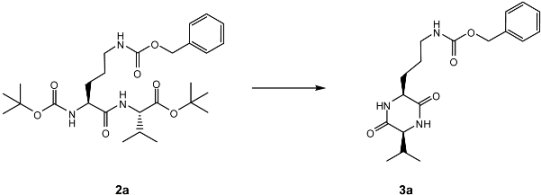
3.3. General procedure for the syntheses of dipeptides 2k-2l
A mixture of Boc-amino acid (2.4 mmol), amino ester hydrochloride (2 mmol), EDAC (2.4 mmol), HOBt (2.4 mmol) and DMAP (0.1 mmol) were dissolved in dry CH2Cl2 (5 mL). Mixture was cooled to 5 °C and then TEA (2.4 mmol) was added. Reaction was stirred at 5 °C for further 30 min, then allowed to warm up to room temperature and stirred for two days. Reaction mixture was treated with sat. NH4Cl soln. (20 mL). The organic phase was separated and the aqueous layer was extracted with CH2Cl2 (3 × 15 mL). Combined organic layers were washed with brine (2 × 15 mL) and with water (2 × 15 mL) and dried over Na2SO4. Solvent was removed under vacuum. The residue was purified by FCC.
Boc-Orn(Cbz)-Val-OtBu (2a): Colorless syrup; [α] + 8.5 (c 1.09, CHCl3); IR: 3,336, 2,973, 2,935, 2,878, 1,712, 1,666, 1,532, 1,454, 1,368, 1,254, 1,162, 1,018 cm-1; 1H-NMR (CDCl3) δ 7.28-7.20 (5H, m, Ar), 6.85 (1H, bs, NH-Val), 5.29 (1H, bs, NH-Orn), 5.18 (1H, bs, NHδ-Orn), 5.04 (1H, d, J = 12.4 Hz, CH2-Cbz), 5.01 (1H, d, J = 12.4 Hz, CH2-Cbz), 4.33 (1H, dd, J = 9.2, 4.8 Hz, Hα-Val), 4.24 (1H, bs, Hα-Orn), 3.29 (1H, bs, Hδ-Orn), 3.08 (1H, bd, J = 13.2 Hz, Hδ’-Orn), 2.09 (1H, dh, J = 9.2, 7.2 Hz, Hβ-Val), 1.78 (1H, m, Hβ-Orn), 1.51 (3H, m, Hβ’-Orn, Hγ-Orn), 1.37 (9H, s, CH3Boc), 1.35 (9H, s, CH3tBu), 0.86 (3H, d, J = 7.2 Hz, Hγ-Val), 0.84 (3H, d, J = 7.2 Hz, Hγ’-Val); 13C-NMR (CDCl3) δ 172.26 (s, CO-Orn), 170.84 (s, CO-Val), 156.97 (s, CO-Cbz), 155.79 (s, CO-Boc), 136.63 (s, Ar), 128.49 (d, Ar), 128.11 (d, Ar), 128.07 (d, Ar), 81.87 (s, C-Boc), 79.89 (s, C-tBu), 66.80 (t, CH2-Cbz), 57.65 (d, Cα-Val), 53.41 (d, Cα-Orn), 40.02 (t, Cδ-Orn), 31.28 (d, Cβ-Val), 30.10 (t, Cβ-Orn), 28.50 and 28.20 (q, CH3-Boc, CH3-tBu), 26.35 (t, Cγ-Orn), 19.17 (q, Cγ-Val), 17.25 (q, Cγ’-Val); FAB+MS m/z: 522 (53) [M + H]+, 466 (11) [M + H - C4H8]+, 422 (13) [M + H - C5H8O2]+, 414 (15) [M - C7H7O]+, 366 (100) [M + H - C5H8O2 - C4H8]+, 258 (13), 213 (20), 91 (100) [C7H7]+, 57 (44) [C4H9]+; HRFAB+MS: observed 522.3176 [M + H]+, (calcd. for C27H44N3O7, 522.3179).
Boc-D-Orn(Cbz)-Val-OtBu (2b): Colorless syrup; [α] + 15.7 (c 1.04, CHCl3); IR: 3,339, 2,973, 2,935, 2,877, 1,712, 1,666, 1,525, 1,456, 1,391, 1,368, 1,254, 1,165, 1,022, 738, 699 cm-1; 1H-NMR (CDCl3) δ 7.36-7.25 (5H, m, Ar), 6.89 (1H, bs, NH-Val), 5.36 (1H, bd, J = 6.6 Hz, NH-Orn), 5.25 (1H, bs, NHδ-Orn), 5.08 (2H, s, CH2-Cbz), 4.39 (1H, dd, J = 8.8, 4.4 Hz, Hα-Val), 4.23 (1H, bs, Hα-Orn), 3.21 (2H, m, Hδ-Orn), 2.10 (1H, dh, J = 9.2, 7.2 Hz, Hβ-Val), 1.85 (1H, m, Hβ-Orn), 1.60 (3H, m, Hβ’-Orn, Hγ-Orn), 1.45 (9H, s, CH3Boc), 1.43 (9H, s, CH3tBu), 0.92 (3H, d, J = 7.0 Hz, Hγ-Val), 0.89 (3H, d, J = 7.0 Hz, Hγ’-Val); 13C-NMR (CDCl3) δ 172.06 (s, CO-Orn), 170.78 (s, CO-Val), 156.74 (s, CO-Cbz), 155.71 (s, CO-Boc), 136.69 (s, Ar), 128.50 (d, Ar), 128.06 (d, Ar), 82.00 (s, C-Boc), 80.06 (s, C-tBu), 66.68 (t, CH2-Cbz), 57.60 (d, Cα-Val), 54.09 (d, Cα-Orn), 40.37 (t, Cδ-Orn), 31.46 (d, Cβ-Val), 30.07 (t, Cβ-Orn), 28.47 and 28.19 (q, CH3-tBu, CH3-tBu), 26.39 (t, Cγ-Orn), 19.11 (q, Cγ-Val), 17.77 (q, Cγ’-Val); FAB+MS m/z: 522 (9) [M + H]+, 466 (3) [M + H - C4H8]+, 422 (4) [M + H - C5H8O2]+, 410 (9), 366 (39) [M + H - C9H17O2]+, 258 (8), 213 (15), 91 (100) [C7H7]+, 72 (28) [C4H8O] +, 57 (38) [C4H9]+; HRFAB+MS: observed 522.3176 [M + H]+, (calcd. for C27H44N3O7, 522.3179).
Boc-Orn(Cbz)-Phe-OtBu (
2c): White solid; Mp 104-105 °C {lit [
30] 102 °C}; [α] + 25.8 (c 1.01, CHCl
3); IR: 3,364, 2,977, 2,880, 1,732, 1,690, 1,668, 1,524, 1,452, 1,368, 1,280, 1,240, 1,164, 1,027 cm
-1;
1H-NMR (CDCl
3): δ 7.35-7.15 (10H, m, Ar), 6.82 (1H, d,
J = 6.8 Hz, NH-Phe), 5.18 (1H, d,
J = 6.8 Hz, NH-Orn), 5.06 (2H, d,
J = 12.8 Hz, CH
2-Cbz, NHδ-Orn), 5.02 (1H, d,
J = 12.8 Hz, CH
2-Cbz), 4.70 (1H, dd,
J = 14.0, 6.0 Hz, Hα-Phe), 4.24 (1H, bs, Hα-Orn), 3.33 (1H, bs, Hδ-Orn), 3.13 (1H, m, Hδ’-Orn), 3.08 (1H, dd,
J = 13.6, 14.0 Hz, Hβ-Phe), 3.04 (1H, dd,
J = 13.6, 6.0 Hz, Hβ’-Phe), 2.14 (1H, m, Hγ-Orn), 1.81 (1H, m, Hγ’-Orn), 1.53 (2H, m, Hβ, Hβ’-Orn), 1.43 (9H, s, CH
3tBu), 1.37 (9H, s, CH
3Boc);
13C-NMR (CDCl
3): δ 171.80 (s, CO-Orn), 170.48 (s, CO-Phe), 156.94 (s, CO-Cbz), 155.66 (s, CO-Boc), 136.61 (s, Ar-Cbz), 136.23 (s, Ar), 129.58 (d, Ar), 128.58 (d, Ar), 128.49 (d, Ar), 128.16 (d, Ar), 127.03 (d, Ar), 82.41 (s, C-Boc), 80.07 (s, C-
tBu), 66.88 (t, CH
2-Cbz), 53.81 (d, Cα-Phe), 53.35 (d, Cα-Orn), 39.97 (t, Cδ-Orn), 38.16 (t, Cβ-Phe), 30.25 (t, Cγ-Orn), 28.44 and 28.04 (q, CH
3-
tBu, CH
3-
tBu), 26.27 (t, Cβ-Orn); FAB
+MS
m/z: 570 (56) [M + H]
+, 556 (9) [M + H - CH
2]
+, 514 (8) [M + H - C
4H
8]
+, 470 (56) [M + H - C
5H
8O
2]
+, 414 (90) [M + H - C
5H
8O
2 - C
4H
8]
+, 306 (12) [M + H - C
14H
19NO
3 - CH
3]
+, 261 (17), 204 (14), 154 (27), 120 (27), 91 (100) [C
7H
7]
+, 57 (44) [C
4H
9]
+; HRFAB
+MS: observed 570.3152 [M + H]
+, (calcd. for C
31H
44N
3O
7, 570.3179).
Boc-Orn(Cbz)-Phe-OMe (
2d): White solid; Mp 118-120 °C {lit [
31] 106-118 °C}; [α] - 7.5 (c 1.01, MeOH) {lit [
31] - 7.6 (c 1.1, MeOH)}; IR: 3,339, 2,974, 2,939, 2,876, 1,743, 1,677, 1,531, 1,449, 1,369, 1,278, 1,249, 1,172, 1,032 cm
-1;
1H-NMR (CDCl
3) δ 7.36-7.10 (10H, m, Ar), 7.00 (1H, d,
J = 7.6 Hz, NH-Phe), 5.24 (1H, d,
J = 8.0 Hz, NH-Orn), 5.12 (1H, t,
J = 6.0 Hz, NHδ-Orn), 5.03 (1H, d,
J = 12.8 Hz, CH
2-Cbz), 4.99 (1H, d,
J = 12.8 Hz, CH
2-Cbz), 4.83 (1H, dd,
J = 13.2, 6.4 Hz, Hα-Phe), 4.25 (1H, bs, Hα-Orn), 3.67 (3H, s, OCH
3), 3.33 (1H, m, Hδ-Orn), 3.12 (1H, m, Hδ’-Orn), 3.11 (1H, dd,
J = 13.6, 5.6 Hz, Hβ-Phe), 3.05 (1H, dd,
J = 13.6, 6.8 Hz, Hβ’-Phe), 1.78 (1H, m, Hγ-Orn), 1.52 (3H, m, Hβ, Hβ’, Hγ-Orn), 1.42 (9H, s, CH
3Boc);
13C-NMR (CDCl
3) δ 172.10 (s, CO-Phe), 171.91 (s, CO-Orn), 156.98 (s, CO-Cbz), 155.68 (s, CO-Boc), 136.58 (s, Ar-Cbz), 135.95 (s, Ar), 129.31 (d, Ar), 128.64 (d, Ar), 128.55 (d, Ar), 128.14 (d, Ar), 127.14 (d, Ar), 80.04 (s, C-Boc), 66.82 (t, CH
2-Cbz), 53.48 (d, Cα-Phe), 53.28 (d, Cα-Orn), 52.47 (q, OCH
3), 39.95 (t, Cδ-Orn), 38.05 (t, Cβ-Phe), 30.26 (t, Cγ-Orn), 28.52 (q, CH
3Boc), 26.24 (t, Cβ-Orn); FAB
+MS
m/z: 528 (11) [M + H]
+, 472 (7) [M + H - C
4H
9]
+, 428 (62) [M + H - C
5H
8O
2]
+, 320 (9) [C
17H
24N
2O
4]
+, 275(10), 180 (19) [C
10H
14NO
2]
+, 120 (31) [C
8H
10N]
+, 91 (100) [C
7H
7]
+, 57 (44) [C
4H
9]
+; HRFAB
+MS: observed 528.2686 [M + H]
+, (calcd. for C
28H
38N
3O
7, 528.2710).
Boc-Gly-Phe-OtBu (2e): Colorless syrup; [α] + 47.2 (c 1.1, CHCl3); IR: 3,414, 3,336, 2,979, 2,934, 1,727, 1,671, 1,521, 1,452, 1,369, 1,220, 1,160, 1,044, 942, 850, 743, 701 cm-1; 1H-NMR (CDCl3): δ 7.30-7.13 (5H, m, Ar), 6.72 (1H, d, J = 8.0 Hz, NH-Phe), 5.32 (1H, dd, J = 4.8, 4.8 Hz, NH-Gly), 4.75 (1H, dd, J = 14.0, 6.0 Hz, Hα-Phe), 3.83 (1H, dd, J = 16.4, 5.2 Hz, Hα-Gly), 3.74 (1H, dd, J = 16.4, 5.2 Hz, Hα’-Gly), 3.08 (2H, d, J = 6.0 Hz, Hβ-Phe), 1.44 (9H, s, CH3-tBu), 1.39 (9H, s, CH3-Boc); 13C-NMR (CDCl3) δ 170.39 (s, CO-Phe), 169.07 (s, CO-Gly), 155.97 (s, CO-tBu), 136.63 (s, Ar), 129.54 (d, Ar), 128.43 (d, Ar), 127.00 (d, Ar), 82.54 (s, C-Boc), 80.21 (s, C-tBu), 53.66 (d, Cα-Phe), 44.32 (t, Cα-Gly), 38.22 (t, Cβ-Phe), 28.47 and 28.09 (q, CH3-tBu, CH3-tBu); FAB+MS m/z: 379 (41) [M + H]+, 323 (16) [M + H - C4H8]+, 267 (100) [M + H - C4H8 - C4H8]+, 223 (25) [M + H - C7H10NO3]+, 166 (13), 154 (57), 120 (28), 57 (38) [C4H9]+; HRFAB+MS: observed 379.2267 [M + H]+, (calcd. for C20H31N2O5, 379.2233).
Boc-Phe-Phe-OtBu (
2f): Colorless crystals; Mp 125-127 °C; [α] + 34.1 (c 1.0, CHCl
3); IR,
1H-NMR and
13C-NMR (CDCl
3) are in agreement with previously reported data [
32]; FAB
+MS
m/z: 469 (27) [M + H]
+, 413 (10) [M + H - C
4H
8]
+, 357 (58) [M + H - C
4H
8 - C
4H
8]
+, 313 (78) [M + H - C
4H
8 - C
5H
8O
2]
+, 166 (15), 120 (100), 57 (45) [C
4H
9]
+; HRFAB
+MS: observed 469.2724 [M + H]
+, (calcd. for C
27H
37N
2O
5, 469.2702).
Boc-Val-Phe-OtBu (2g): Colorless crystals; Mp 112-115 °C; [α] + 29.0 (c 1.01, CHCl3); IR: 3,337, 3,282, 2,972, 2,936, 2,874, 1,733, 1,689, 1,659, 1,525, 1,457, 1,371, 1,248, 1,162, 1,020, 848, 752, 696 cm-1; 1H-NMR (CDCl3): δ 7.30-7.15 (5H, m, Ar), 6.50 (1H, d, J = 7.2 Hz, NH-Phe), 5.16 (1H, d, J = 8.8 Hz, NH-Val), 4.74 (1H, dd, J = 14.0, 6.4 Hz, Hα-Phe), 3.94 (1H, dd, J = 8.4, 6.8Hz, Hα-Val), 3.08 (2H, dd, J = 6.4, 4.4 Hz, Hβ-Phe), 2.09 (1H, m, Hβ-Val), 1.45 (9H, s, CH3-tBu), 1.38 (9H, s, CH3-Boc), 0.93 (3H, d, J = 6.8 Hz, Hγ-Val), 0.88 (3H, d, J = 6.4 Hz, Hγ’-Val); 13C-NMR (CDCl3) δ 171.16 (s, CO-Val), 170.46 (s, CO-Phe), 155.79 (s, CO-Boc), 136.10 (s, Ar), 129.57 (d, Ar), 128.45 (d, Ar), 127.02 (d, C4), 82.42 (s, C-Boc), 79.89 (s, C-tBu), 60.03 (d, Cα-Val), 53.78 (d, Cα-Phe), 38.34 (t, Cβ-Phe), 31.16 (d, Cβ-Val), 28.52 and 28.09 (q, CH3-tBu, CH3-Boc), 19.44 (q, Cγ-Val), 17.97 (q, Cγ’-Val); FAB+MS m/z: 421 (59) [M + H]+, 365 (17) [M + H - C4H8]+, 309 (100) [M + H - C4H8 - C4H8]+, 265 (95) [M + H - C4H8 - C5H8O2]+, 166 (30), 120 (60), 72 (53), 57 (48) [C4H9]+; HRFAB+MS: observed 421.2666 [M + H]+, (calcd. for C23H37N2O5, 421.2702).
Boc-Gly-Val-OtBu (2h): Colorless oil; [α] + 23.8 (c 1.02, CHCl3); IR: 3,333, 2,975, 2,935, 2,878, 1,726, 1,671, 1,524, 1,458, 1,391, 1,369, 1,281, 1,251, 1,166, 1,052, 943, 848, 786 cm-1; 1H-NMR (CDCl3) δ 6.80 (1H, bs, NH-Val), 5.48 (1H, bd, J = 5.6 Hz, NH-Gly), 4.46 (1H, dd, J = 9.2, 4.4 Hz, Hα-Val), 3.87 (1H, dd, J = 16.4, 5.6 Hz, Hα-Gly), 3.80 (1H, dd, J = 16.4, 5.6 Hz, Hα’-Gly), 2.17 (1H, hd, J = 7.2, 4.4 Hβ-Val), 1.47 (9H, s, CH3Boc), 1.46 (9H, s, CH3tBu), 0.94 (3H, d, J = 7.2 Hz, Hγ-Val), 0.89 (3H, d, J = 7.2 Hz, Hγ’-Val); 13C-NMR (CDCl3) δ 170.95 (s, CO-Val), 169.52 (s, CO-Gly), 156.12 (s, CO-Boc), 82.16 (s, C-Boc), 80.22 (s, C-tBu), 57.42 (d, Cα-Val), 44.50 (t, Cα-Gly), 31.58 (d, Cβ-Val), 28.46 and 28.20 (q, CH3-Boc, CH3-tBu), 19.08 (q, Cγ-Val), 17.68 (q, Cγ’-Val); FAB+MS m/z: 331 (28) [M + H]+, 275 (18) [M + H - C4H8]+, 219 (100) [M + H - C4H8 - C4H8]+, 175 (20) [M + H - C4H8 - C5H8O2]+, 72 (17) [C4H8O]+, 57 (20) [C4H9]+; HRFAB+MS: observed 331.2251 [M + H]+, (calcd. for C16H31N2O5, 331.2233).
Boc-Phe-Val-OtBu (2i): White solid; Mp 119-121 °C; [α]DHg (365 nm) + 9.0 (c 0.5, CHCl3); IR: 3,327, 2,975, 2,933, 1,735, 1,687, 1,651, 1,538, 1,367, 1,252, 1,165, 1,025, 855 cm-1; 1H-NMR (CDCl3) δ 7.32 - 7.10 (5H, Ar), 6.56 (1H, d, J = 8.4 Hz, NH-Phe), 5.20 (1H, d, J = 8.0 Hz, NH-Val), 4.41 (1H, m, Hα-Phe), 4.36 (1H, dd, J = 8.0, 4.8 Hz, Hα-Val), 3.10 (2H, dd, J = 13.6, 6.4 Hz, Hβ-Phe), 3.04 (1H, dd, J = 13.6, 6.8 Hz, Hβ’-Phe), 2.17 (1H, hd, J = 6.8, 4.8 Hβ-Val), 1.45 (9H, s, CH3Boc), 1.41 (9H, s, CH3tBu), 0.88 (3H, d, J = 6.8 Hz, Hγ-Val), 0.89 (3H, d, J = 6.8 Hz, Hγ’-Val); 13C-NMR (CDCl3) δ 171.07 (s, CO-Phe), 170.45 (s, CO-Val), 155.39 (s, CO-Boc), 136.69 (s, C1), 129.36 (d, C2 and C6), 128.58 (d, C3 and C5), 126.85 (d, C4), 81.98 (s, C-Boc), 80.10 (s, C-tBu), 57.65 (d, Cα-Val), 55.95 (d, Cα-Phe), 38.23 (t, Cβ-Phe), 31.62 (d, Cβ-Val), 28.43 and 28.26 (q, CH3-Boc, CH3-tBu), 18.92 (q, Cγ-Val), 17.91 (q, Cγ’-Val); FAB+MS m/z: 421 (46) [M + H]+, 365 (15) [M + H - C4H8]+, 309 (100) [M + H - C4H8 - C4H8]+, 265 (96) [M + H - C4H8 - C5H8O2]+, 120 (40), 72 (32) [C4H8O]+, 57 (44) [C4H9]+; HRFAB+MS: observed 421.2688 [M + H]+, (calcd. for C23H37N2O5, 421.2702).
Boc-Val-Val-OtBu (2j): White solid; Mp 132-134 °C; [α] - 6.8 (c 1.1, CHCl3); IR: 3,309, 2,972, 2,934, 2,888, 1,744, 1,685, 1,651, 1,536, 1,464, 1,372, 1,301, 1,254, 1,219, 1,157, 1,017, 855 cm-1; 1H-NMR (CDCl3) δ 6.35 (1H, d, J = 7.6 Hz, NH-Val), 5.09 (1H, d, J = 8.8 Hz, NH-Val), 4.36 (1H, dd, J = 8.4, 4.4 Hz, Hα-Val), 3.86 (1H, dd, J = 7.6, 7.6 Hz, Hα-Val), 2.08 (2H, m, Hβ-Val, Hβ-Val), 1.40 (9H, s, CH3Boc), 1.38 (9H, s, CH3tBu), 0.90, 0.87, 0.86, 0.84 (3H each, d, J = 6.8 Hz, Hγ-Val, Hγ’-Val, Hγ-Val, Hγ’-Val); 13C-NMR (CDCl3) δ 171.58 (s, CO-Val), 170.81 (s, CO-Val), 155.93 (s, CO-Boc), 81.16 (s, C-Boc), 80.00 (s, C-tBu), 60.38 (d, Cα-Val), 57.70 (d, Cα-Val), 31.64 (d, Cβ-Val), 31.06 (d, Cβ-Val), 28.56 and 28.29 (q, CH3-Boc, CH3-tBu), 19.57, 19.16, 18.21, 17.96 (q, Cγ-Val, Cγ’-Val, Cγ-Val, Cγ’-Val); FAB+MS m/z: 373 (46) [M + H]+, 317 (28) [M + H - C4H8]+, 261 (89) [M + H - C4H8 - C4H8]+, 217 (94) [M + H - C4H8 - C5H8O2]+, 116 (26) [C5H10NO2]+, 72 (100) [C4H8O]+, 57 (47) [C4H9]+; HRFAB+MS: observed 373.2711 [M + H]+, (calcd. for C19H37N2O5, 373.2702).
Boc-Phe-Sar-OtBu (2k): Colorless syrup (73:27 rotamer mixture); [α] - 22.5 (c 1.58, CHCl3); IR: 3,427, 3,322, 2,978, 2,933, 1,741, 1,710, 1,652, 1,491, 1,367, 1,236, 1,164, 1,049, 1,020, 952, 850, 759 and 701 cm-1; 1H-NMR (CDCl3) δ 7.30-7.00 (5H, m, Ar), 5.31 (1H, d, J = 8.8 Hz, NH-Phe), 4.80 (1H, dd, J = 15.4, 6.6 Hz, Hα-Phe), 3.92 (1H, d, J = 17.2 Hz, Hα-Sar), 3.84 (1H, d, J = 17.2 Hz, Hα’-Sar), 2.83 (3H, s, NCH3-Sar), 2.97 (1H, dd, J = 13.6, 7.2 Hz, Hβ-Phe), 2.90-2.86 (1H, m, Hβ’-Phe), 1.38 (9H, s, CH3-Boc), 1.31 (9H, s, CH3-tBu); 13C-NMR (CDCl3) δ 172.11 (s, CO-Phe), 167.89 (s, CO-Sar), 155.09 (s, CO-Boc), 136.42 (s, C1), 129.65 (d, C2,C6), 128.38 (d, C3,C5), 126.85 (d, C4), 82.03 (s, C-tBu), 79.67 (s, C-Boc), 51.52 (d, Cα-Phe), 50.45 (t, Cα-Sar), 39.57 (t, Cβ-Phe), 36.34 (q, NCH3-Sar), 28.49 and 28.24 (q, CH3-Boc, CH3-tBu); FAB+MS m/z: 393 (27) [M + H]+, 337 (15) [M + H - C4H8]+, 281 (76) [M + H - 2 C4H8]+, 263 (15) [M + H - C4H8 - C4H8O]+, 237 (100) [M + H - C4H8 - C5H8O2]+, 164 (18), 120 (81), 90 (43) [C7H6]+, 57 (78) [C4H9]+; HRFAB+MS: observed 393.2409 [M + H]+, (calcd. for C21H33N2O5, 393.2389).
Boc-Phe-Sar-OMe (2l): Colorless syrup (77:23 rotamer mixture); [α] + 19.1 (c 0.54, CHCl3); IR: 3,427, 3,322, 2,977, 2,943, 1,751, 1,708, 1,652, 1,490, 1,407, 1,365, 1,250, 1,212, 1,171, 1,047, 1,021, 751 and 702 cm-1; 1H-NMR (CDCl3) δ 7.32-7.18 (5H, m, Ar), 5.34 (1H, d, J = 8.8 Hz, NH-Phe), 4.88 (1H, dd, J = 14.8, 6.8 Hz, Hα-Phe), 4.14 (1H, d, J = 17.2 Hz, Hα-Sar), 3.99 (1H, d, J = 17.2 Hz, Hα'-Sar), 3.73 (3H, s, OCH3), 3.04 (1H, dd, J = 13.6, 7.2 Hz, Hβ-Phe), 2.95 (1H, dd, J = 13.6, 6.4 Hz, Hβ’-Phe), 2.87 (3H, s, NCH3-Sar), 1.40 (9H, s, CH3-Boc); 13C-NMR (CDCl3) δ 172.35 (s, CO-Phe), 169.28 (s, CO-Sar), 155.07 (s, CO-Boc), 136.33 (s, Ar), 129.65 (d, Ar), 128.45 (d, Ar), 126.93 (d, C4), 79.81 (s, C-Boc), 52.38 (q, OCH3), 51.59 (d, Cα-Phe), 49.66 (t, Cα-Sar), 39.80 (t, Cβ-Phe), 36.43 (q, NCH3-Sar), 28.55 (q, CH3-Boc); FAB+MS m/z: 351 (33) [M + H]+, 295 (55) [M + H - C4H8]+, 251 (100) [M + H - C5H9O2]+, 164 (16), 120 (67), 104 (70) [C4H10NO2]+, 57 (44) [C4H9]+, 44 (18); HRFAB+MS: observed 351.1907 [M + H]+, (calcd. for C18H27N2O5, 351.1920).
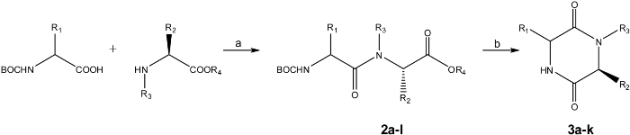
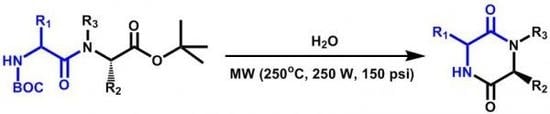
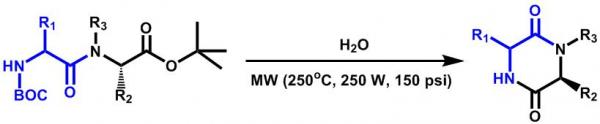
3.4. General procedure for the syntheses of 2,5-diketopiperazines 3a-3k
Each
Nα-Boc-dipeptidyl ester (0.25 mmol) was dissolved or suspended in water (1 mL) and heated during 10 minutes at 250 °C and 150 psi, using a monomode CEM Discover microwave apparatus at 250 W. The resulting suspension was filtered through a Hirsch funnel and washed with water (5 mL), the solid was dried under high vacuum and analyzed without further purification by NMR. Compounds
3h and
3k were water soluble, and in these cases, resulting solutions were lyophilized and the solids purified as indicated in
Table 2 and analyzed by NMR.
Cyclo[Val-Orn(Cbz)] (
3a): White solid; Mp 202-204 °C {lit [
33] 206-208 °C}; [α] - 26.0 (c 0.27, DMSO) {lit [
33] - 47.4 (c 1 %)}; IR: 3,434, 3,333, 3,201, 3,094, 3,052, 2,965, 2,878, 1,678, 1,531, 1,444, 1,258, 1,142, 1,025, 774, 696, 629 cm
-1;
1H-NMR (DMSO): δ 8.14 (1H, s, NH-Orn), 8.04 (1H, s, NH-Val), 7.39-7.26 (6H, m, Ar, NHδ-Orn), 5.00 (2H, s, CH
2-Cbz), 3.81 (1H, t,
J = 4.8 Hz, Hα-Orn), 3.66 (1H, bs, Hα-Val), 2.97 (1H, dd,
J = 12.8, 6.4 Hz, Hδ-Orn), 2.14 (1H, m, Hβ-Val), 1.69 (1H, m, Hβ-Orn), 1.62 (1H, m, Hβ’-Orn), 1.46 (2H, m, Hγ-Orn), 0.93 (3H, d,
J = 7.2 Hz, Hγ-Val), 0.82 (3H, m, d,
J = 7.2 Hz, Hγ’-Val);
13C-NMR (DMSO): δ 167.83 (s, CO-Orn), 166.88 (s, CO-Val), 156.06 (s, CO-Cbz), 137.21 (s, Ar), 128.33 (d, Ar), 127.64 (d, Ar), 65.18 (t, CH
2-Cbz), 59.43 (d, Cα-Val), 53.75 (d, Cα-Orn), 40.18 (t, Cδ-Orn), 31.33 (d, Cβ-Val), 31.16 (t, Cβ-Orn), 25.35 (t, Cγ-Orn), 18.74 (q, Cγ-Val), 17.29 (q, Cγ’-Val); FAB
+MS
m/z: 348 (87) [M + H]
+, 307 (100) [M + H - C
3H
6]
+, 289 (61), 240 (22) [M + H - C
7H
8O]
+, 219 (25) [C
12H
15N
2O
2]
+, 214 (12), 195 (12), 165 (12); HRFAB
+MS: observed 348.1887 [M + H]
+, (calcd. for C
18H
26N
3O
4, 348.1923).
Cyclo[Val-D-Orn(Cbz)] (3b): White solid; Mp 212-214 °C;[α] + 11.7 (c 0.5, MeOH);IR: 3,347, 3,192, 3,054, 2,962, 1,675, 1,540, 1,460, 1,265, 1,143, 1,031, 852, 696 cm-1; 1H-NMR (DMSO): δ 8.11 (1H, s, NH-Orn), 7.32 (6H, m, Ar, NH-Val), 4.99 (2H, s, CH2-Cbz), 4.03 (1H, bs, Hα-Val), 3.88 (1H, bt, Hα-Orn), 2.96 (1H, d, J = 5.6 Hz, Hδ-Orn), 2.09 (1H, m, Hβ-Val), 1.65 (1H, m, Hβ-Orn), 1.62 (1H, m, Hβ’-Orn), 1.41 (2H, m, Hγ-Orn), 0.92 (3H, d, J = 6.8 Hz, Hγ-Val), 0.83 (3H, m, d, J = 7.0 Hz, Hγ’-Val); 13C-NMR (DMSO): δ 168.13 (s, CO-Orn), 167.67 (s, CO-Val), 156.17 (s, CO-Cbz), 137.25 (s, Ar), 128.44 (d, Ar), 127.77 (d, Ar), 65.30 (t, CH2-Cbz), 59.79 (d, Cα-Val), 53.27 (d, Cα-Orn), 40.18 (t, Cδ-Orn), 32.19 (d, Cβ-Val), 29.64 (t, Cβ-Orn), 24.41 (t, Cγ-Orn), 18.54 (q, Cγ-Val), 17.08 (q, Cγ’-Val); FAB+MS m/z: 348 (3) [M + H]+, 219 (5), 154 (12), 130 (30), 107 (8), 91 (100), 85 (28), 72 (25); HRFAB+MS: observed 348.1949 [M + H]+, (calcd. for C18H26N3O4, 348.1923).
Cyclo[Phe-Orn(Cbz)] (
3c): White solid; Mp 200-202 °C {lit [
33] 210-212 °C}; [α] - 24.8 (c 0.24, DMSO) {lit [
33] - 12.9 (c 1 %)}; IR: 3,322, 3,189, 3,039, 2,965, 2,894, 1,669, 1,532, 1,458, 1,338, 1,246, 1,134, 1,101, 1,016, 852, 753, 669 cm
-1;
1H-NMR (DMSO): δ 8.16 (1H, s, NH-Phe), 8.05 (1H, s, NH-Orn), 7.40 - 7.05 (10H, m, Ar), 5.00 (2H, s, CH
2-Cbz), 4.17 (1H, s, Hα-Phe), 3.57 (1H, bs, Hα- Orn), 3.13 (1H, dd,
J = 13.6, 4.0 Hz, Hβ-Phe), 2.83 (1H, dd,
J = 13.6, 5.2 Hz, Hβ’-Phe), 2.68 (1H, dd,
J = 12.8, 6.4 Hz, Hδ-Orn), 1.04 (1H, m, Hβ-Orn), 0.85 (2H, m, Hγ-Orn), 0.65 (2H, m, Hβ’-Orn);
13C-NMR (DMSO): δ 166.70 (s, CO-Orn), 166.00 (s, CO-Phe), 155.79 (s, CO-Cbz), 137.16 (s, Ar-Cbz), 135.92 (s, Ar), 130.16 (d, Ar), 128.27 (d, Ar), 127.92 (d, Ar), 127.69 (d, Ar), 126.57 (d, Ar), 65.10 (t, CH
2-Cbz), 55.30 (d, Cα-Phe), 53.61 (d, Cα-Orn), 39.95 (t, Cδ-Orn), 38.16 (t, Cβ-Phe), 30.66 (t, Cβ-Orn), 24.33 (t, Cγ-Orn); FAB
+MS
m/z: 396 (100) [M + H]
+, 352 (32) [M + H - H
2NCO]
+, 335 (9), 307 (53) [M + H - C
7H
6]
+, 289 (32) [M + H - C
7H
7O]
+, 243 (16) [M + H - C
14H
16NO
2]
+, 219 (18) [M + H - C
10H
12NO
2]
+, 165 (14); HRFAB
+MS: observed 396.1949 [M + H]
+, (calcd. for C
22H
26N
3O
4, 396.1923).
Cyclo(Phe-Gly) (
3e): White solid; Mp 266-268 °C {lit [
34] 271-273 °C }; [α] + 26.6 (c 0.95, DMSO) {lit [
34] + 7.3 (c 0.95, DMSO)}; IR: 3,426, 3,190, 3,057, 2,977, 2,921, 2,878, 1,676, 1,462, 1,332, 1,086, 1,004, 847, 794, 758, 702 cm
-1;
1H- and
13CNMR (DMSO) are in agreement with previously reported data [
35]; FAB
+MS
m/z: 205 (13) [M + H]
+, 169 (15), 154 (24), 130 (43) [M + H - C
4H
4]
+, 85 (100) [C
3H
3NO
2]
+; HRFAB
+MS: observed 205.1058 [M + H]
+, (calcd. for C
11H
13N
2O
2, 205.0977).
Cyclo(Phe-Phe) (
3f): White solid; [α] - 42.8 (c 0.2, AcOH);
1H- and
13C-NMR (DMSO), and HRFAB
+MS are in agreement with previously reported data [
36]; IR: 3,440, 3,318, 3,198, 3,056, 2,970, 2,929, 1,669, 1,458, 1,338, 1,197, 1,088, 1,013, 759 and 700 cm
-1.
Cyclo(Val-Phe) (
3g): White solid; Mp 264-266 °C {lit [
36] 263-265 °C}; [α] - 66.0 (c 0.28, DMSO) {lit [
37] - 64 (c 0.2 AcOH); [
34] - 43.3 (c 0.27 DMSO)}; IR: 3,440, 3,316, 3,192, 3,056, 2,967, 2,885, 1,668, 1,454, 1,341, 1,090, 859, 758, 699, cm
-1;
1H- and
13C-NMR (DMSO) are in agreement with previously reported data [
34,
37]; FAB
+MS
m/z: 247 (100) [M + H]
+, 219 (13) [M + H - CO]
+, 217 (10), 203 (7), 165 (5); HRFAB
+MS: observed 247.1446 [M + H]
+, (calcd. for C
14H
19N
2O
2, 247.1447).
Cyclo(Val-Gly) (
3h): White solid; Mp 210-212 °C {lit [
38] 256 °C}; [α] + 32.9 (c 0.46, H
2O) {lit [
38] + 23.7 (c 1.0, H
2O)}; IR: 3,431, 3,199, 3,055, 2,925, 2,859, 1,670, 1,461, 1,381, 1,346, 1,109, 1,047, 808, 621 cm
-1;
1H- and
13C-NMR (DMSO) are in agreement with previously reported data [
38,
39]; FAB
+MS
m/z: 157 (94) [M + H]
+, 154 (100) [M - H
2], 136 (78), 120 (12), 107 (25), 85 (58) [C
5H
9O]
+, 77 (25), 55 (32), 43 (27) [C
3H
7]
+, 41 (25); HRFAB
+MS: observed 157.0957 [M + H]
+, (calcd. for C
7H
13N
2O
2, 157.0957).
Cyclo(Val-Val) (
3j): Colorless needless; Mp 268 °C with sublimation {lit [
40] 268 °C}; [α] - 54.8 (c 0.5, AcOH) {lit [
15] - 62 (c 0.5, AcOH); IR: 3,434, 3,325, 3,193, 3,100, 3,057, 2,966, 2,880, 1,664, 1,449, 1,346, 1,293, 847 cm
-1;
1H-NMR (DMSO): δ 7.96 (1H, s, NH-Val), 3.69 (1H, s, Hα-Val), 2.18 (1H, m, Hβ-Val), 0.95 (3H, d,
J = 7.6 Hz, Hγ-Val), 0.83 (3H, d,
J = 7.6 Hz, Hγ’-Val);
13C-NMR (DMSO): δ 167.26 (s, CO), 59.09 (d, Cα), 31.06 (d, Cβ), 18.73 (q, Cγ), 17.33 (q, Cγ’); FAB
+MS
m/z: 199 (100) [M + H]
+, 197 (19), 169 (8); HRFAB
+MS: observed 199.1463 [M + H]
+, (calcd. for C
10H
19N
2O
2, 199.1447).
Cyclo(Sar-Phe) (
3k): Colorless needles; Mp 180-183 °C {lit [
41] 184-185 °C}; [α] + 56.0 (c 1.01, MeOH) {lit [
41] + 47.5 (c 2.3 MeOH)}; IR: 3,440, 3,253, 2,963, 2,929, 2,895, 1,656, 1,470, 1,441, 1,322, 1,102, 1,033, 870, 754, 699, cm
-1;
1H-NMR (CDCl
3): δ 7.60 (1H, bs, NH-Phe), 7.35-7.16 (5H, m, Ar), 4.31 (1H, bs, Hα-Phe), 3.48 (1H, d,
J = 17.6 Hz, Hα-Sar), 3.24 (1H, dd,
J = 13.6, 5.2 Hz, Hβ-Phe), 3.07 (1H, dd,
J = 13.6, 4.4 Hz, Hβ’-Phe), 2.81 (3H, s, NCH
3-Sar), 2.79 (1H, d,
J = 17.6 Hz, Hα’-Sar);
13C-NMR (CDCl
3): δ 166.32 (s, CO-Sar), 165.52 (s, CO-Phe), 134.99 (s, Ar), 130.07 (d, Ar), 128.63 (d, Ar), 127.58 (d, C
4), 56.53 (d, Cα-Phe), 50.92 (t, Cα-Sar), 40.94 (t, Cβ-Phe), 33.66 (s, NCH
3-Sar); FAB
+MS
m/z: 219 (100) [M + H]
+, 154 (50) [M + H - CO]
+, 136 (56), 91 (35) [C
7H
7]
+, 73 (58); HRFAB
+MS: observed 219.1134 [M + H]
+, (calcd. for C
12H
15N
2O
2, 219.1134).


{kind=link}
{kind=link}