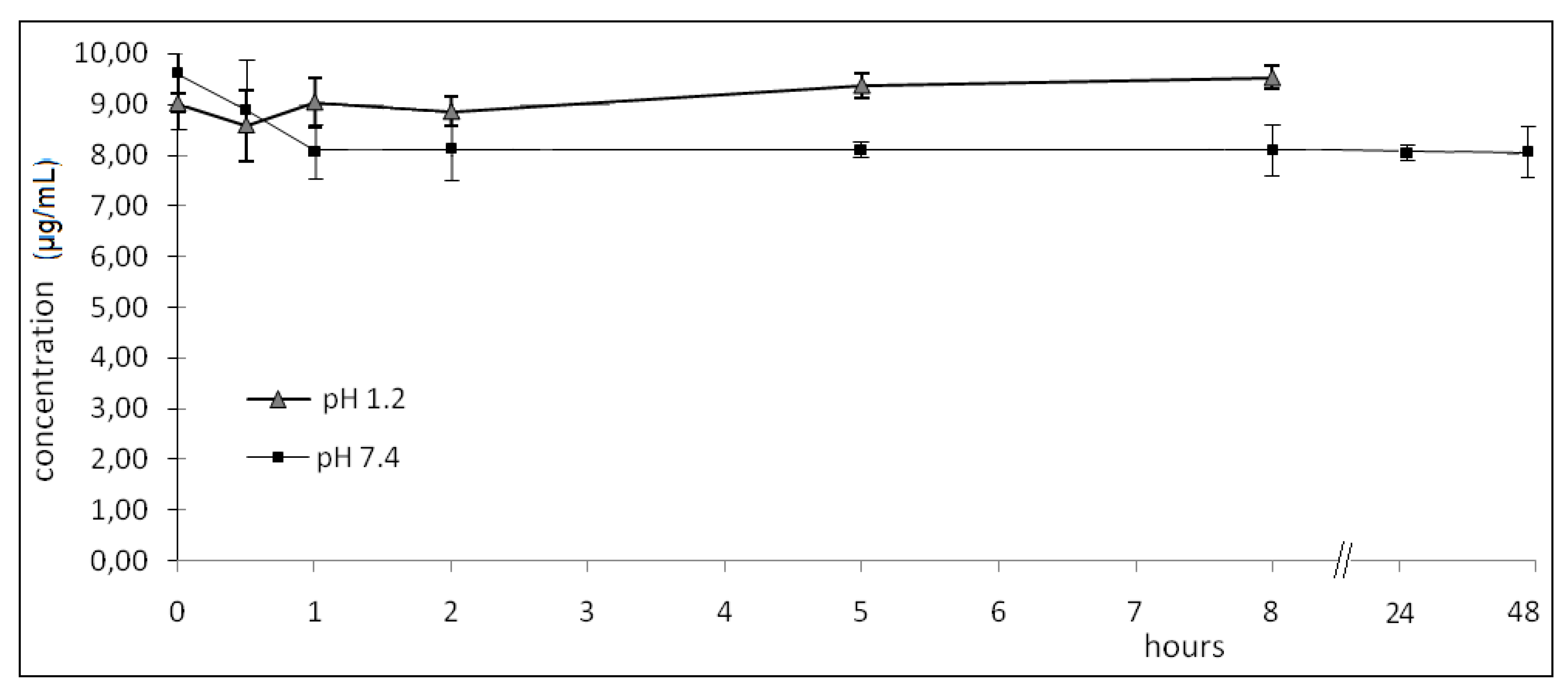
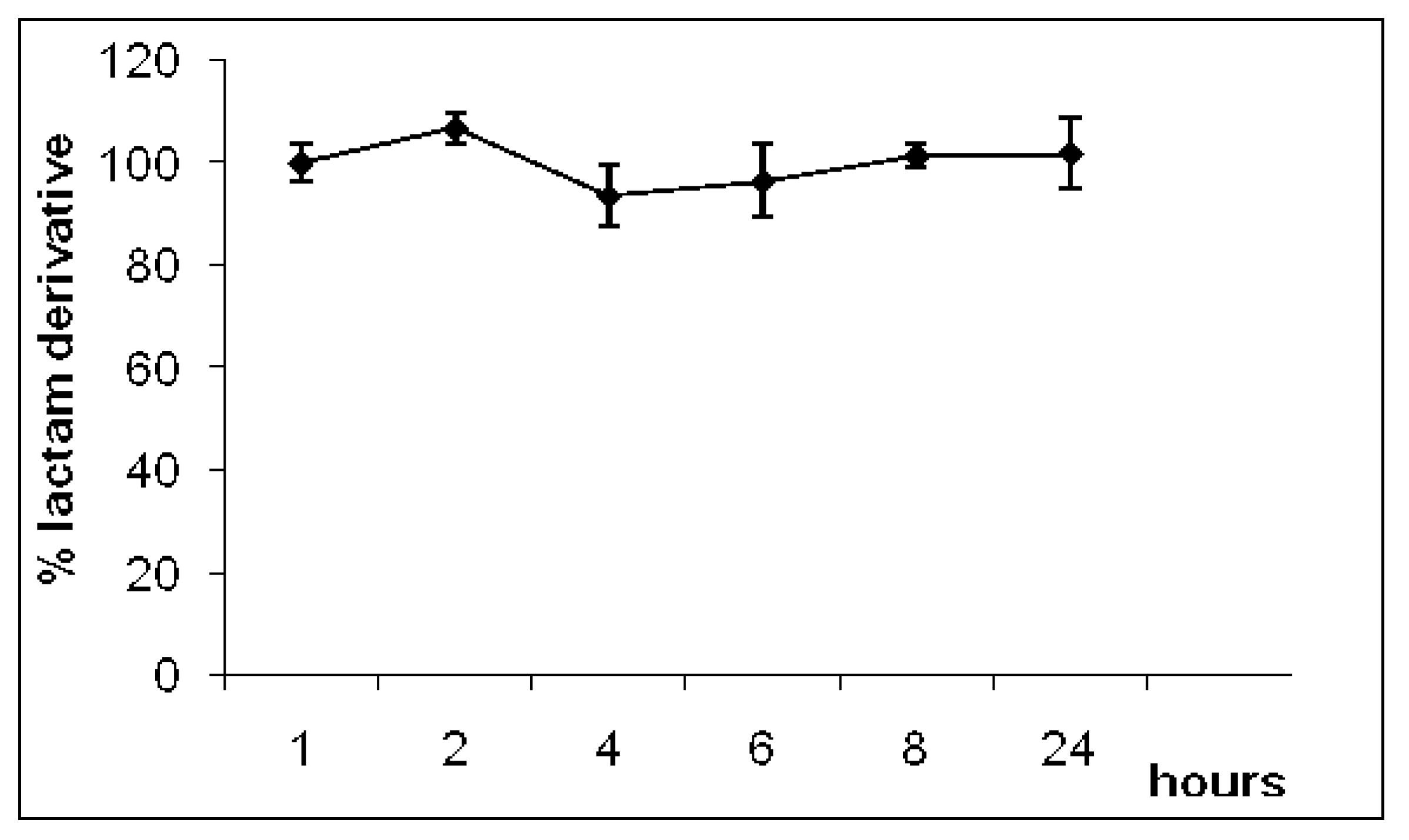
2.2. Hydrolysis
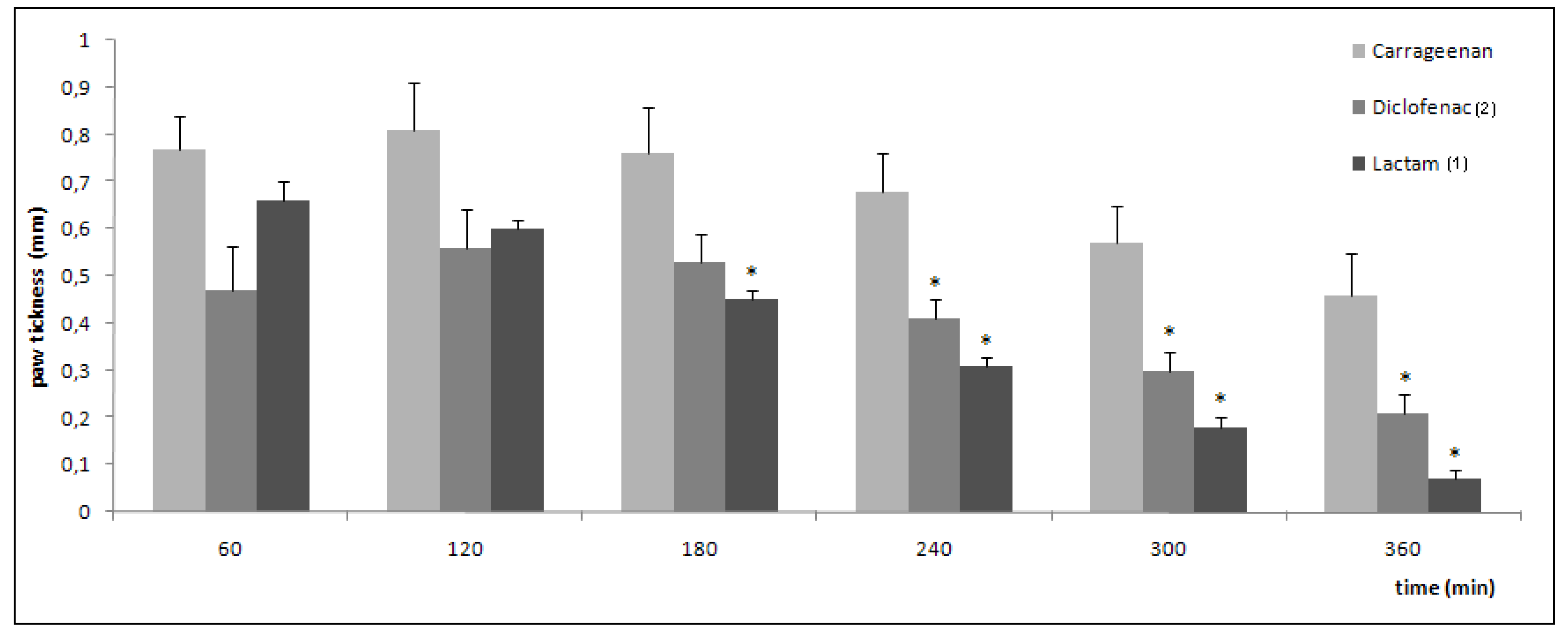
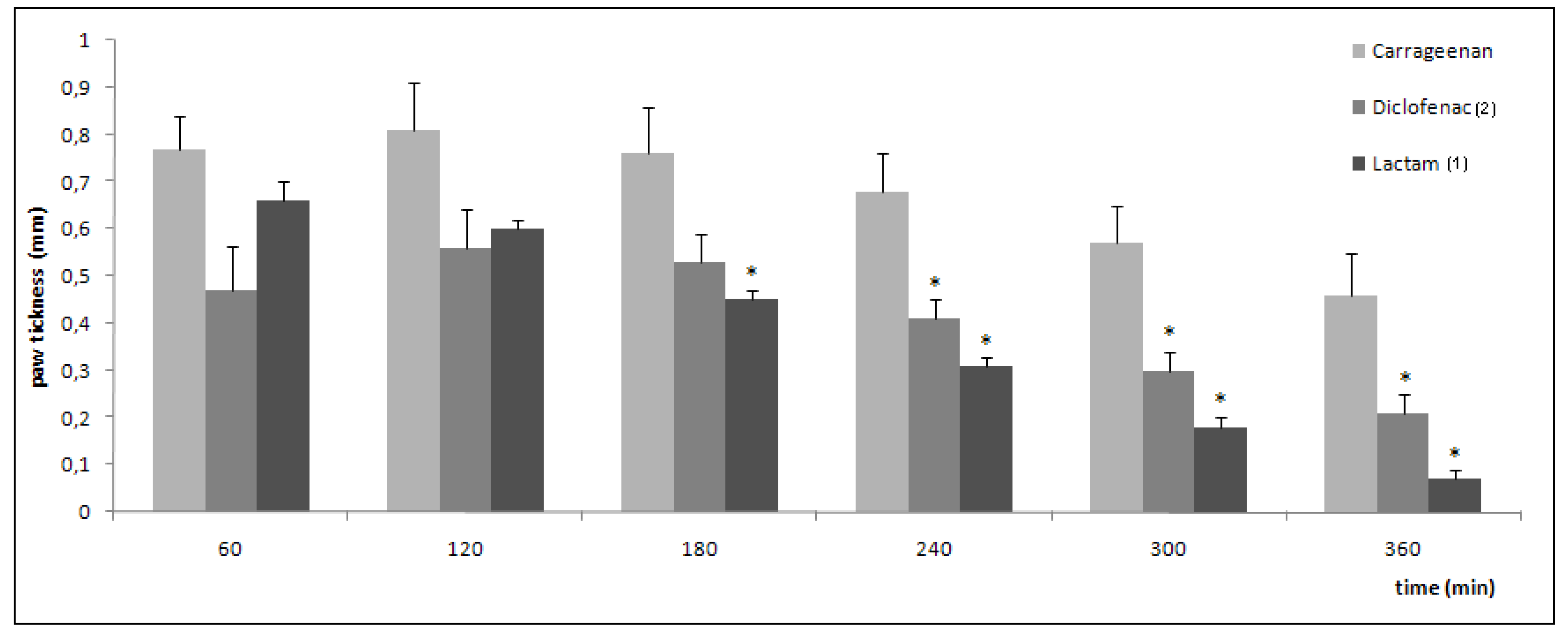
2.3. Pharmacological evaluation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Number of writhings (average) | % protection |
|---|---|---|
| control | 62 ± 1.9 | - |
| diclofenac | 21.6 ± 0.8≠ | 65.2 |
| lactam 1 | 29.7 ± 0.5≠* | 48.1 |
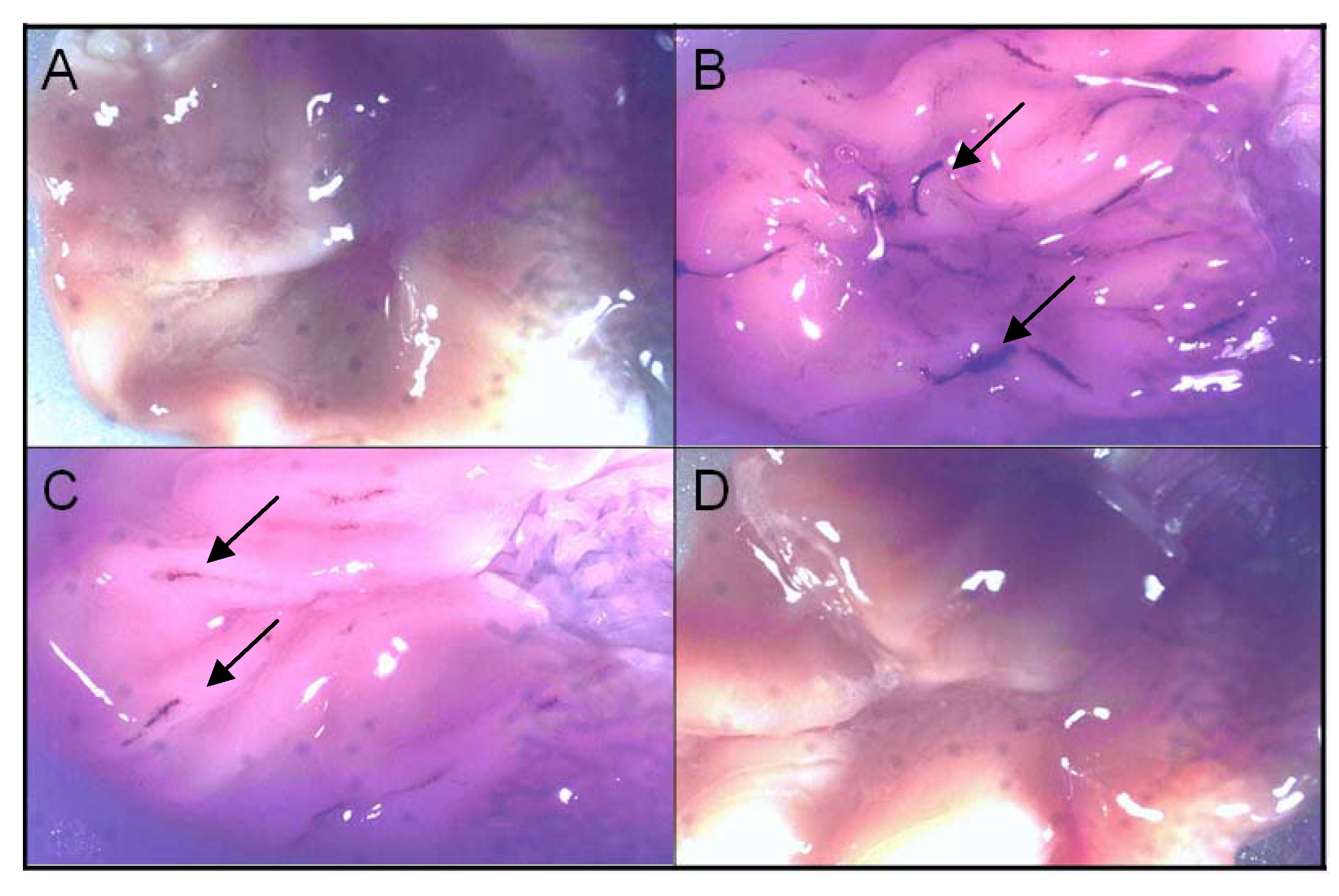
| compound | number of ulcers | < 1 mm | 1-2 mm | >2 mm |
|---|---|---|---|---|
| diclofenac | 69 ± 6.15 | 62 ± 7.5 (89%) | 2.9 ± 2.5 (4.3%) | 4.6 ± 1.9 (6.7%) |
| celecoxib | 6 ± 1.1* | 3 ± 1.5 (50%) | 3 ± 0.9 (50%) | - |
| lactam 1 | 0* | - | - | - |
3. Experimental
3.1. General
3.2. Materials
3.3. Animals
3.4. Synthesis of 1-(2,6-dichlorophenyl)indolin-2-one (1)
3.5. In vitro hydrolyses of 1-(2,6-dichlorophenyl)indolin-2-one (1) in buffer and human plasma
3.5.1. Analytical protocol
3.5.2. 1-(2,6-dichlorophenyl)indolin-2-one (1) in vitro hydrolyses in buffer and human plasma
- pH 1.2: zero; 0.50; 1.00; 2.00; 5.00; 8.00 h (08 h, considered to be the maximum time for this drug to be in gastrointestinal tract).
- pH 7.4: zero; 0.50; 1.00; 2.00; 5.00; 8.00; 24.0; 48.0 h.
- Plasma: zero; 0.25; 0.50; 1.00; 2.00; 4.00; 8.00; 24.0 h.
3.5.3. Statistical analysis
3.6. Anti-inflammatory activity
3.7. Analgesic activity
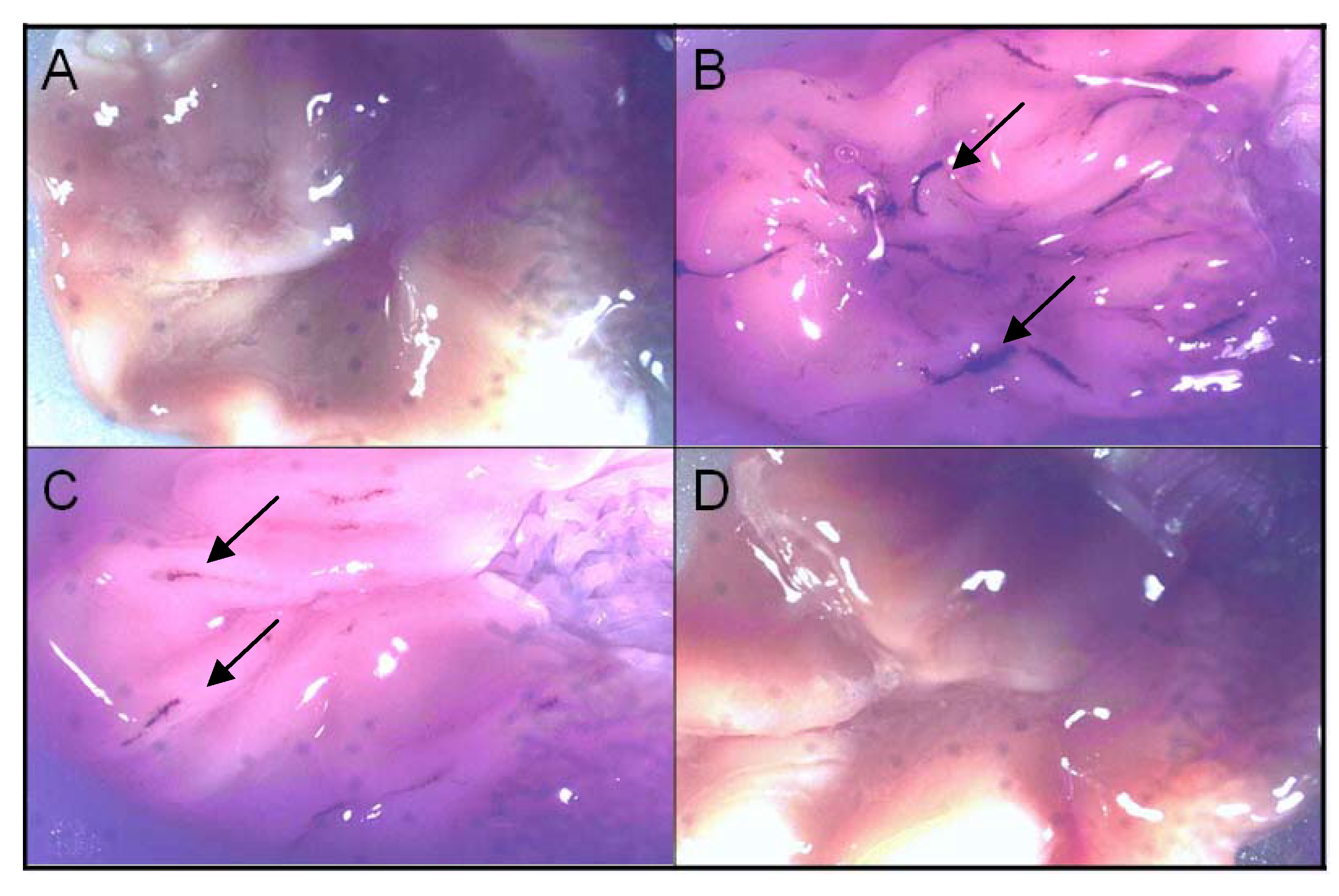
3.8. Ulcerogenicity
4. Conclusions
Acknowledgements
- Samples Availability: Not available.
References and Notes
- Cena, C.; Lolli, M.L.; Lazzarato, L.; Guaita, E.; Morini, G.; Coruzzi, G.; McElroy, S.P.; Megson, I.L.; Fruttero, R.; Gasco, A. Antiinflammatory, gastrosparing and antiplatelet properties of new NO-donor esters of aspirin. J. Med. Chem. 2003, 46, 747–754. [Google Scholar] [CrossRef]
- Bandarage, U.K.; Chen, L.; Fang, X.; Garvey, D.S.; Glavin, A.; Janero, D.R.; Letts, L.G.; Mercer, G.J.; Saha, J.K.; Schroeder, J.D.; Shumway, M.J.; Tam, S.W. Nitrosothiol esters of diclofenac: Synthesis and pharmacological characterization as gastrointestinal-sparing prodrugs. J. Med. Chem. 2000, 43, 4005–4016. [Google Scholar] [CrossRef]
- Wermuth, C.G. Selective optimization of side activities: another way for drug discovery. J. Med. Chem. 2004, 47, 1303–1314. [Google Scholar] [CrossRef]
- Silva, A.T.A.; Chung, M.C.; Castro, L.F.; Guido, R.V.; Ferreira, E.I. Advances in prodrug design. Mini Rev. Med. Chem. 2005, 10, 893–914. [Google Scholar]
- Zhao, X.; Tao, X.; Wei, D.; Song, Q. Pharmacological activity and hydrolysis behavior of novel ibuprofen glucopyranoside conjugates. Eur. J. Med. Chem. 2006, 41, 1352–1358. [Google Scholar] [CrossRef]
- Shanbhag, V.R.; Crider, A.M.; Gokhale, R.; Harpalani, A.; Dick, R.M. Ester and amide prodrugs of ibuprofen and naproxen: Synthesis, anti-inflammatory activity, and gastrointestinal toxicity. J. Pharm. Sci. 1992, 81, 149–154. [Google Scholar] [CrossRef]
- Kumakura, S.; Mishima, M.; Kobayashi, S.; Shirota, H.; Abe, S.; Yamada, K.; Tsurufuji, S. Inhibitory effect of indomethacin farnesil, a novel antiinflammatory prodrug, on carrageenin-induced inflammation in rats. Agent. Action. 1990, 29, 286–291. [Google Scholar] [CrossRef]
- Ribeiro, L.; Silva, N.; Iley, J.; Rautio, J.; Jarvinen, T.; Mota-Filipe, H.; Moreira, R.; Mendes, E. Aminocarbonyloxymethyl ester prodrugs of flufenamic acid and diclofenac: Suppressing the rearrangement pathway in aqueous media. Arch. Pharm. 2007, 340, 32–40. [Google Scholar]
- Halen, P.; Kuldeep, K.; Chagti, K.K.; Giridhar, R.; Yadav, M.R. Synthesis and pharmacological evaluation of some dual-acting amino-alcohol ester derivatives of flurbiprofen and 2-[1,1'-biphenyl-4-yl]acetic acid: A potential approach to reduce local gastrointestinal toxicity. Chem. Biodiv. 2006, 3, 1238–1248. [Google Scholar]
- Wallace, J.L. The 1994 Merck Frosst Award. Mechanisms of nonsteroidal anti-inflammatory drug (NSAID) induced gastrointestinal damage—potential for development of gastrointestinal tract safe NSAIDs. Can. J. Physiol. Pharmacol. 1994, 72, 1493–1498. [Google Scholar] [CrossRef]
- Kim, H.; Jeon, H.; Kong, H.; Yang, Y.; Choi, B.; Kim, Y.M.; Neckers, L.; Jung, Y. A molecular mechanism for the anti-inflammatory effect of taurine-conjugated 5-amino-salicylic acid in inflamed colon. Mol. Pharmacol. 2006, 69, 1405–1412. [Google Scholar] [CrossRef]
- Gairola, N.; Nagpal, D.; Dhaneshwar, S.S.; Dhaneshwar, S.R.; Chaturvedi, S.C. Synthesis, hydrolysis kinetics and pharmacodynamic profile of novel prodrugs of flurbiprofen. Indian J. Pharm. Sci. 2005, 67, 369–373. [Google Scholar]
- Winter, C.; Risley, E.; Nuss, G. Carrageenin-induced edema in hind paw of the rat as an assay for anti-inflammatory drugs. Proc. Soc. Exp. Biol. Med. 1962, 111, 544–547. [Google Scholar]
- Seigmund, E.; Cadmus, R.; Lu, G. A method for evaluating both non-narcotic and narcotic analgesics. Proc. Soc. Exp. Biol. Med. 1957, 95, 729–733. [Google Scholar]
- Cioli, V.; Putzolu, S.; Rossi, V.; Corza, B.P.; Corradino, C. The role of direct tissue contact in the production of gastrointestinal ulcers by anti-inflammatory drugs in rats. Toxicol. Appl. Pharmacol. 1979, 50, 283–289. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chung, M.C.; Dos Santos, J.L.; Oliveira, E.V.; Blau, L.; Menegon, R.F.; Peccinini, R.G. Synthesis, ex Vivo and in Vitro Hydrolysis Study of an Indoline Derivative Designed as an Anti-Inflammatory with Reduced Gastric Ulceration Properties. Molecules 2009, 14, 3187-3197. https://doi.org/10.3390/molecules14093187
Chung MC, Dos Santos JL, Oliveira EV, Blau L, Menegon RF, Peccinini RG. Synthesis, ex Vivo and in Vitro Hydrolysis Study of an Indoline Derivative Designed as an Anti-Inflammatory with Reduced Gastric Ulceration Properties. Molecules. 2009; 14(9):3187-3197. https://doi.org/10.3390/molecules14093187
Chicago/Turabian StyleChung, Man Chin, Jean Leandro Dos Santos, Ednir Vizioli Oliveira, Lorena Blau, Renato Farina Menegon, and Rosângela Gonçalves Peccinini. 2009. "Synthesis, ex Vivo and in Vitro Hydrolysis Study of an Indoline Derivative Designed as an Anti-Inflammatory with Reduced Gastric Ulceration Properties" Molecules 14, no. 9: 3187-3197. https://doi.org/10.3390/molecules14093187
APA StyleChung, M. C., Dos Santos, J. L., Oliveira, E. V., Blau, L., Menegon, R. F., & Peccinini, R. G. (2009). Synthesis, ex Vivo and in Vitro Hydrolysis Study of an Indoline Derivative Designed as an Anti-Inflammatory with Reduced Gastric Ulceration Properties. Molecules, 14(9), 3187-3197. https://doi.org/10.3390/molecules14093187