Synthesis and Total 1H- and 13C-NMR Assignment of Cephem Derivatives for Use in ADEPT Approaches

Abstract
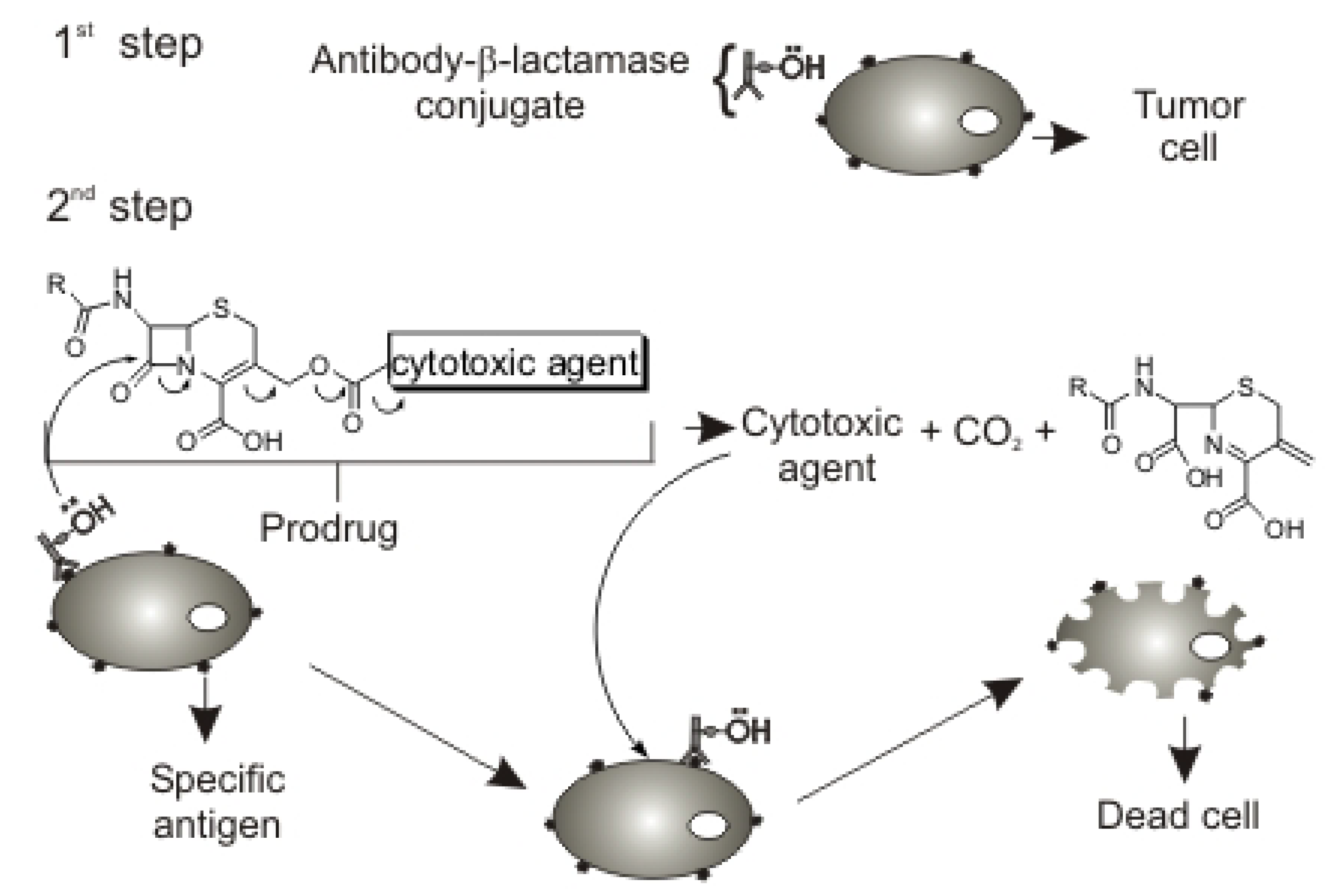
:Introduction
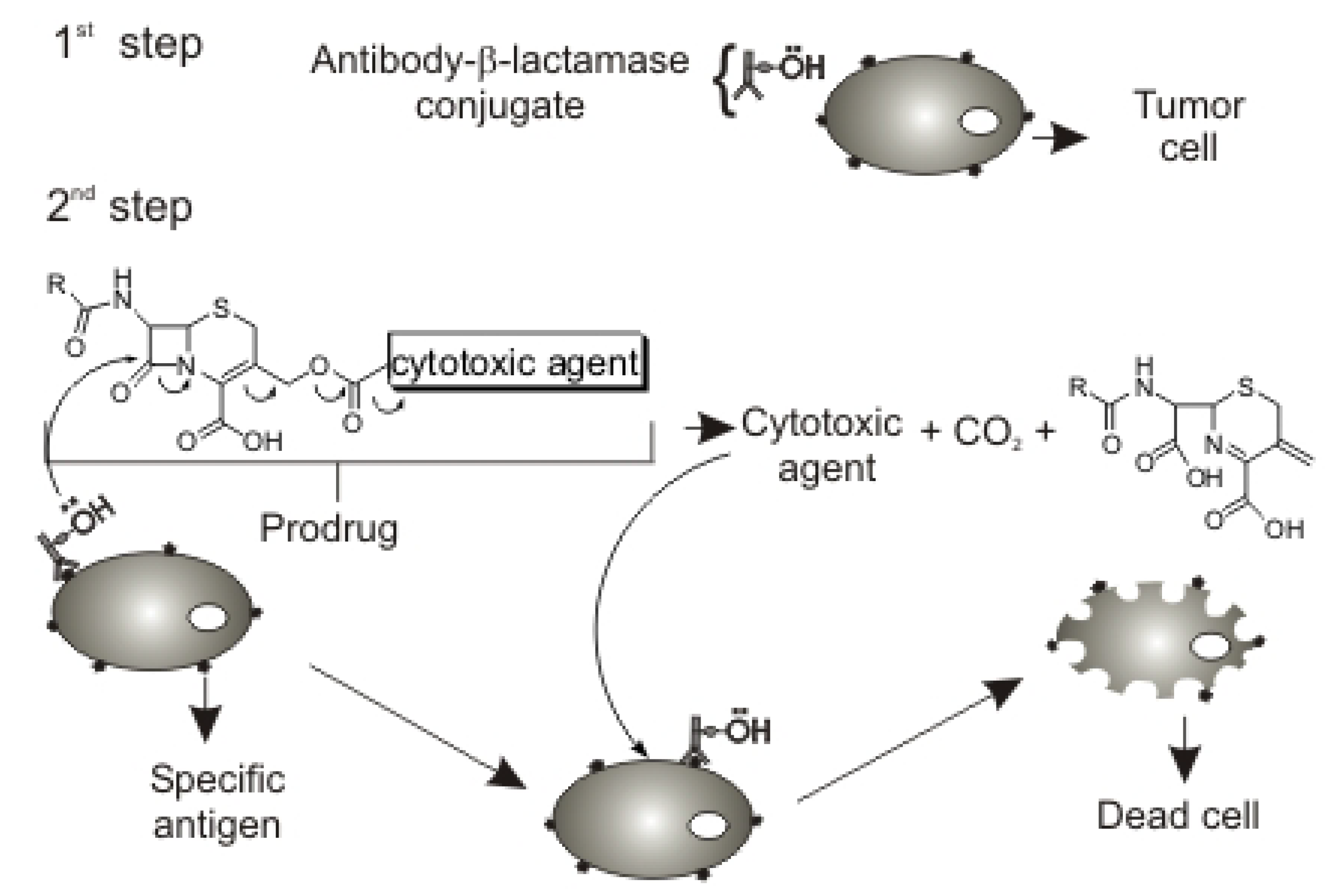
Results and Discussion
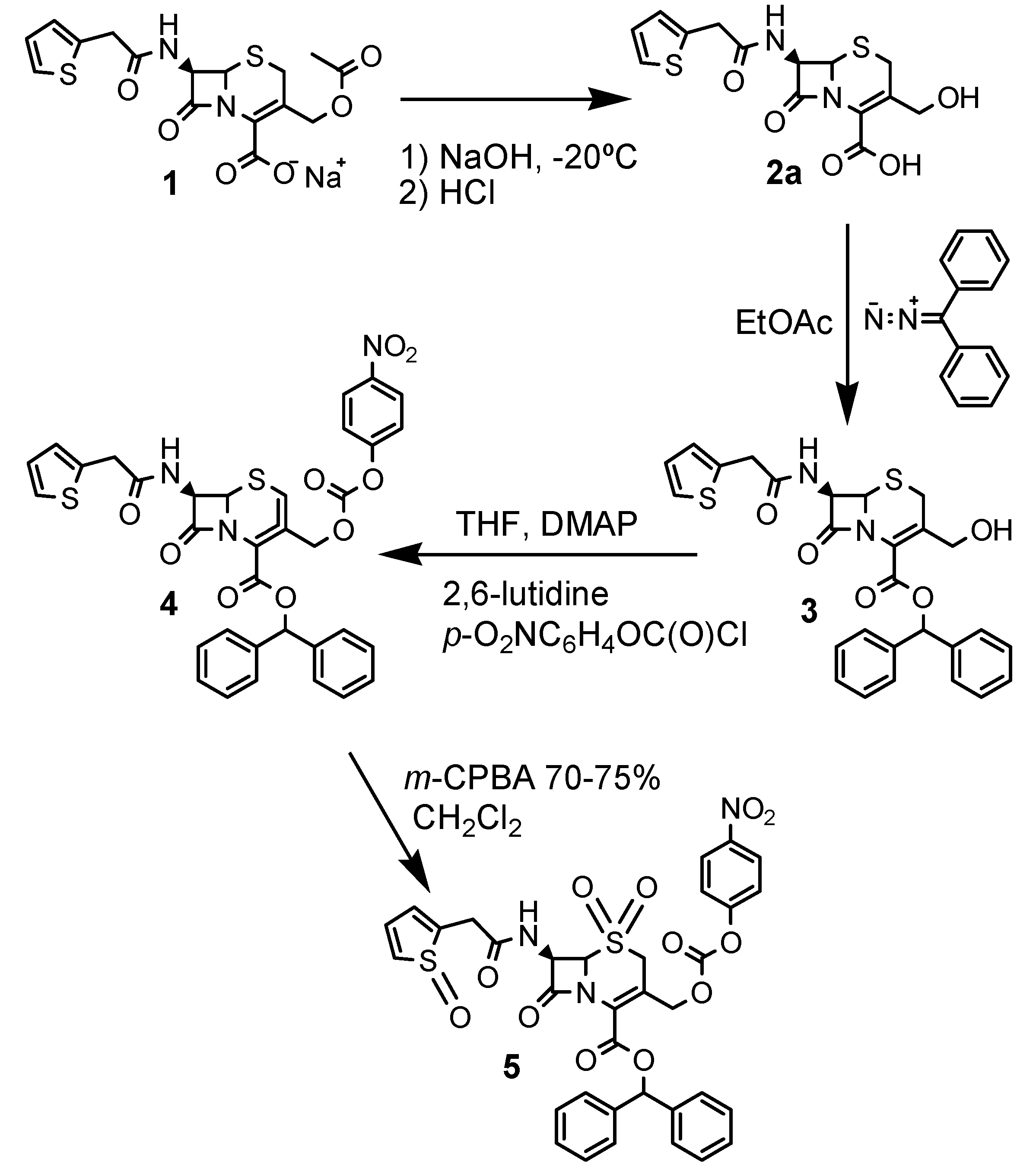
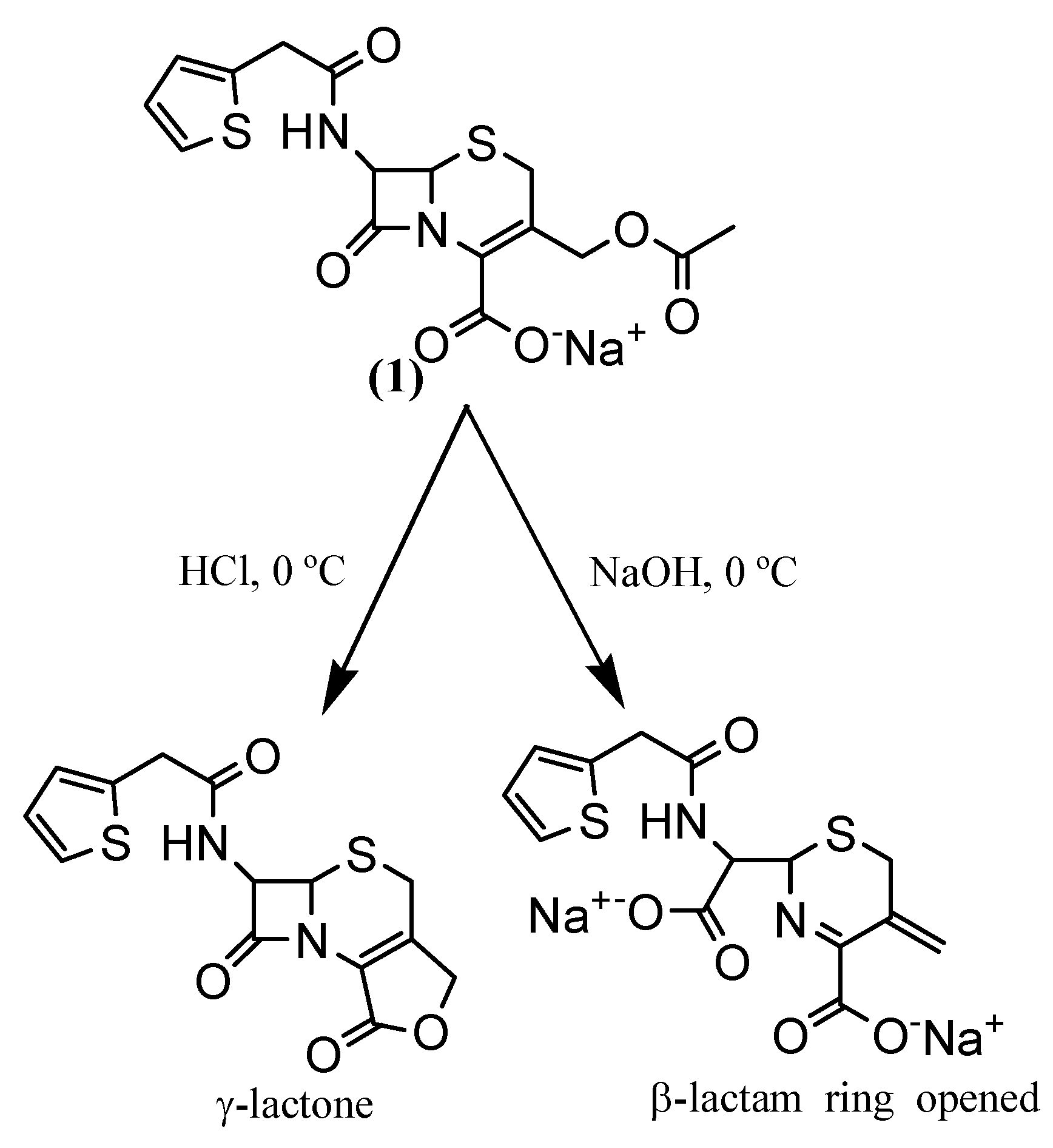
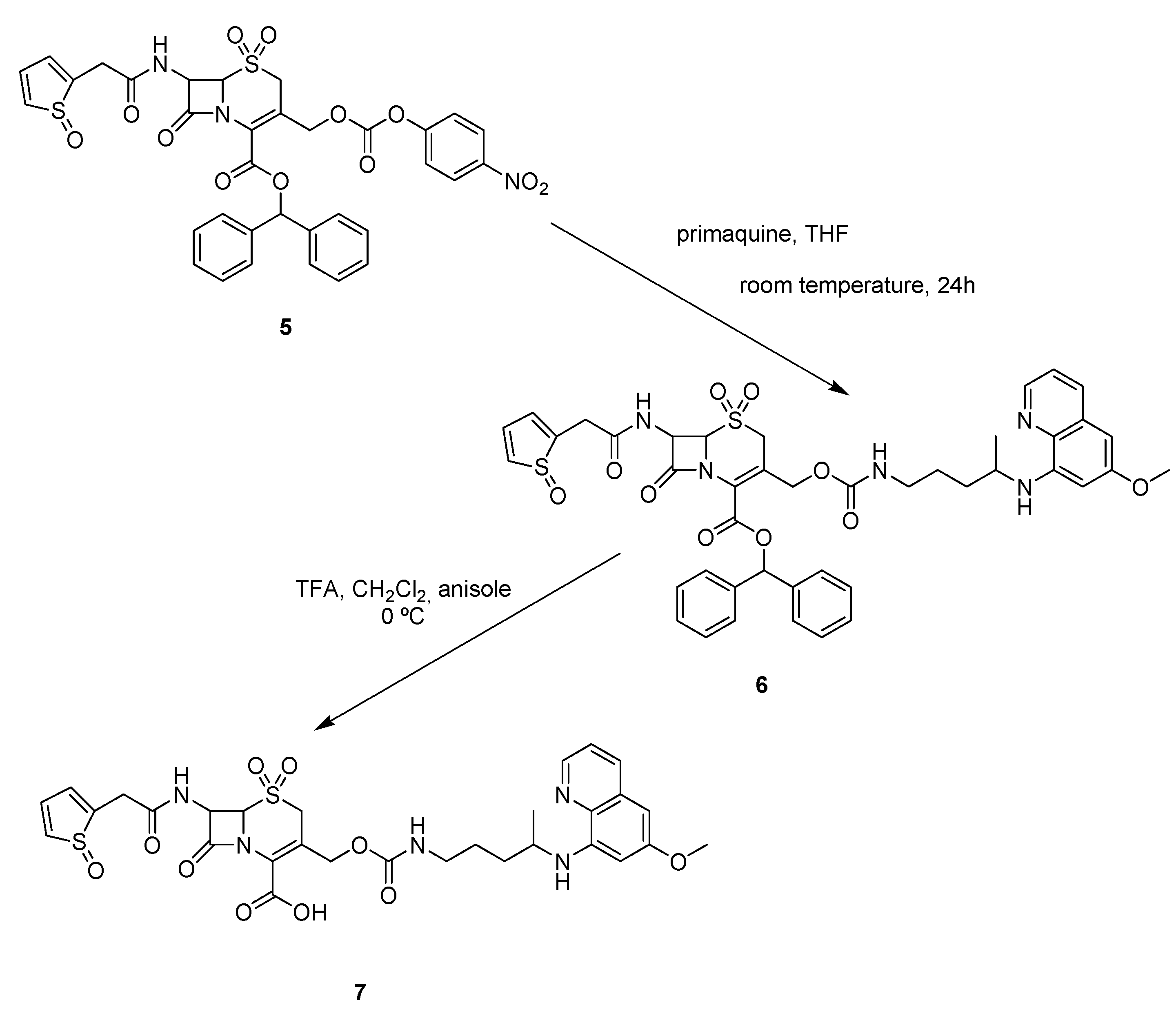
Synthesis

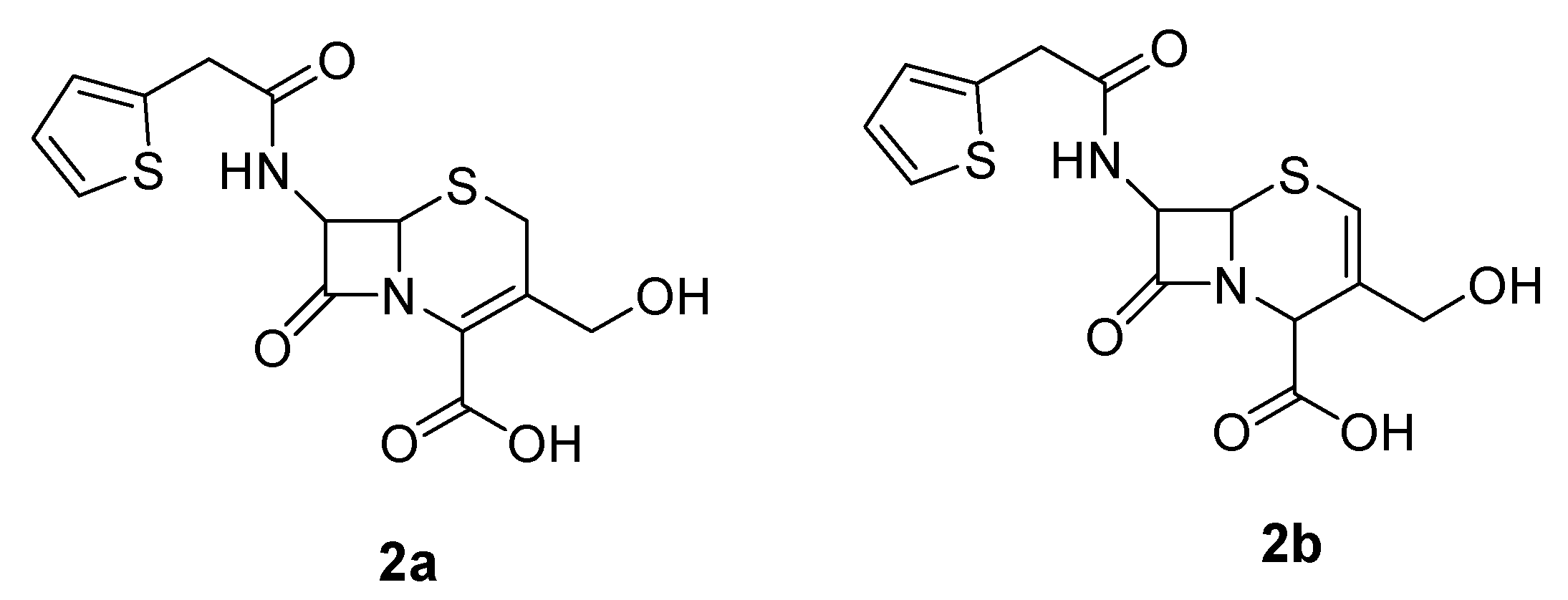
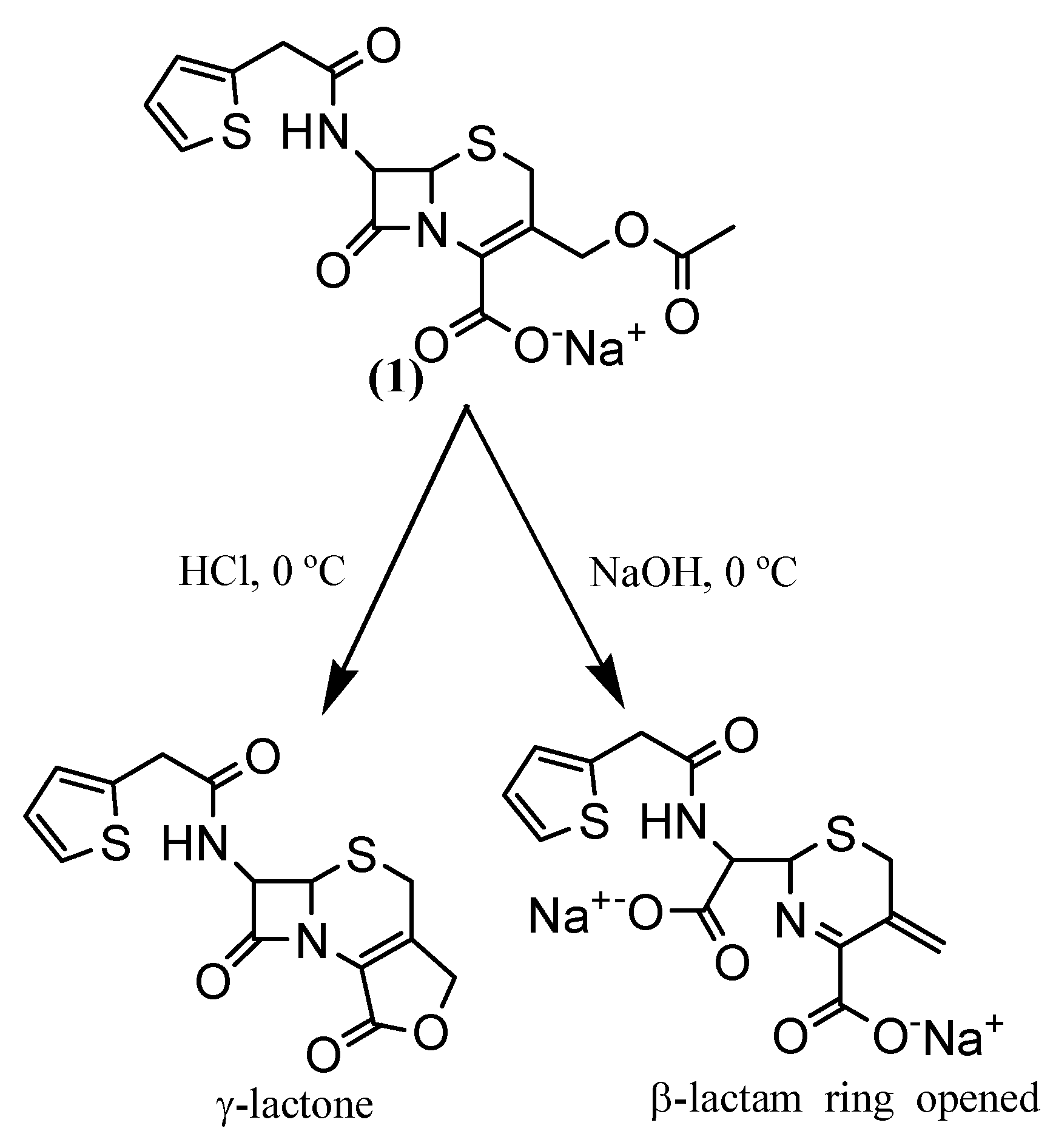

NMR Assignments

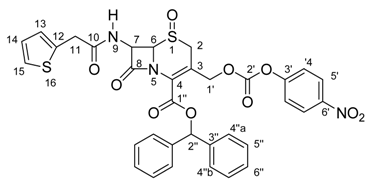
| Position | δ 13C (ppm) | δ 1H (ppm); (int., mult.); J (Hz) | gHMBC |
|---|---|---|---|
| 2 | 45.4 | a – 4.08; (1H, d); J=18.6 | C3; C4; C6 |
| b – 3.71; (1H, d); J=18.6 | |||
| 3 | 120.1 | --- | --- |
| 4 | 125.3 | --- | --- |
| 6 | 66.6 | 4.99; (1H, d); J=4.5 | C8 |
| 7 | 58.2 | 5.98; (1H, dd); J=8.3; J=4.5 | C6; C8; C10 |
| 8 | 164.5 | --- | --- |
| 9 | --- | 8.47; (1H, d); J=8.3 | C7; C8; C10 |
| 10 | 170.2 | --- | --- |
| 11 | 35.7 | a – 3.92; (1H, d); J=15.5 | C10; C12; C13 |
| b – 3.83; (1H, d); J=15.5 | |||
| 12 | 136.7 | --- | --- |
| 13 | 126.9 | 7.29; (1H, d); J=5.5 | --- |
| 14 | 126.7 | 7.28; (1H, dd); J=6.7; J=5.5 | --- |
| 15 | 125.1 | 7.54; (1H, d); J=6.7 | --- |
| 1’ | 67.4 | a – 5.30 ; (1H, d); J=13,0 | C2; C3; C4; C2’ |
| b – 4.91; (1H, d); J=13,0 | |||
| 2’ | 151.5 | --- | --- |
| 3’ | 155.1 | --- | --- |
| 4’ | 122.4 | 7.52; (2H, d); J=9.0 | C3’; C5’; C6’ |
| 5’ | 125.4 | 8.31; (2H, d); J=9.0 | C3’; C5’; C6’ |
| 6’ | 145.2 | --- | --- |
| 1” | 159.7 | --- | --- |
| 2” | 79.3 | 6.96; (1H, s) | C1’’; C3’’; C4’’ |
| 3” | 139.6 | --- | --- |
| 4” | 126.6 | a – 7.44; (2H, d); J=7.4 | C2’’ |
| b – 6.98; (2H, d); J=3.5 | |||
| 5” | 128.5 | 7.34; (4H, dd); J=7.4; J=6.7 | C3’’; C6’’ |
| 6” | 128.4 | 7.38; (2H, dd); J=6.7; J=3.5 | --- |
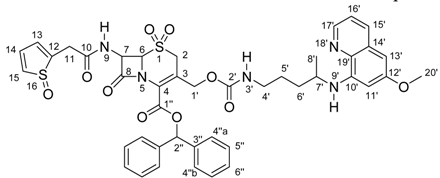
| Position | δ 1H (ppm), (int., mult.), J (Hz) | δ 13C (ppm) | gHSQC | gHMBC |
|---|---|---|---|---|
| 2 | a – 3.62; (1H, d); J=18.8 | 45.6 | C2 | C3; C4; C6; C1’ |
| b – 2.94; (1H, d); J=18.8 | ||||
| 3 | --- | 123.4 | --- | --- |
| 4 | --- | 124.6 | --- | --- |
| 6 | 4.17 – 4.26; (1H, m) | 66.7 | C6 | C8 |
| 7 | 5.81 – 5.96; (1H, m) | 59.0 | C7 | C6; C8; C10 |
| 8 | --- | 164.5 | --- | --- |
| 9 | 6.99; (1H, d); J=9.8 | --- | --- | C7; C10 |
| 10 | --- | 170.5 | --- | --- |
| 11 | 3.72; (2H, s) | 37.2 | C11 | C10; C11; C12 |
| 12 | --- | 135.0 | --- | --- |
| 13 14 | 6.83 – 6.92; (2H, m) | 127.2 127.4 | C13; C14 | C12; C15 |
| 15 | 7.08 – 7.40; (1H, m) | 125.9 | C15 | --- |
| 1’ | a – 5.05; (1H, d); J=14.3 | 63.4 | C1’ | C2; C3; C4; C2’ |
| b – 4.52; (1H, d); J=14.3 | ||||
| 2’ | --- | 156.0 | --- | --- |
| 4’ | 2.99 – 3.13; (2H, m) | 41.2 | C4’ | --- |
| 5’, 6’ | 1.38 – 1.66; (4H, m) | 26.6; 33.9 | --- | C4’; C5’; C6’; C7’; C8’ |
| 7’ | 3.43 – 3.55; (1H, m) | 47.9 | --- | C5’; C6’ |
| 8’ | 1.18; (3H, d); J=6.0 | 20.6 | C8’ | C6’; C7’ |
| 10’ | --- | 144.9 | --- | --- |
| 11’ | 6.25; (1H, brs) | 92.0 | C11’ | C12’; C13’; C19’ |
| 12’ | --- | 159.6 | --- | --- |
| 13’ | 6.19; (1H, brs) | 97.1 | C13’ | C11’; C12’; C19’ |
| 14’ | --- | 130.1 | --- | --- |
| 15’ | 7.83; (1H, d); J=8.1 | 135.1 | --- | --- |
| 16’ | 7.07 – 7.41; (1H, m) | 122.1 | --- | --- |
| 17’ | 8.43; (1H, dd); J=5.8; J=1.6 | 144.4 | C17’ | C16’; C19’ |
| 19’ | --- | 135.3 | --- | --- |
| 20’ | 3.79; (3H, s) | 55.5 | C20’ | C12’; C20’ |
| 1’’ | --- | 160.1 | --- | --- |
| 2’’ | 6.81; (1H, s) | 80.4 | C2’’ | C1’’; C3’’; C4’’a; C4’’b |
| 3’’ | --- | 139.2 | --- | --- |
| 4’’a, 4’’b 5’’, 6’’ | 7.08 – 7.40; (10H, m) | 127.7; 127.8 128.6; 128.8; 128.3; 128.4 | --- | --- |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
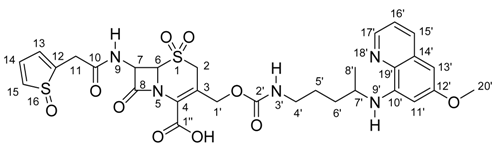
| Position | δ 1H (ppm); (int., mult.); J (Hz) | δ 13C (ppm) | gCOSY | gHSQC | gHMBC |
|---|---|---|---|---|---|
| 2 | 3.75 – 3.94; (2H, m) | 45.2 | --- | C2 | C3; C4; C6 |
| 3 | --- | 119.7 | --- | --- | --- |
| 4 | --- | 125.3 | --- | --- | --- |
| 6 | 4.88; (1H, d); J=4.8 | 66.2 | H7 | C6 | C8 |
| 7 | 5.80; (1H, dd); J=8.4; J=4.8 | 58.0 | H6; H9 | C7 | C6; C8; C10 |
| 8 | --- | 164.1 | --- | --- | --- |
| 9 | 8.42; (1H, d); J=8.4 | --- | H7 | --- | C7; C8; C10 |
| 10 | --- | 170.0 | --- | --- | --- |
| 11 | 3.75 – 3.94; (2H, m) | 35.6 | --- | C11 | C10; C12; C13 |
| 12 | --- | 136.8 | --- | --- | --- |
| 13; 14 | 6.91 –6.97; (2H, m) | 126.4; 126.7 | H15 | C13; C14 | C12; C15 |
| 15 | 7.36; (1H, dd); J=4.8; J=1.8 | 125.1 | H13; H14 | C15 | C12; C13 |
| 1’ | a – 5.11; (1H, d); J=13.1 b – 4.52; (1H, d); J=13.1 | 62.7 | H1’a; H1’b | C1’ | C2; C3; C4; C2’ |
| 2’ | --- | 155.8 | --- | --- | --- |
| 3’ | 7.29; (1H, t); J=5.9 | --- | H4’ | --- | --- |
| 4’ | 2.98; (2H, d); J=5.9 | 40.3 | H3’; H5’; H6’ | C4’ | --- |
| 5’; 6’ | 1.41 – 1.70; (4H, m) | 26.1; 33.2 | H4’; H7’ | C5’; C6’ | C8’ |
| 7’ | 3.55 – 3.66; (1H, m) | 47.0 | H8’ | C7’ | --- |
| 8’ | 1.19; (3H, d); J=6.3 | 20.1 | H7’ | C8’ | C6’; C7’ |
| 10’ | --- | 144.5 | --- | --- | --- |
| 11’ | 6.48; (1H, d); J=2.6 | 91.7 | H13’ | C11’ | C12’; C13’; C15’ |
| 12’ | --- | 159.0 | --- | --- | --- |
| 13’ | 6.25; (1H, d); J=2.6 | 96.3 | H11’ | C13’ | C12’; C15’ |
| 14’ | --- | 129.6 | --- | --- | --- |
| 15’ | 8.08; (1H, dd); J=8.4; J=1.5 | 135.0 | H16’ | C15’ | C11’; C15’; C17’ |
| 16’ | 7.43; (1H, dd); J=8.4; J=4.2 | 122.1 | H15’; H17’ | C16’ | C14’; C17’ |
| 17’ | 8.53; (1H, dd); J=4.2; J=1.5 | 144.1 | H16’ | C17’ | C15’; C16’ |
| 19’ | --- | 134.3 | --- | --- | --- |
| 20’ | 3.75 – 3.94; (3H, m) | 55.0 | --- | C20’ | C12’; C20’ |
| 1’’ | --- | 162.0 | --- | --- | --- |
Conclusions
Experimental
General
NMR Experiments
Synthesis
Cephalothin derivative 5
Diphenyldiazomethane
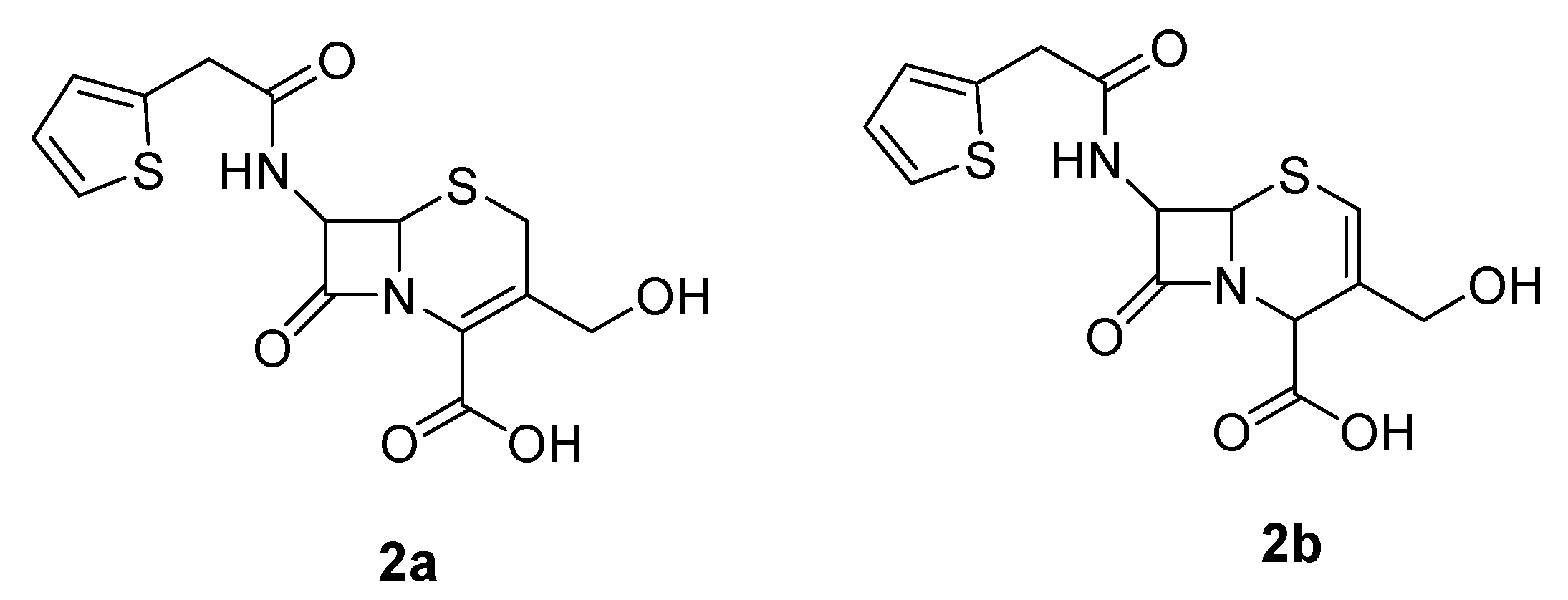
3-Hydroxymethyl-8-oxo-7-[(2-thienylacetyl)-amino]-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (2a)
3-Hydroxymethyl-8-oxo-7-[(2-thienylacetyl)amino]-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic aciddiphenylmethyl ester (3)
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid-3-[[[(4’’-nitrophenoxy)carbonyl]oxy]methyl]-8-oxo-7-[(2-thienylacetyl)amino]-diphenylmethyl ester and 5-thia-1-azabicyclo[4.2.0]oct-3-ene-2-carboxylic acid-3-[[[(4’’-nitrophenoxy)carbonyl]oxy]methyl]-8-oxo-7-[(2-thienylacetyl)amino]- diphenylmethyl ester (4)
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid-3-[[[(4’’-nitrophenoxy)carbonyl]oxy]methyl]-8-oxo-7-[(2-thienyloxoacetyl)amino]-diphenylmethyl ester-5-dioxide (5)
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid-3-[methyl 4-(6-methoxyquinolin-8-ylamino)-pentylcarbamate]-8-oxo-7-[(2-thienyloxoacetyl)amino]-diphenylmethyl ester-5-dioxide (6)
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 3-[methyl 4-(6-methoxyquinolin-8-ylamino)-pentylcarbamate]-8-oxo-7-[(2-thienyloxoacetyl)amino]- 5-dioxide (7)
Acknowledgements
References
- Vangapandu, S.; Sachdeva, S.; Jain, M.; Singh, S.; Singh, P. P.; Kaul, C. L.; Jain, R. 8-Quinolinamines conjugated with amino acids are exhibiting potent blood-schizontocidal antimalarial activities. Bioorg. Med. Chem. 2004, 12, 239–247. [Google Scholar] [CrossRef]
- Gomes, P.; Araújo, M. J.; Rodrigues, M.; Vale, N.; Azevedo, Z.; Iley, J.; Chambel, P.; Morais, J.; Moreira, R. Synthesis of imidazolidin-4-one and 1H-imidazo[2,1-a]isoindole-2,5(3H,9bH)-dione derivatives of primaquine: scope and limitations. Tetrahedron 2004, 60, 5551–5562. [Google Scholar] [CrossRef]
- Araújo, M. J.; Bom, J.; Capela, R.; Casimiro, C.; Chambel, P.; Gomes, P.; Iley, J.; Lopes, F.; Morais, J.; Moreira, R.; De Oliveira, E.; Do Rosário, V.; Vale, N. Imidazolidin-4-one Derivatives of Primaquine as Novel Transmission-Blocking Antimalarials. J. Med. Chem 2005, 48, 888–892. [Google Scholar] [CrossRef]
- Mihaly, G. W.; Ward, S. A.; Edwards, G.; Orme, M. L.; Breckenridge, A. M. Pharmacokinetics of primaquine in man: identification of the carboxylic acid derivative as a major plasma metabolite. Br. J. Clin. Pharmacol. 1984, 17, 441–446. [Google Scholar] [CrossRef]
- Constantino, L.; Paixão, P.; Moreira, R.; Portela, M. J.; Rosário, V. E.; Iley, J. Metabolism of primaquine by liver homogenate fractions. Evidence for monoamine oxidase and cytochrome P450 involvement in the oxidative deamination of primaquine to carboxyprimaquine. Exp. Toxicol. Pathol. 1999, 51, 299–303. [Google Scholar] [CrossRef]
- Tsalic, M.; Bar, S. G.; Beny, A.; Visel, B.; Haim, N. Severe Toxicity Related to the 5-Fluorouracil/Leucovorin Combination (The Mayo Clinic Regimen): A Prospective Study in Colorectal Cancer Patients. Am. J. Clin. Oncol. 2003, 23, 103–106. [Google Scholar] Extermann, M.; Chen, H.; Cantor, A. B.; Corcoran, M. B.; Meyer, J. Predictors of tolerance to chemotherapy in older cancer patients: a prospective pilot study. Eur. J. Cancer 2002, 38, 1466–1473. [Google Scholar]
- Blagosklonny, M. V. Drug-resistance enables selective killing of resistant leukemia cells: exploiting of drug resistance instead of reversal. Leukemia 1999, 13, 2031–2035. [Google Scholar] [CrossRef] Fracasso, P. M.; Westerveldt, P.; Fears, C. A.; Rosen, D. M.; Zuhowski, E. G.; Cazenave, L. A.; Litchman, M.; Egorin, M. J. Phase I Study of Paclitaxel in Combination With a Multidrug Resistance Modulator, PSC 833 (Valspodar), in Refractory Malignancies. J. Clin. Oncol. 2000, 18, 1124–1134. [Google Scholar]
- Blau, L.; Menegon, R. F.; Chung, M. C. Pró-fármaco ativado por enzima, uma estratégia promissora na quimioterapia. Quim. Nova 2006, 29, 1307–1317. [Google Scholar] [CrossRef]
- Stijilemans, B.; Conrath, K.; Cortez-Retamozo, V.; Van Xong, H.; Wyns, L.; Senter, P.; Revets, H.; De Baetselier, P.; Muyldermans, S.; Magez, S. Efficient Targeting of Conserved Cryptic Epitopes of Infectious Agents by Single Domain Antibodies: African Trypanosomes as Paradigm*. J. Biol. Chem. 2004, 279, 1256–1261. [Google Scholar]
- Heleno, V. C. G.; Silva, R.; Pedersoli, S.; Albuquerque, S.; Bastos, J. K.; Silva, M. L. A.; Donate, P. M.; Silva, G. V. J.; Lopes, J. L. C. Detailed 1H and 13C NMR structural assignment of three biologically active lignan lactones. Spectrochim. Acta A 2006, 63, 234–239. [Google Scholar] [CrossRef]
- Heleno, V. C. G.; Crotti, A. E. M.; Constantino, M. G.; Lopes, N. P.; Lopes, J. L. C. Total assignment of 1H and 13C NMR data for the sesquiterpene lactone 15-deoxygoyazensolide. Magn. Reson. Chem. 2004, 42, 364–367. [Google Scholar] [CrossRef]
- Jungheim, L. N.; Shepherd, T. A.; Meyer, D. L. Synthesis of acylhydrazido-substituted cephems. Design of cephalosporin-vinca alkaloid prodrugs: substrates for an antibody targeted enzyme. J. Org. Chem. 1992, 57, 2334–2340. [Google Scholar] [CrossRef]
- Nomura, H.; Fugono, T.; Hitaka, T.; Minami, I.; Azuma, T.; Morimoto, S.; Masuda, T. Semisynthetic β-lactam antibiotics. 6. Sulfocephalosporins and their antipseudomonal activities. J. Med. Chem. 1974, 17, 1312–1315. [Google Scholar] [CrossRef]
- Suzuki, N.; Sowa, T.; Murakami, M. Process for preparing 7-aminocephalosporanic acid derivatives. US Pat 4,079,180, 1978. [Google Scholar]
- Mobashery, S.; Johnston, M. Reactions of Escherichia coli TEM β-lactamase with cephalothin and with C10-dipeptidyl cephalosporin esters. J. Biol. Chem. 1986, 261, 7879–7887. [Google Scholar]
- Somerfield, G. A.; Wycombe, H.; Chagouri, H. Cephemoic acids and process for preparing same. US Pat 3,532,694, 1970. [Google Scholar]
- Letsinger, R. L.; Ogilvie, K. K. Use of p-nitrophenyl chloroformate in blocking hydroxyl groups in nucleosides. J. Org. Chem. 1967, 32, 296–300. [Google Scholar] [CrossRef]
- Alexander, R. P.; Bates, R. W.; Pratt, A. J.; Kraunsoe, J. A. E. AN-nitrosochloroethyl-cephalosporin carbamate prodrug for antibody-directed enzyme prodrug therapy (ADEPT). Tetrahedron 1996, 52, 5983–5988. [Google Scholar] [CrossRef]
- Kaiser, G. V.; Cooper, D. G.; Koehler, R. E.; Murphy, C. F.; Webber, J. A.; Wright, I. G.; Van Heyningen, E. M. Chemistry of cephalosporin antibiotics. XIX. Transformation of Δ2-cephem to Δ3-cephem by oxidation-reduction at sulfur. J. Org. Chem. 1970, 35, 2430–2433. [Google Scholar] [CrossRef]
- Sample Availability: No compound samples are available.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Blau, L.; Menegon, R.F.; Ferreira, E.I.; Ferreira, A.G.; Boffo, E.F.; Tavares, L.A.; Heleno, V.C.G.; Chung, M.-C. Synthesis and Total 1H- and 13C-NMR Assignment of Cephem Derivatives for Use in ADEPT Approaches. Molecules 2008, 13, 841-854. https://doi.org/10.3390/molecules13040841
Blau L, Menegon RF, Ferreira EI, Ferreira AG, Boffo EF, Tavares LA, Heleno VCG, Chung M-C. Synthesis and Total 1H- and 13C-NMR Assignment of Cephem Derivatives for Use in ADEPT Approaches. Molecules. 2008; 13(4):841-854. https://doi.org/10.3390/molecules13040841
Chicago/Turabian StyleBlau, Lorena, Renato Farina Menegon, Elizabeth Igne Ferreira, Antonio Gilberto Ferreira, Elisangela Fabiana Boffo, Leila Aley Tavares, Vladimir Constantino Gomes Heleno, and Man-Chin Chung. 2008. "Synthesis and Total 1H- and 13C-NMR Assignment of Cephem Derivatives for Use in ADEPT Approaches" Molecules 13, no. 4: 841-854. https://doi.org/10.3390/molecules13040841
APA StyleBlau, L., Menegon, R. F., Ferreira, E. I., Ferreira, A. G., Boffo, E. F., Tavares, L. A., Heleno, V. C. G., & Chung, M.-C. (2008). Synthesis and Total 1H- and 13C-NMR Assignment of Cephem Derivatives for Use in ADEPT Approaches. Molecules, 13(4), 841-854. https://doi.org/10.3390/molecules13040841