Abstract
X-ray structures of two compounds isolated from wood knots of coniferous trees, namely dihydrokaempferol (3,5,8,13-tetrahydroxyflavanon) and lariciresinol (3,14-dimetoxy-7,10-epoxylignan-4,15,19-triol), are presented here. Diffraction data for the dihydrokaempferol crystals were collected on a CAD4 diffractometer and on a synchrotron for the lariciresinol crystal. The investigated compounds inhibit lipid peroxidation and lariciresinol is additionally a good scavenger of superoxide radicals. The structural data presented in this work provide a useful basis for designing more active compounds with potential use as antioxidants.
Introduction
Large amounts of bioactive phenolic compounds are present in the wood knots of several tree species. The amount of lignans in the knots can be up to several hundred times larger than in the adjacent stemwood [1,2,3,4,5]. The amount of extractable phenolic compounds is on average around 15% (w/w) in Picea abies, while Populus tremula and Abies balsamea can contain considerable amounts of interesting polyphenols. Those phenolic compounds can be potentially used as antioxidants in food, pharmaceuticals, and natural biocides such as bactericides, pesticides and fungicides [6]. Additionally, lignans are of great interest in the search for antitumor agents and have potential as chemotherapeutics [7,8,9,10].
The phenolic compounds are extracted from wood knots and purified by chromatographic methods [11,12]. The extract obtained from heartwood, foliage, phloem, bark, and cork of several species is a good resource of natural phenolic antioxidants [13,14,15,16] but it contains a mixture of different phenolic and nonphenolic compounds in the form of both glycosides and free aglycones. Glycosylation is not desirable, since it affects the antioxidant properties of phenolic compounds [17]. In comparison, the hydrophilic compounds in knots contain mainly free aglycones of flavonoids and lignans [1,2,3,4,5,18].
Wood knots of P. tremula and A. balsamea growing in Europe are rich in dihydrokaempferol (1) and lariciresinol (2) (Figure 1). Dihydrokaempferol - belonging to the flavanones group - shows a capacity to scavenge peroxyl radicals in vitro. The trapping capacity of that compound (expressed as the number of peroxy radicals in millimoles that are scavenged per gram of extract) is 0.78 mmol/g [19]. Lignans - among them lariciresinol - also inhibit lipid peroxidation. The trapping capacity of that compound in one of the test series was shown to be 7.3 mmol/g. In comparison, the trapping capacity of a well known antioxidant Trolox® was reported as 6.8 mmol/g in the same test series [12]. Lariciresinol also reveals a capacity to scavenge superoxide radicals. Scavenging of superoxide radicals in vitro expressed as IC50 values (i.e., concentration of extract required for scavenging of 50% of the radicals) for this compound is 13 μg/L. A X-ray crystallography structural investigation of dihydrokaempferol and lariciresinol is presented in this paper.
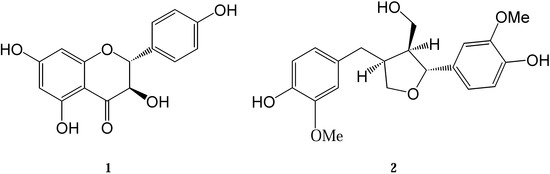
Figure 1.
Structural formula of compounds 1 and 2.
Results and Discussion
The crystal structures of two compounds isolated from the European tree species P. tremula and A. balsamea are presented here: dihydrokaempferol (3,5,8,13-tetrahydroxyflavanone) (1) and lariciresinol (3,14-dimethoxy-7,10-epoxylignan-4,15,19-triol) (2). Their chemical structures are depicted in Figure 1. The isolation and purification procedures of compound 1 and 2 and their spectroscopic characterization were described earlier [12,20]. Crystal data and experimental details for compound 1 and 2 are shown in Table 1. ORTEP views of the investigated molecules with the atom numbering schemes prepared using the program XP are shown in Figure 2 [21].
Two diffraction data sets were collected for 1 with different crystallization solvents and the structure was solved twice: 1a with molecules of ethanol and 1b with molecules of methanol trapped in the crystal lattices. Those are typical "host - guest" type inclusion crystals. The cell parameters a, b and c are similar for both 1a and 1b, which is typical for isostructural solvatomorphs [22]. The difference in cell volume is 34.5 A3, which well correlates with the volume of the two methylene groups that distinguish these two structures. The Robs factor for 1a is significantly higher than that of 1b (Table 1), so only 1b was further analyzed.
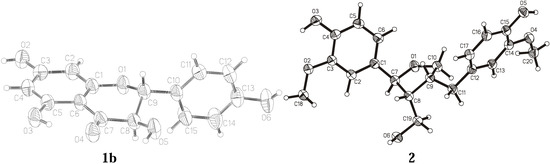
Figure 2.
Thermal ellipsoidal view with the atom numbering scheme of the molecules of 1b and 2.
The analysis of bond lengths of compound 1 and 2 shows that their values do not differ significantly from typical values for compounds deposited in Cambridge Crystallographic Data Centre [23]. Elongation of bonds C7-C8, C8-C9, O1-C9 and C9-C10 observed in 1 is typical and caused by asymmetry of heterocyclic ring O1,C1,C6,C7,C8,C9. That ring adopts a conformation halfway between a half-chair and a sofa. In comparison, the five-membered ring of 2 is in a half-chair conformation. The asymmetry parameters indicating the lowest discrepancy from the dominant symmetry elements are shown in Table 2. The C-C and C-O bonds in the five-membered ring of 2 and also the bonds between carbon atoms (C7, C11) and m-methoxy-p-hydroxyphenyl groups of 2 are elongated. The aromatic ring of the hydroxyphenyl substituent can rotate around the C9-C10 bond in the molecule of 1. Free rotation of the aromatic ring of the m-methoxy-p-hydroxyphenyl substituent in 2 can occur around only one bond (C1-C7), whilst the second substituent of that type can rotate around two bonds C9-C11 and C11-C12. The values of selected torsion angles of 1 and 2 are presented in Table 3.

Table 1.
Crystal data and experimental details for compound 1 and 2.
| Compound | 1a | 1b | 2 | ||||
|---|---|---|---|---|---|---|---|
| Molecular formula | C15H12O6*CH3CH2OH | C15H12O6*CH3OH | C20H24O6 | ||||
| Formula weight | 334.31 | 320.29 | 360.39 | ||||
| CCDC No. | 719360 | 719361 | 719362 | ||||
| Crystallographic system | triclinic | triclinic | monoclinic | ||||
| Space group | P1 | P1 | P21 | ||||
| a [Å] | 7.617(5) | 7.581(2) | 10.718(6) | ||||
| b [Å] | 10.349(3) | 10.275(2) | 5.656(3) | ||||
| c [Å] | 11.488(3) | 11.120(2) | 14.264(8) | ||||
| α [o] | 63.92(2) | 65.28(3) | |||||
| β [o] | 85.36(4) | 81.80(3) | 92.75(5) | ||||
| γ [o] | 79.18(3) | 76.61(3) | |||||
| V [Å3] | 798.9(6) | 764.4(3) | 863.7(8) | ||||
| Z | 2 | 2 | 2 | ||||
| Dc [g/cm3] | 1.390 | 1.392 | 1.386 | ||||
| μ [mm-1] | 0.918 | 0.936 | 0.102 | ||||
| Crystal dimensions [mm] | 0.60x0.40x0.02 | 0.56x0.12x0.1 | 1.00x0.06x0.02 | ||||
| Radiation, λ (Å) | CuKα, 1.54178 | CuKα, 1.54178 | synchrotron, 0.80420 | ||||
| hkl ranges: | h = | -9 | 0 | 0 | 9 | -14 | 14 |
| k = | -12 | 12 | -12 | 12 | -6 | 6 | |
| l = | -14 | 14 | -13 | 13 | -19 | 19 | |
| EAC correction: | min. | 0.8867 | 0.9392 | NA | |||
| max. | 0.9933 | 0.9980 | |||||
| ave. | 0.9294 | 0.9679 | |||||
| No. of reflections: | unique | 3545 | 3396 | 4342 | |||
| with I>0σ(I) | 3353 | 3210 | 3372 | ||||
| obs. with I>2σ(I) | 2982 | 2982 | 4007 | ||||
| No. of parameters refined | 472 | 454 | 332 | ||||
| Robs | 0.0691 | 0.0430 | 0.0431 | ||||
| wRobs | 0.1871 | 0.1376 | 0.1137 | ||||
| Rint | 0.0000 | 0.0000 | 0.0000 | ||||
| Sobs | 1.098 | 1.094 | 1.051 | ||||
Robs=Σ||Fo|-|Fc||/Σ|Fo|; wRobs=[Σ[w(Fo2-Fc2)2]/ [Σ[w(Fo2)2]]1/2; Rint=Σ|hi-heq|/Σhave ; Sobs=[Σ[w(Fo2-Fc2)2]/(n-p)]1/2, where n – no of reflections, p – no of parameters.
Table 2.
Asymmetry parameters [24] for heteroatom rings for compound 1b and 2.
| 1b | |||||
| molecule | 1 | 1’ | molecule | 1 | 1’ |
| ΔCsC6=ΔCsC9 | 12.2(8) | 12.4(8) | ΔC2C1-C6=ΔC2C8-C9 | 13.3(9) | 16.6(9) |
| 2 | |||||
| ΔCsC8 | 11.2(3) | ΔC2C8-C9 | 4.6(3) | ||
| ΔCsC9 | 17.8(3) | ΔC2C9-C10 | 40.2(3) | ||
1 and 1’ – molecules in the asymmetric unit.
Table 3.
Selected torsion angles (°) for compounds 1b and 2.
| 1b | ||||||||||||
| molecule | 1 | 1’ | molecule | 1 | 1’ | |||||||
| C1 | C2 | C3 | O2 | -177.3(3) | -179.2(4) | C1 | O1 | C9 | C10 | 172.7(3) | -179.7(3) | |
| O2 | C3 | C4 | C5 | 176.5(3) | -179.7(4) | O5 | C8 | C9 | C10 | 60.4(4) | 54.0(4) | |
| C3 | C4 | C5 | O3 | -178.7(3) | 179.1(3) | C7 | C8 | C9 | C10 | -176.4(3) | 176.8(3) | |
| O3 | C5 | C6 | C1 | 180.0(3) | -178.8(3) | O1 | C9 | C10 | C15 | -65.8(4) | -66.5(4) | |
| O3 | C5 | C6 | C7 | 2.9(5) | 4.4(5) | C8 | C9 | C10 | C15 | 52.8(5) | 53.1(5) | |
| C6 | C7 | C8 | O5 | 160.4(3) | 161.4(3) | O1 | C9 | C10 | C11 | 118.9(4) | 114.5(4) | |
| C5 | C6 | C7 | O4 | -7.3(6) | -7.6(6) | C8 | C9 | C10 | C11 | -122.4(4) | -125.9(4) | |
| O4 | C7 | C8 | O5 | -22.5(5) | -20.0(5) | O6 | C13 | C14 | C15 | 179.4(5) | -179.5(4) | |
| O4 | C7 | C8 | C9 | -144.9(3) | -142.1(3) | |||||||
| 2 | ||||||||||||
| C18 | O2 | C3 | C2 | 0.4(2) | O1 | C7 | C8 | C19 | -88.0(1) | |||
| C18 | O2 | C3 | C4 | 179.8(1) | C1 | C7 | C8 | C19 | 149.7(1) | |||
| O2 | C3 | C4 | O3 | -0.1(2) | O1 | C7 | C8 | C9 | 33.6(1) | |||
| C2 | C3 | C4 | O3 | 179.3(1) | C19 | C8 | C9 | C11 | -43.2(2) | |||
| O2 | C3 | C4 | C5 | 180.0(1) | C9 | C11 | C12 | C17 | 84.7(2) | |||
| O3 | C4 | C5 | C6 | -178.6(1) | C9 | C11 | C12 | C13 | -92.5(2) | |||
| C10 | O1 | C7 | C1 | 107.9(1) | C20 | O4 | C14 | C13 | -0.2(2) | |||
| C10 | O1 | C7 | C8 | -15.4(1) | C20 | O4 | C14 | C15 | 177.7(1) | |||
| C6 | C1 | C7 | O1 | -19.2(2) | C12 | C13 | C14 | O4 | -179.6(1) | |||
| C2 | C1 | C7 | O1 | 162.0(1) | O4 | C14 | C15 | O5 | -1.1(2) | |||
| C6 | C1 | C7 | C8 | 99.3(2) | C7 | C8 | C19 | O6 | -67.7(2) | |||
| C2 | C1 | C7 | C8 | -79.4(2) | ||||||||
1 and 1’ – molecules in the asymmetric unit.
The values of dihedral angles between the planes of the rings of 1 and 2 are presented in Table 4. Plane 2 passing through the atoms of the hydroxyphenyl substituent is almost perpendicular to the plane of the heterocyclic ring in 1.
Table 4.
Dihedral angles between the planes passing through selected atoms for compounds 1b and 2.
| 1b | 2 | |||
|---|---|---|---|---|
| Plane 1 C1, C2, C3, C4, C5, C6 | Plane 1 C1, C2, C3, C4, C5, C6 | |||
| Plane 2 C10, C11, C12, C13, C14, C15 | Plane 2 C12, C13, C14, C15, C16, C17 | |||
| Plane 3 C7, C8, C9 | Plane 3 O1, C7, C9, C10 | |||
| Plane 4 O1, C8, C9 | Plane 4 C7, C8, C9 | |||
| Plane 5 O1, C1, C6, C7 | Plane 5 O1, C7, C10 | |||
| molecule | 1 | 1’ | ||
| 1 / 2 | 85.76(14) | 87.22(16) | 1 / 2 | 38.61(4) |
| 1 / 3 | 38.63(38) | 40.55(22) | 1 / 3 | 86.53(5) |
| 2 / 3 | 56.54(36) | 52.61(25) | 2 / 3 | 55.71(5) |
| 1 / 4 | 44.27(39) | 50.10(21) | 1 / 4 | 68.13(7) |
| 2 / 4 | 88.23(19) | 89.68(23) | 2 / 4 | 75.63(8) |
| 3 / 4 | 58.58(42) | 63.41(28) | 3 / 4 | 36.70(10) |
| 1 / 5 | 3.06(20) | 3.02(18) | 1 / 5 | 80.48(8) |
| 2 / 5 | 88.57(20) | 89.78(17) | 2 / 5 | 61.55(8) |
| 3 / 5 | 35.64(42) | 37.53(28) | 3 / 5 | 6.16(9) |
| 4 / 5 | 45.21(38) | 50.32(20) | 4 / 5 | 32.72(12) |
1 and 1’ – molecules in the asymmetric unit.
Similarly, Plane 1 (passing through the atoms of one of the m-methoxy-p-hydroxyphenyl group) is almost perpendicular to Plane 3 (passing through atoms O1, C7, C9, C10) in 2. In comparison Plane 2 (passing through the atoms of the second m-methoxy-p-hydroxyphenyl group) is inclined to Plane 3 at an angle of 55.71(5)° in 2.
Figure 3 shows the crystal packing and Figure 4 presents the intermolecular interactions in the crystal lattices of 1a, 1b, and 2. The conformations of the molecules depend on the net of hydrogen bonds and π-stacking hydrophobic interactions influenced by the presence of solvent molecules. The hydrogen-bonding geometry for compounds 1 and 2 is shown in Table 5. Molecules of 1 create strong hydrogen bonds with the solvent molecules (1a with methanol and 1b with ethanol, respectively). There are also two intramolecular hydrogen bonds O3−H3O···O4, O5−H5O···O4 and a few intermolecular hydrogen bonds in the crystal lattice of 1. π-stacking interactions between aromatic rings of the molecules from neighboring unit cells are important factors determining the crystal packing of compound 2. There are also two intramolecular hydrogen bonds O3−H3O···O2, O5−H5O···O4, and three intermolecular hydrogen bonds: O6−H6O···O1, O5−H5O···O6 in the crystal lattice of compound 2.
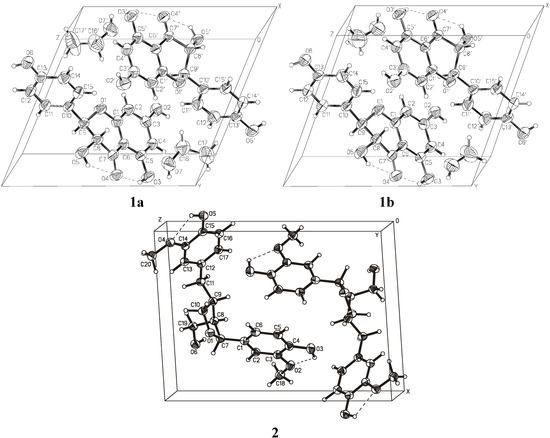
Figure 3.
Crystal packing diagram for 1 and 2.
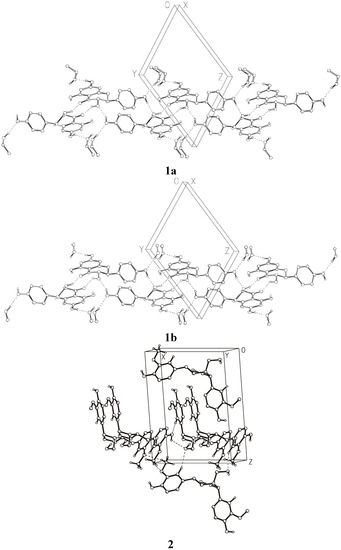
Figure 4.
Intermolecular interactions in the crystal lattices of 1 and 2 (hydrogens attached to carbon atoms are omitted for clarity).
Table 5.
Hydrogen-bonding geometry (Å, °) (H···A not greater then 2.55Å) for 1 and 2.
| D―H···A | D―H | H···A | D···A | D―H···A |
| 1a | ||||
| O3―H3O···O4 | 0.820(27) | 1.926(30) | 2.646(5) | 146.0(36) |
| O5―H5O···O4 | 0.820(26) | 2.265(28) | 2.698(5) | 113.4(28) |
| O3'―H3'O···O4' | 0.820(14) | 1.955(34) | 2.656(5) | 143.0(35) |
| O5'―H5'O···O4' | 0.820(23) | 2.231(20) | 2.701(4) | 116.7(23) |
| O5―H5O···O4' i | 0.820(26) | 2.068(33) | 2.767(5) | 142.9(31) |
| O6―H6O···O5' ii | 0.820(36) | 1.895(39) | 2.698(6) | 165.7(45) |
| O5'―H5'O···O4 iii | 0.820(23) | 2.088(20) | 2.751(4) | 137.7(25) |
| O6'―H6'O···O5 iv | 0.820(43) | 1.929(44) | 2.663(7) | 148.7(40) |
| O2―H2O···O7 v | 0.820(42) | 1.814(46) | 2.616(6) | 165.6(59) |
| O7―H7O···O6vi | 0.820(18) | 2.005(32) | 2.796(7) | 161.8(63) |
| O2'―H2'O···O7' vii | 0.820(19) | 1.934(56) | 2.630(6) | 142.1(42) |
| O7'―H7'O···O6' viii | 0.820(16) | 2.008(22) | 2.817(7) | 168.7(38) |
| C8―H8···O2 vii | 0.980(8) | 2.406(6) | 3.133(7) | 130.5(5) |
| 1b | ||||
| O3―H3O···O4 | 0.820(18) | 1.925(22) | 2.646(4) | 146.3(28) |
| O5―H5O···O4 | 0.820(23) | 2.303(27) | 2.689(4) | 109.4(28) |
| O3'―H3'O···O4' | 0.820(14) | 1.945(23) | 2.665(4) | 146.0(28) |
| O5'―H5'O···O4' | 0.820(21) | 2.257(23) | 2.698(3) | 114.1(23) |
| O5―H5O···O4' i | 0.820(23) | 2.104(35) | 2.773(4) | 138.5(32) |
| O6―H6O···O5' ii | 0.820(25) | 1.871(26) | 2.676(4) | 166.5(30) |
| O5'―H5'O···O4 iii | 0.820(21) | 2.066(13) | 2.762(3) | 142.5(26) |
| O6'―H6'O···O5 iv | 0.820(52) | 1.893(52) | 2.651(5) | 153.2(51) |
| O2―H2O···O7 v | 0.820(18) | 1.815(19) | 2.627(4) | 169.9(20) |
| O7―H7O···O6 vi | 0.820(38) | 1.979(33) | 2.769(5) | 161.4(43) |
| O2'―H2'O···O7' vii | 0.820(49) | 1.809(49) | 2.626(5) | 174.3(57) |
| O7'―H7'O···O6' viii | 0.820(18) | 1.995(19) | 2.789(6) | 162.6(33) |
| C8―H8···O2 vii | 0.980(6) | 2.441(5) | 3.124(5) | 126.5(4) |
| 2 | ||||
| O3―H3O···O2 | 0.889(30) | 2.128(30) | 2.652(2) | 117.0(24) |
| O5―H5O···O4 | 0.874(35) | 2.187(27) | 2.660(2) | 113.6(26) |
| O6―H6O···O1ix | 0.975(31) | 1.848(30) | 2.787(2) | 160.8(27) |
| O5―H5O···O6 i | 0.874(35) | 2.312(28) | 2.880(2) | 122.7(27) |
| C2―H2···O5 iii | 1.075(24) | 2.386(24) | 3.454(2) | 172.3(18) |
| C13―H13···O6 x | 1.018(23) | 2.546(24) | 3.452(2) | 148.1(19) |
| C20―H201···O4 xi | 0.965(30) | 2.544(28) | 3.464(3) | 159.5(22) |
Symmetry operators: (i) -1 + x, 1 + y, z; (ii) -1 + x, y, 1 + z; (iii) 1 + x, -1 + y, z; (iv) 1 + x, y, -1 + z; (v) 1 + x, y, z; (vi) x, 1 + y, -1 + z; (vii) -1 + x, y, z; (viii) x, -1 + y, 1 + z; (ix) x, -1 + y, z; (x) 1 - x, 0.5 + y, 2 – z; (xi) –x , 0.5 + y, 2 – z;
Experimental
General
Crystallization of the investigated compounds − dihydrokaempferol and lariciresinol − was carried out by the vapor diffusion method using organic solvents. Compound 1 (dihydrokaempferol) crystallizes in a triclinic system in space group P1 and compound 2 (lariciresinol) in a monoclinic system w space group P21 with the unit cell consisting of two molecules. Crystal data and experimental details of both compounds are shown in Table 1. The overall view of all molecules with the atom numbering scheme is seen in Figure 2, the crystal packing diagram in Figure 3 and hydrogen bonding in Figure 4. The crystal structure of compound 1 was determined using data collected at room temperature on a CAD4 diffractometer with graphite monochromatized CuKα radiation (λ = 1.54184 Å). Maximum 2θ was 150º and the scan mode: ω/2θ. The diffraction data set for a crystal of compound 2 was collected with synchrotron radiation at EMBL beamline X13 (DESY Hamburg). Diffraction images were recorded using a Mar 165 mm CCD detector at 100 K. Compound 1 was crystallized separately from ethanol (1a) and from methanol (1b). The structure of 1 was determined with the molecules of both solvents trapped in the crystal lattices. Compound 2 was crystallized from methanol, but in this case the solvent did not trap in the unit cell of the crystal. For compound 2, data was collected on a synchrotron (EMBL Hamburg) with two runs corresponding to low and high resolution. The high resolution run consisted of 90 images with oscillation 4º and the low resolution run consisted of 60 images with oscillation 6º. To avoid overloaded reflections, the exposure time for the low resolution run was ten times shorter than for the high resolution run. The diffraction data were processed with Denzo and scaled with Scalepack from the HKL program package [25].
An empirical absorption correction was applied for 1 by the use of the ψ-scan method (EAC program) [26,27]. All observed reflections with I > 0σ(I) were used to solve the structures by direct methods and to refine them by full matrix least-squares using F2 [27,28]. Anisotropic thermal parameters were refined for all nonhydrogen atoms. Hydrogen atoms were found on the difference Fourier map and refined isotropically except for H atoms attached to carbon atoms of solvent in the crystal lattice of compound 1. These hydrogen atoms were placed geometrically at idealized positions and set as riding with fixed thermal parameters equal to 1.33 times the equivalent isotropic thermal parameter of the parent-atom. The final calculation of compound 1a, 1b, 2 converged to R = 6.91%, R = 4.30%, R = 4.31% for 472, 454, 332 refined parameters and 2982, 2982, 4007 reflections with I ≥ 2σ(I), respectively.
Data correction was carried out with the Enraf-Nonius SDP crystallographic computing package [26]; structure solution with SHELXS [28,29] and structure refinement with SHELXL [29,30]. The torsion angles and the dihedral angles between planes of aromatic rings of molecules were calculated by CSU [31]. CCDC 719360 (1a), CCDC 719361 (1b) and CCDC 719362 (2) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre [23] via www.ccdc.cam.ac.uk/data_request/cif.
Conclusions
The investigated compounds are natural phenolic compounds. Dihydrokaempferol (1) is a member of the flavanones group, whilst lariciresinol (2) belongs to the lignans group − a group that consists of phenylpropane dimers enzymatically coupled through β-β-linkages between the propane chains. These phenolic compounds and their derivatives are antioxidants and should be investigated for their potential as antitumor agents [32,33]. The structural data presented in this work are a good basis for designing more biologically active inhibitors of lipid peroxidation and scavengers of superoxide radicals.
- Sample Availability: Samples of the compounds are available from the authors.
References
- Ekman, R.; Willför, S.; Sjöholm, R.; Reunanen, M.; Mäki, J.; Lehtilä, R.; Eckerman, C. Identification of the lignan Nortrachelogenin in knot and branch heartwood of Scots pine (Pinus sylvestris L.). Holzforschung 2002, 56, 253–256. [Google Scholar]
- Willför, S.; Hemming, J.; Reunanen, M.; Eckerman, C.; Holmbom, B. Lignans and lipophilic extractives in Norway spruce knots and stemwood. Holzforschung 2003, 57, 27–36. [Google Scholar]
- Willför, S.; Hemming, J.; Reunanen, M.; Holmbom, B. Phenolic and lipophilic extractives in scots pine knots and stemwood. Holzforschung 2003, 57, 359–372. [Google Scholar]
- Holmbom, B.; Eckerman, C.; Eklund, P.; Hemming, J.; Reunanen, M.; Sjöholm, R.; Sundberg, A.; Willför, S. Knots in trees – a new rich source of lignans. Phytochem. Rev. 2003, 2, 331–340. [Google Scholar]
- Pietarinen, S.P.; Willför, S.M.; Vikström, F.A.; Holmbom, B.R. Aspenknots, a rich source of flavonoids. J. Wood Chem. Technol. 2006, 26, 245–258. [Google Scholar] [CrossRef]
- Välimaa, A.L.; Honkalampi-Hämäläinen, U.; Pietarinen, S.; Willför, S.; Holmbom, B.; von Wright, A. Antimicrobial and cytotoxic knotwood extracts and related pure compounds and their effects on food-associated microorganisms. Int. J. Food Microbiol. 2007, 115, 235–243. [Google Scholar]
- Saarinen, N.M.; Power, K.A.; Chen, J.; Thompson, L.U. Lignans are accessible to human breast cancer xenografts in athymic mice. Nutr. Cancer 2008, 60, 245–250. [Google Scholar]
- Kangas, L.; Saarinen, N.; Mutanen, M.; Ahotupa, M.; Hirsinummi, R.; Unkila, M.; Perälä, M.; Soininen, P.; Laatikainen, R.; Korte, H.; Santti, R. Antioxidant and antitumor effects of hydroxymatairesinol (HM-3000, HMR), a lignan isolated from the knots of spruce. Eur. J. Cancer Prev. 2002, 11 (Suppl. 2), S48–S57. [Google Scholar]
- Saarinen, N.M.; Wärri, A.; Dings, R.P.M.; Airio, M.; Smeds, A.I.; Mäkelä, S. Dietary lariciresinol attenuates mammary tumor growth and reduces blood vessel density in human MCF-7 breast cancer xenografts and carcinogen-induced mammary tumors in rats. Int. J. Cancer 2008, 123, 1196–1204. [Google Scholar]
- Yang, X.W.; Li, S.M.; Shen, Y.H.; Zhang, W.D. Phytochemical and biological studies of Abies species. Chem. Biodivers. 2008, 5, 56–81. [Google Scholar] [CrossRef]
- Holmbom, B.; Eckerman, C.; Hemming, J.; Reunanen, M.; Sundberg, K.; Willför, S. Method for isolating chemical substances from wood. Pat. Appl. PCT/FI02/00418, 2002. [Google Scholar]
- Willför, S.M.; Ahotupa, M.O.; Hemming, J.E.; Reunanen, M.H.T.; Eklund, P.C.; Sjöholm, R.E.; Eckerman, C.S.E.; Pohjamo, S.P.; Holmbom, B.R. Antioxidant activity of knotwood extractives and phenolic compounds of selected tree species. J. Agric. Food Chem. 2003, 51, 7600–7606. [Google Scholar] [CrossRef]
- Arima, Y.; Hatanaka, A.; Fujimoto, K.; Fukuda, K.; Sakurai, H. Scavenging activities of α-, β- and γ-thujaplicins against active oxygen species. Chem. Pharm. Bull. 1997, 45, 1881–1886. [Google Scholar]
- Kähkönen, M.P.; Hopia, A.I.; Vuorela, H.J.; Rauha, J.P.; Pihlaja, K.; Kujala, T.S.; Heinonen, M. Antioxidant activity of plant extracts containing phenolic compounds. J. Agric. Food Chem. 1999, 47, 3954–3962. [Google Scholar] [CrossRef]
- Chang, S.T.; Wu, J.H.; Wang, S.Y.; Kang, P.L.; Yang, N.S.; Shyur, L.F. Antioxidant activity of extracts from Acacia confusa bark and heartwood. J. Agric. Food Chem. 2001, 49, 3420–3424. [Google Scholar] [CrossRef]
- Shimizu, K.; Kondo, R.; Sakai, K. Antioxidant activity of heartwood extracts of Papua New Guinean woods. J. Wood Sci. 2002, 48, 446–450. [Google Scholar] [CrossRef]
- Hopia, A.I.; Heinonen, M.J. Comparison of antioxidant activity of flavonoid aglycones and their glycosides in methyl linoleate. J. Am. Oil Chem. Soc. 1999, 76, 139–144. [Google Scholar]
- Willför, S.; Reunanen, M.; Eklund, P.; Sjöholm, R.; Kronberg, L.; Fardim, P.; Pietarinen, S.; Holmbom, B. Oligolignans in Norway spruce and Scots pine knots and Norway spruce stemwood. Holzforschung 2004, 58, 345–354. [Google Scholar]
- Pietarinen, S.P.; Willför, S.M.; Ahotupa, M.O.; Hemming, J.E.; Holmbom, B.R. Knotwood and bark extracts: strong antioxidants from waste materials. J. Wood Sci. 2006, 52, 436–444. [Google Scholar]
- Neacsu, M.; Eklund, P.C.; Sjöholm, R.E.; Pietarinen, S.P.; Ahotupa, M.O.; Holmbom, B.R.; Willför, S.M. Antioxidant flavonoids from knotwood of Jack pine and European aspen. Holz Roh. Werkst. 2007, 65, 1–6. [Google Scholar] [CrossRef]
- Siemens Analytical Xray Inst, XP - Interactive Molecular Grafics. 1992.
- Fábián, L.; Kálmán, A. Isostructurality in one and two dimensions: isostructurality of polymorphs. Acta Cryst. 2004, B60, 547–558. [Google Scholar]
- Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ. Available online: http://www.ccdc.cam.ac.uk/ accessed on 10 October 2009.
- Duax, W.L.; Weeks, C.M.; Rohrer, D.C. Topics in Stereochemistry; Eliel, E.L., Allinger, N.L., Eds.; Wiley-Interscience: New York, NY, USA, 1976; Volume 9, p. 271. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Frenz, B.A. SDP—Structure Determination Package; Enraf-Nonius: Delft, The Netherland, 1984. [Google Scholar]
- North, A.C.T.; Philips, D.C.; Mathews, F.S. A semi-empirical method of absorption correction. Acta Cryst. 1968, A24, 351–359. [Google Scholar]
- Sheldrick, G.M.; Kruger, G.M.; Goddard, R. SHELXS-86. In Crystallographic Computing 3; Oxford University Press: Oxford, UK, 1985; pp. 175–189. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar]
- Sheldrick, G.M. SHELXL-93, Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1993. [Google Scholar]
- Vickovic, J. CSU, a highly automatic and selective program for the calculation and presentation of geometrical parameters and their e.s.d.`s. J. Appl. Cryst. 1988, 21, 987–990. [Google Scholar]
- Issa, A.Y.; Volate, S.R.; Wargovich, J.M. The role of phytochemicalsin inhibition of cancer and inflammation: New directions and perspectives. J. Food Compos. Anal. 2006, 19, 405–419. [Google Scholar] [CrossRef]
- Fresco, P.; Borges, F.; Diniz, C.; Marques, M.P.M. New insights on the anticancer properties of dietary polyphenols. Med. Res. Rev. 2006, 26, 747–766. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).