Introduction
This study is a continuation of our efforts to evaluate plant extracts as sources of natural antioxidants [
1].
Equisetum arvense L. (Equisetaceae, subgenus
Equisetum) is a well-known and widespread pteridophyte distributed throughout the Northern Hemisphere. Field horsetail contains more than 10% inorganic substances (two-thirds of which are silicic acid and potassium salts). Also, the drug is rich in sterols: β-sitosterol, campasterol, isofucosterol [
2], ascorbic acid, phenolic acids (cinnamic acids, caffeic acid, di-
E-caffeoyl-
meso-tartaric acid and 5-
O-caffeoylshikimic acids), polienic acids, rare dicarboxylic acids (equisetolic acid), flavonoids [
3] and styrylpyrones [
4]. Flavonoid composition reveals the existence of two chemotypes of
E. arvense L., one occuring in Asia and North America and the other in Europe. Material from Asia and North America contains luteolin 5-
O-glucoside and its malonyl ester, whereas European material is lacking these compounds [
5]. Di-
E-caffeoyl-
meso-tartaric acid is a marker for both chemotypes. The dominant compounds in European plants are quercetin 3-
O-glucoside, apigenin 5-
O-glucoside and dicaffeoyl-
meso-tartaric acid [
6]. Sterile stems of
E. arvense are used in herbal medicine in various countries, constituting the “Equiseti herba” of European Pharmacopeias (DAB 10, Ph.Helv. VII, ÖAB 90, Ph. Pol. III, Ph. Ross 9 and Ph. Hung.). Therapeutic use of
Equisetum preparations are related to the reputed aquaretic and antihaemorragic properties of the plant. The most recent experimental data shows that ethanol and aqueous extracts of
E. arvense fertile stems are possessing very high radical scavenging activity towards superoxide anion and hydroxyl radicals [
7], but data regarding to antioxidant properties of sterile stems of
E. arvense are still being scarce. Concerning the complex composition of the plant extracts, their antioxidant activity must be evaluated combining different
in vitro assays to get relevant data.
With respect to this, the antioxidant activity of three separated fractions of methanolic extract (EtOAc, n-BuOH and H2O) of Equisetum arvense sterile stems has been investigated in the present study. The antioxidant activity was evaluated measuring the total reducing power, inhibition of induced lipid peroxidation in liposome and free radical scavenging capacity (RSC) regarding DPPH and NO radicals. In addition, the total flavonoid content (TFC) and HPLC chemical profile of phenolic constituents in each extract were determined.
Experimental
Plant material: The sterile stems of wild growing E. arvense were collected in May of 2002 from the Fruska Gora mountain in the Vojvodina province in Serbia. Aerial parts of the plants were used for the experiment. Voucher specimens (E. arvense L. N° 2-1965, Fruska Gora-Vrdnik, UTM 34TDQ09, det: Goran Anackov) were confirmed and deposited at the Herbarium of the Department of Biology and Ecology (BUNS Herbarium), Faculty of Natural Sciences, University of Novi Sad.
Chemicals: Petroleum ether, methanol, ethyl-acetate, n-buthanol, acetic acid, trichloroacetic acid (TCA), H3PO4, NaH2PO4, Na2HPO4 and ascorbic acid were purchased from Lach-Ner s.r.o. (Neratovice, Czech Republic). AlCl3x6H2O, CH3COONa, FeSO4, HClO4, sodium nitroprusside, K3[Fe(CN)6] and FeCl3 were provided by Reanal (Budapest, Hungary). Phosphoric acid (HPLC purity), methanol (HPLC purity), EDTA, 2-thiobarbituric acid (TBA) and 2,2-diphenyl-1-picrylhydrazyl (DPPH) were obtained from Sigma (St. Louis, MO, USA). 3,5-di-tert-butyl-4-hydroxytoluene (BHT) and propylgallate (PG) were purchased from Aldrich (Taufkirchen, Germany) and 3-tert-butyl-4-hydroxyanisole (BHA) and sulfanilamide from Fluka Chemie GmbH (Buchs, Switzerland). Preparation of liposomes “PRO-LIPO S” pH 5-7 was obtained from Lucas- Meyer (Hamburg, Germany) and N-(1-naphtyl)-ethylenediamine dihydrochloride (NEDA) from Alfa Aesar (Karlsruhe, Germany). Rutin trihydrate was provided by Carl Roth GmbH, Karlsruhe, Germany).
Preparation of extracts and determination of total flavonoid content: 100 g of air-dried and powdered plant material was macerated with petroleum ether overnight and afterwards with 70% MeOH (24 h). After filtration, the methanolic extract was concentrated to dryness using a rotary evaporator. The dry residue was dissolved in hot water and then separated by liquid-liquid extraction into CHCl3, EtOAc and n-BuOH extracts. These extracts were evaporated to dryness and the dry residues were re-dissolved in 70% MeOH to obtain mass concentration: 0.5, 1.0, 5.0 and 10%. The obtained extracts were used in further experiments.
Total flavonoid content: Determination of total flavonoid content (TFC) in the examined extracts was based on flavonoid affinity to form a complex with AlCl
3 [
10]. 100µL of each extract (10%) was made up to a final volume of 10 mL with reaction medium (MeOH/H
2O/CH
3COOH=14:5:1). From this solution, 1 mL was taken and diluted with distilled water to 20 mL.
Prepared solution (10 mL) was mixed with AlCl3 reagent (5 mL, 133 mg of AlCl3x6H2O and 400 mg of CH3COONa dissolved in 100 mL H2O). After 5 min, the absorbance of the resulting solutions was measured in comparison with the blank (containing the same chemicals, except for the sample) at 430 nm (CECIL 2021 spectrophotometer). TFC was calculated on the basis of the calibration curve of rutin and expressed by mg rutin/g dry extract (mg/g DE). For each sample three replicates were carried out.
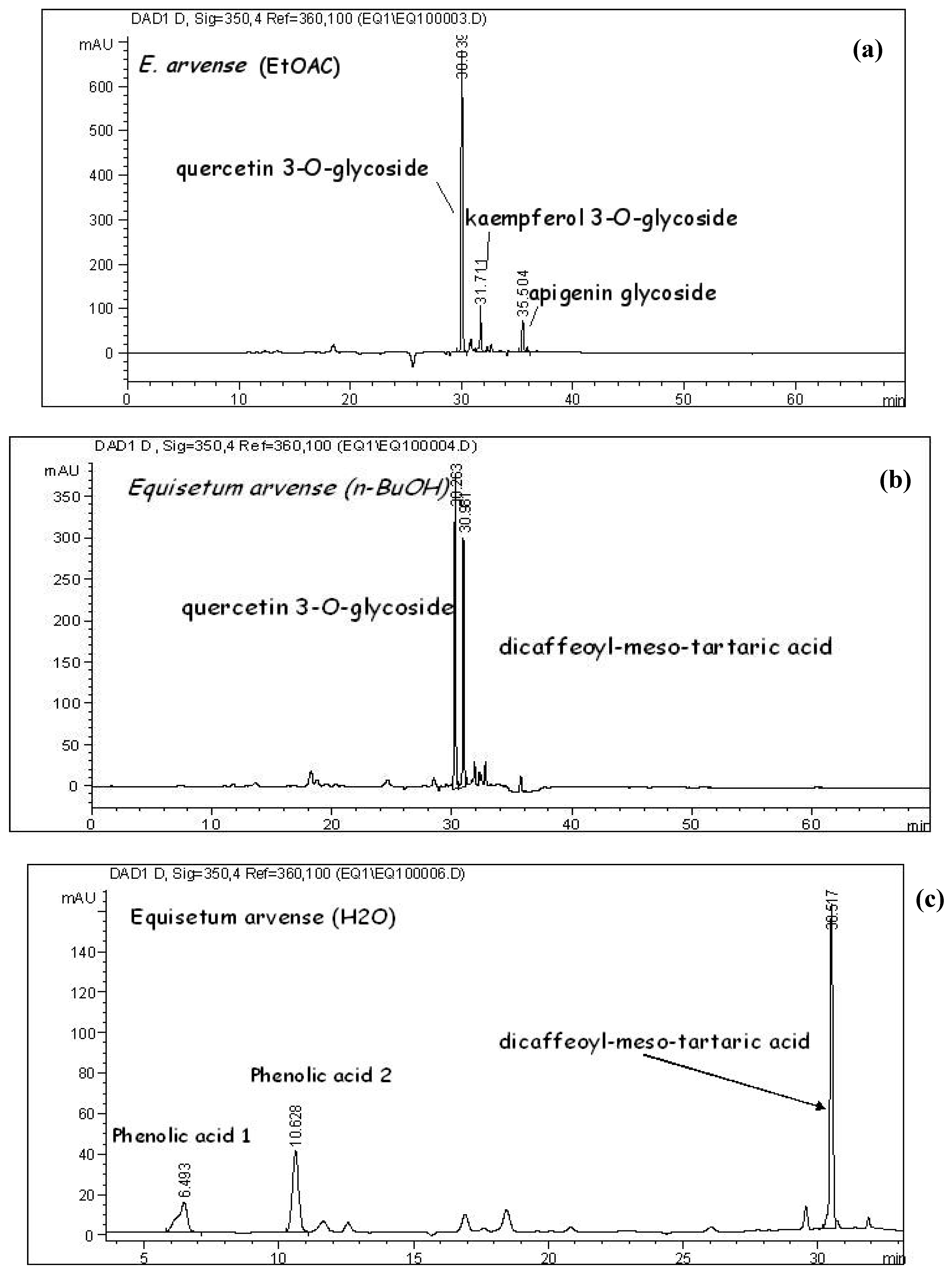
HPLC analysis: The sample was filtrated through 0.45 µm cellulose membrane filter (Econofilters, Agilent Technologies, Germany) and an aliquot (5 µL) of the sample was injected in the HPLC. The reversed-phase HPLC system (Agilent 1100 Series, Germany) consisting of an Agilent Technologies HPLC system (Agilent 1100 Series, Germany), was equipped with a binary pump, UV-diode array detector, autosampler and ChemStation for LC 3D (Rev. A.09.03.) software. The chromatographic conditions were: temperature 15°C thermostated, column Zorbax (SB-C18 4.6 x 150 mm, 5-µm), guard column Zorbax (SB-C18 4.6 x 12.5 mm, 5-µm). Two solvents were used for the gradient elution: (A) 0.15 % phosphoric acid in H2O: MeOH (77:23) (pH = 2) and (B) methanol. The elution profile was: isocratic: 0 - 3.6 min 100A; gradient: 3.6 min. 100 % A - linear - 24.0 min. 80.5% A - isocratic - 30 min - linear - 60 min. 51.8 % A - linear - 67.2 min. 100 % B. The flow rate was 1.0 mL/min.
Detection and quantification were carried out using 330 and 350 nm as preferred wavelengths. Peak purity and identity were checked by comparison of the UV spectra (DAD-detector) with those of the reference substances and with literature data [
6,
11]. Quantification of dominant compounds (mg/g DE) was performed by HPLC method using a single point calibration with an external standard. Quercetin 3-O-rutinoside (rutin) was used as standard compound for quantification of flavonols, apigenine for quantification of flavons and chlorogenic acid for phenolic acids.
Scavenging of the stable radical 1,1-diphenyl-2-picrylhydrasyl – DPPH˙ assay: DPPH˙-assay was performed as described before [
12], with small modifications [1b]. 10 μL of each extract (1.25 - 62.5 µg/mL) was mixed with 90 µmol/L DPPH˙ in methanol (1.0 mL) and made up with pure methanol to a final volume of 4.0 mL. The mixtures were shaken vigorously and were stored in dark for 60 min at room temperature. This is done to reach the steady state of the reaction. After incubation the absorbance was measured at 515 nm (CECIL CE2021 spectrophotometer). All reactions were carried out in triplicate. Commercial synthetic antioxidant: BHT, BHA and PG were used as positive controls. The DPPH˙ scavenging activity was expressed by radical scavenging capacity using the following equation:
where
A0 was the absorbance of the control reaction (full reaction, without the tested extract or compound) and
A1 was the absorbance in the presence of the scavenger.
The EC50 values (concentration of extract in the reaction mixture needed to decrease by 50% the initial DPPH˙ concentration) were determined by polynomial regression analysis of the obtained DPPH-RSC values (software ORIGIN 2001).
Determination of LP inhibition in liposome: The extent of Fe(II)/ascorbate induced LP was determined by TBA-assay [
13]. The commercial liposome preparation “PRO-LIPO S” pH 5-7 was used as a model system of biological membranes. The liposomes, 225-250 nm in diameter, were obtained by dissolving the commercial preparation in distilled water (1:10) in an ultrasonic bath. The reaction mixture contained liposome suspension (60 µL), FeSO4 (20 µL, 0.01 mol/L), ascorbic acid (20 µL, 0.01 mol/L) and examined extract (10 µL, final concentration 9.62 – 192.31 µg/mL) and made up with NaH
2PO4-Na
2HPO4 buffer (2.89 mL, 0.067 mol/L, pH 7.4) to start the peroxidation. After 60 min at 37 ºC the reaction was terminated with TBA-reagent (2.0 mL, 10.4 mL 60% HClO4, 3 g TBA and 120 g 20% trichloroacetic acid (TCA) dissolved in 800 mL dH2O) and EDTA (0.2 mL, 0.1 mol/L). Tubes were heated at 100 ºC for 20 min and cooled immediately. The mixtures were centrifuged at 4000 rpm for 10 min. The content of the MDA (TBARS) was determined by measuring the absorbance at 532 nm (CECIL CE2021 spectrophotometer). The results were compared with commercial synthetic antioxidants: BHT, BHA and PG. The percentage of LP inhibition was calculated by the following equation:
where
Ao is the absorbance of the control reaction (full reaction, without the tested extract or compound) and
A1 is the absorbance in the presence of the inhibitor. A higher percentage indicates a higher LP inhibition.
The EC50 value, which represented the concentration of the extract that caused 50% of LP inhibition, was determined by polynomial regression analysis of the obtained I values (software ORIGIN 2001).
NO scavenging activity: NO-RSC was evaluated by measuring the accumulation of nitrite (formed by the reaction of NO with oxygen), according to the Griess reaction [
14]. NO was generated by sodium nitroprusside in buffered aqueous solution. Each prepared extract (10 μL) was mixed with fresh prepared solution of sodium nitroprusside (1.5 mL, 0.01 mol/L in NaH
2PO4-Na
2HPO4 buffer, 0.067 mol/L, pH 7.4) and NaH
2PO4-Na
2HPO4 buffer (1.5 mL, 0.067 mol/L, pH 7.4). These mixtures were incubated at 25°C for 60 min. Each reaction mixture (1 mL) was mixed with Griess reagent (1 mL, 0.1%
N-(1-naphtyl)-ethylenediamine dihydrochloride [NEDA] in distilled water and 1% sulfanilamide in 5% H
3PO
4, ana partes). Reduction of nitrite by the extracts was determinated spectrophotometrically at 546 nm, by measuring decreased of absorbance of the reaction mixtures regarding the control (containing the same chemicals, except for the sample). For each sample three replicates were carried out. RSC was calculated by following equation:
where
Ao is control and
A1 is a sample solution absorbance. The EC
50 values were determined by polynomial fitting of the inhibition values (RSC) using software ORIGIN 6.1.
Reducing power assay: Reducing power (RP) of all extracts was determined according to the method of Yen and Chen [
15], with some modifications. RP was expressed in relation to the reducing power of ascorbic acid as a positive control (AEAC - Ascorbate Equivalent Antioxidant Capacity). Each prepared extract (10 μL) was mixed with K
3[Fe(CN)
6] (1 mL, 1%) and NaH
2PO4-Na
2HPO4 buffer (1 mL, 0.2 mol/L, pH 6.6). These mixtures were incubated at 50°C for 30 min. Afterwards, trichloroacetic acid (1mL, 10%) was added. Then the mixtures were centrifuged at 3000 rpm for 10 min. Finally, the supernatant fractions (1 mL) were mixed with distilled water (1 mL) and FeCl
3 (0.2 mL, 0.1%). The absorbances of resulting solutions were measured at 700 nm. For each sample three replicates were carried out. Increased absorbances of the reaction mixture indicated increased reduction power. AEAC was calculated by the following equation:
where C
A- final concentration of ascorbic acid in µg/mL, A
S- absorbance of the sample, A
A- absorbance of ascorbic acid.


{kind=link}