Design and Synthesis of Bis-amide and Hydrazide-containing Derivatives of Malonic Acid as Potential HIV-1 Integrase Inhibitors

 ,
,
Abstract
:
Introduction
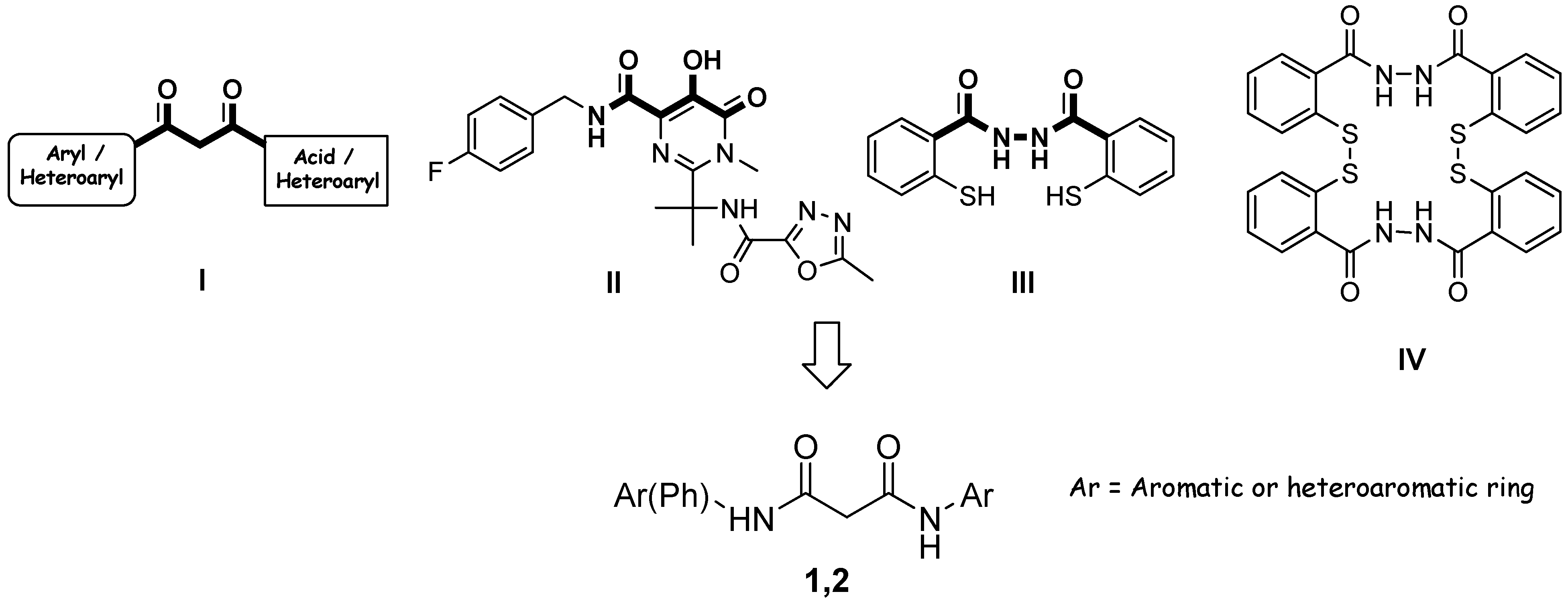
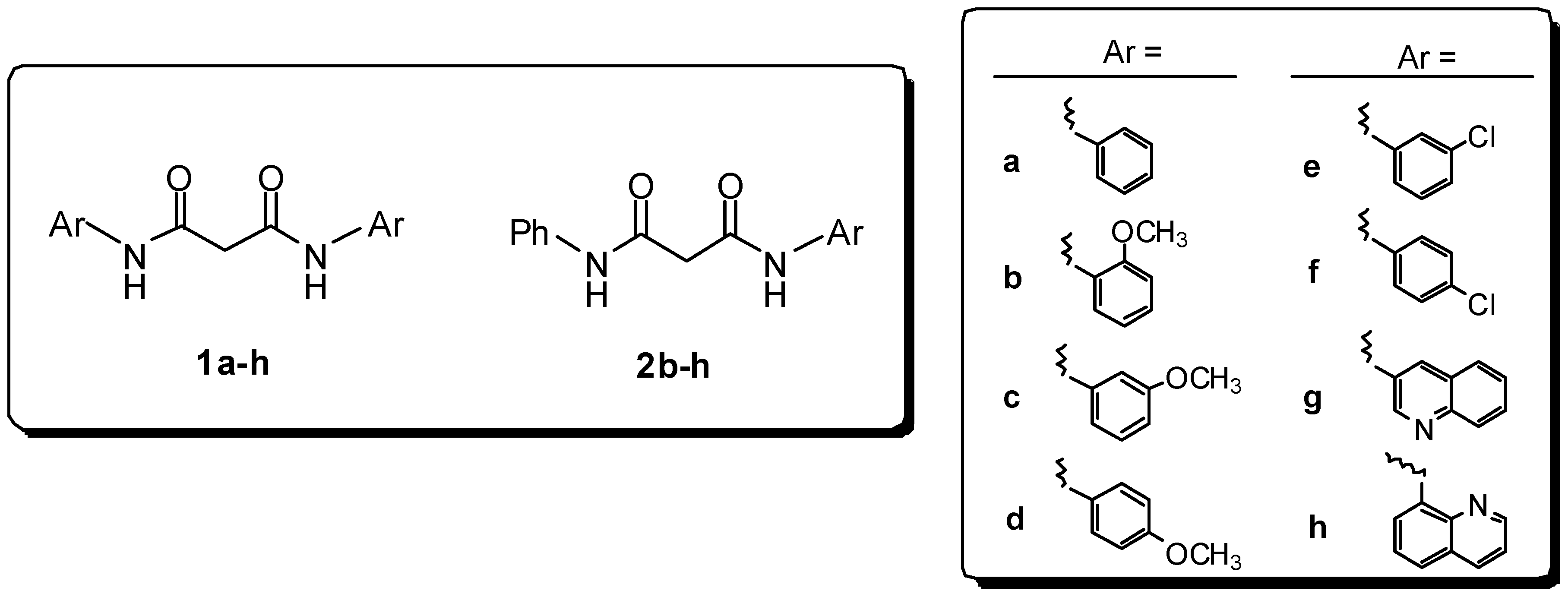
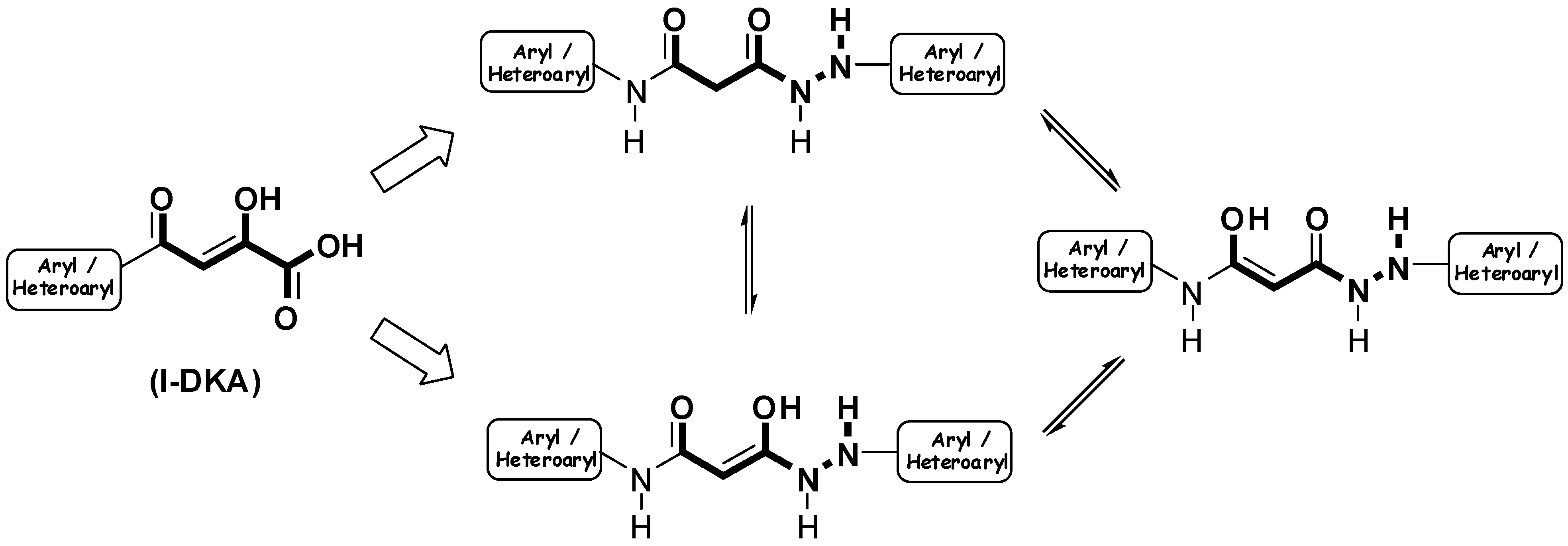
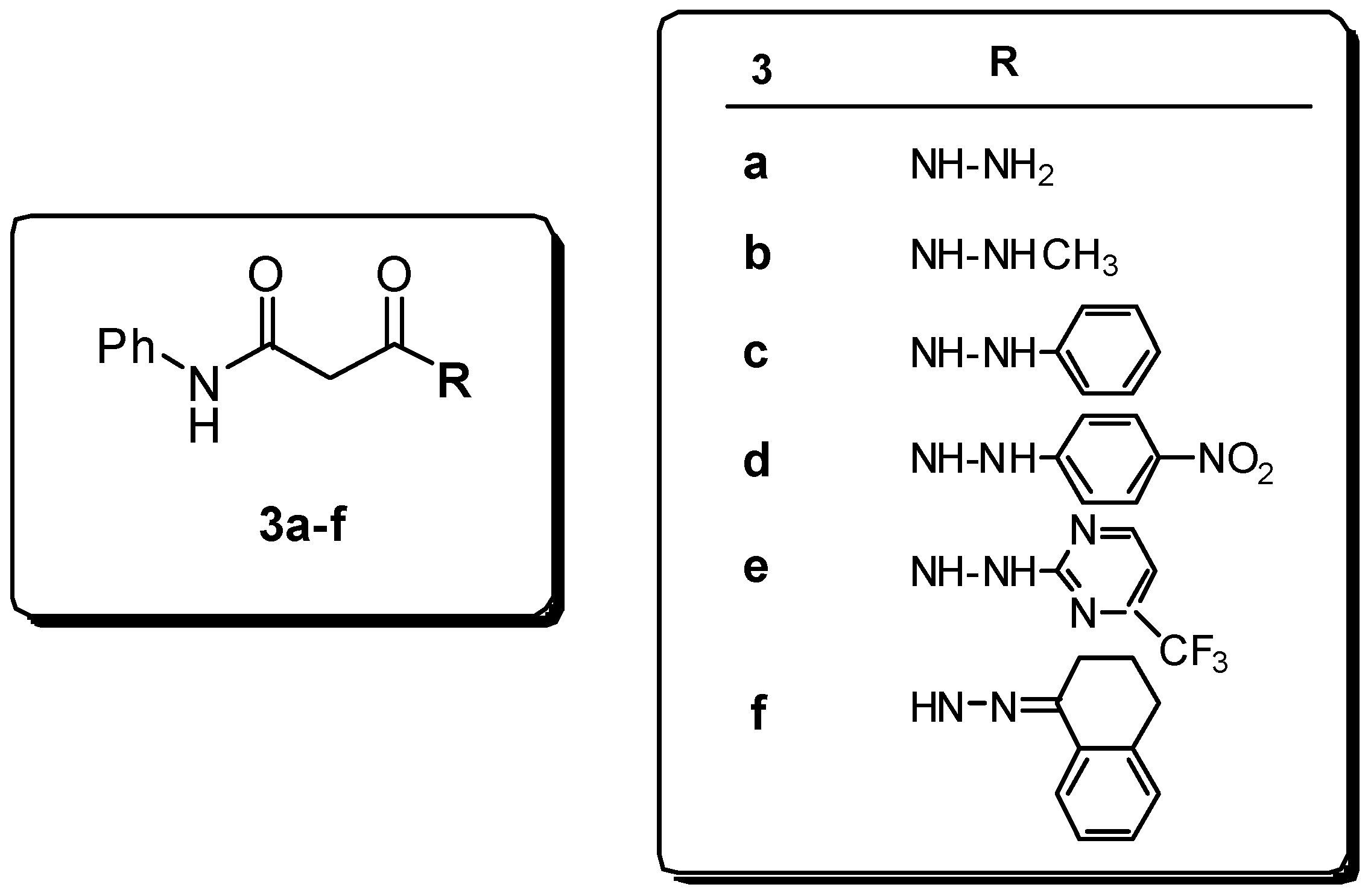
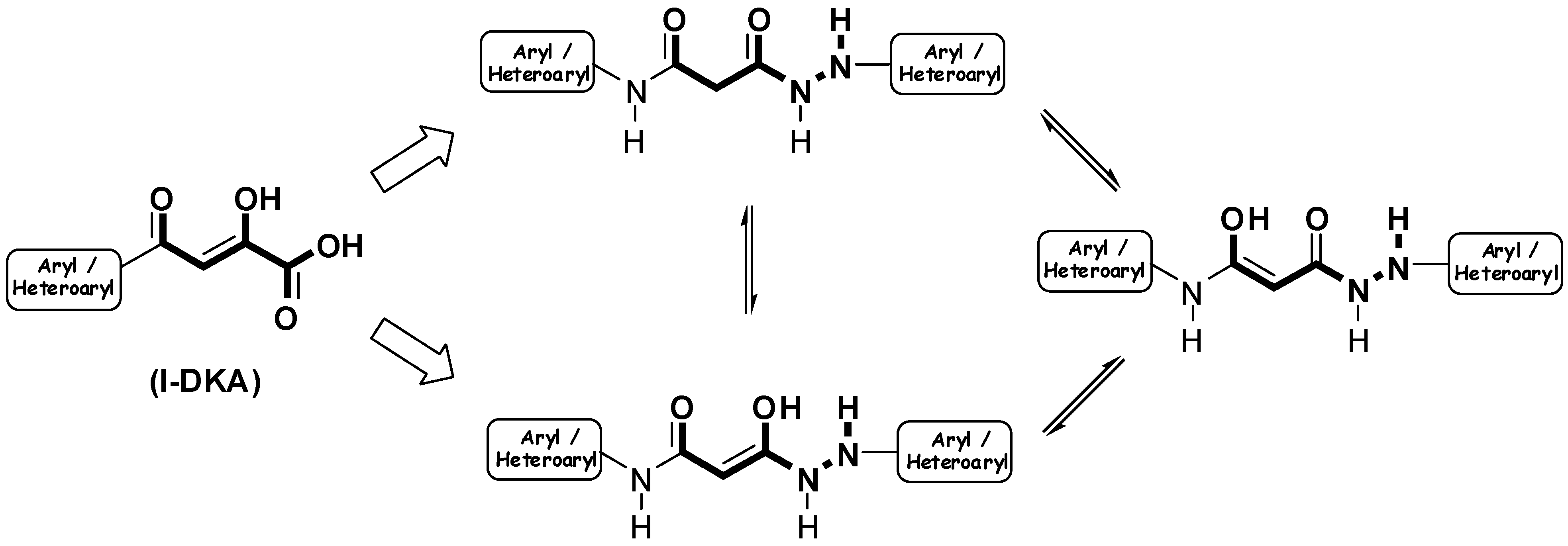
Results and Discussion
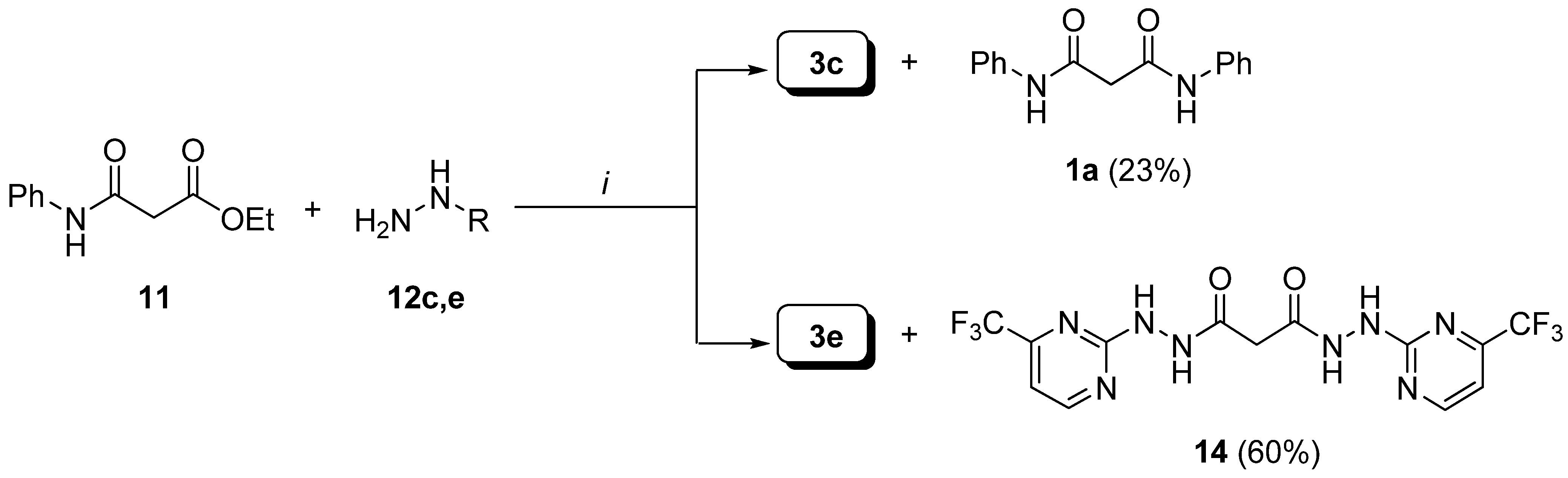
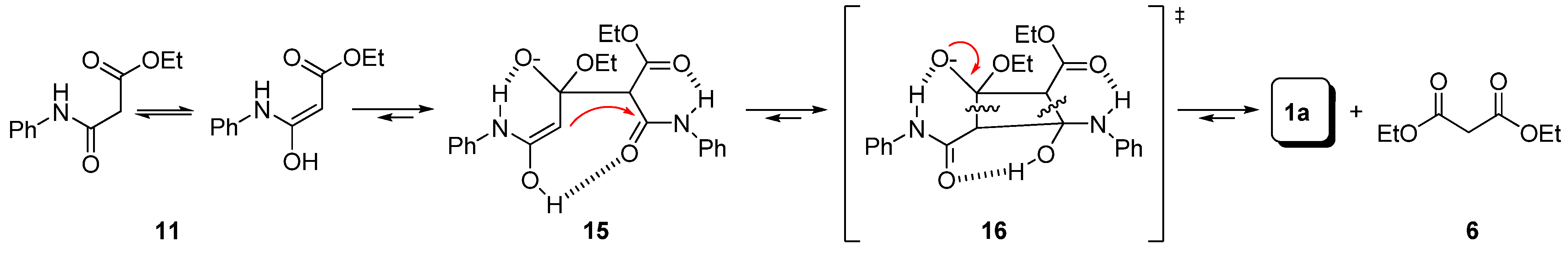
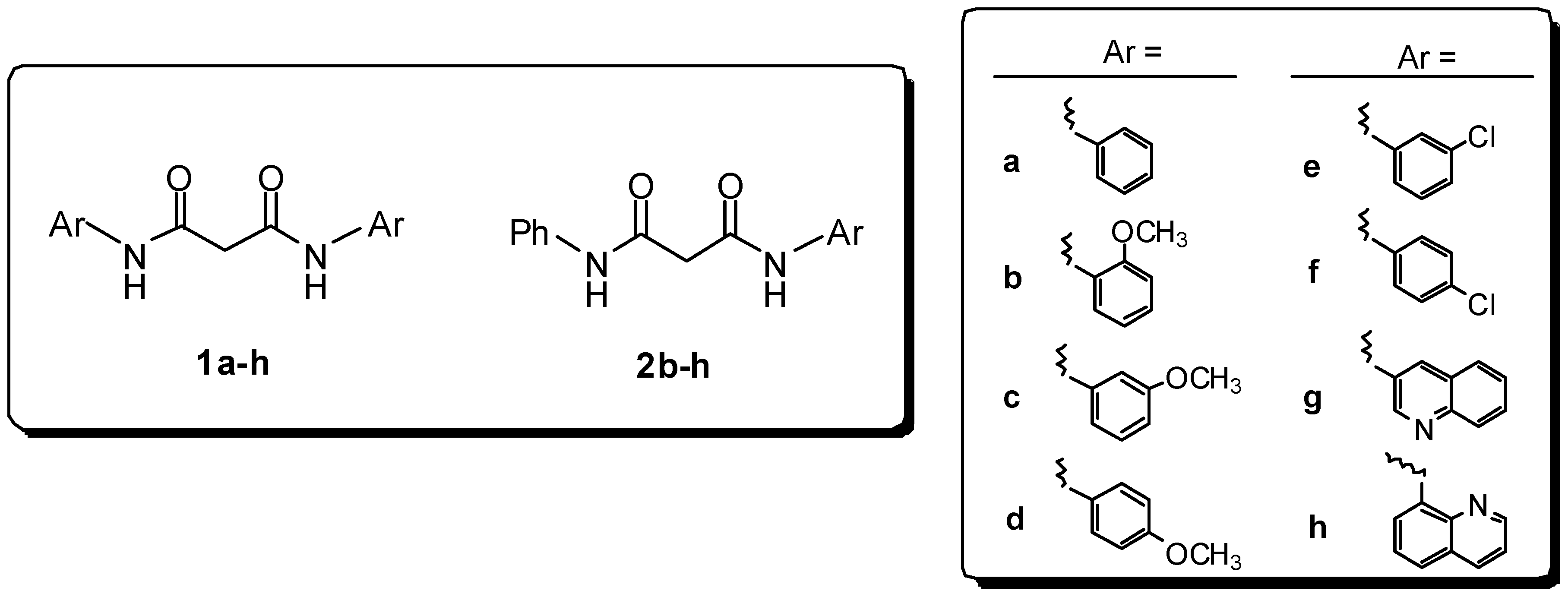
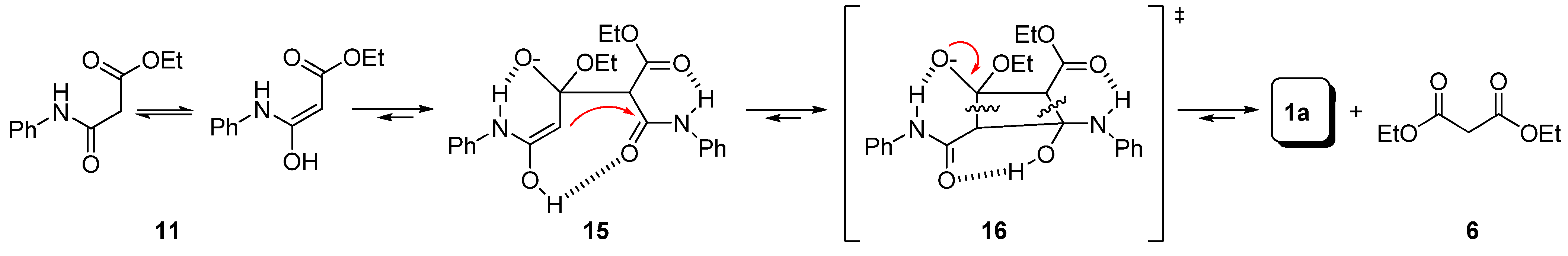
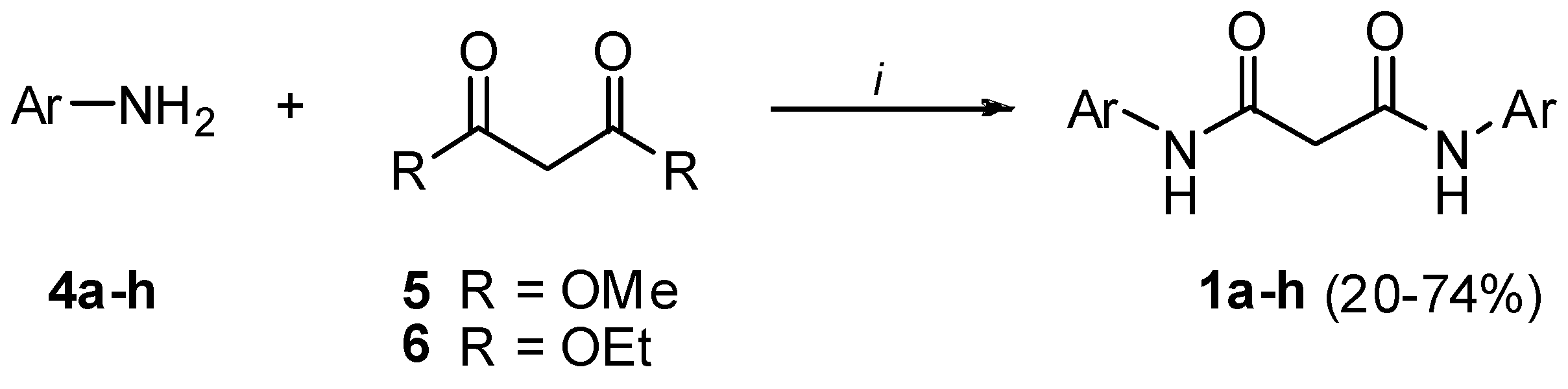
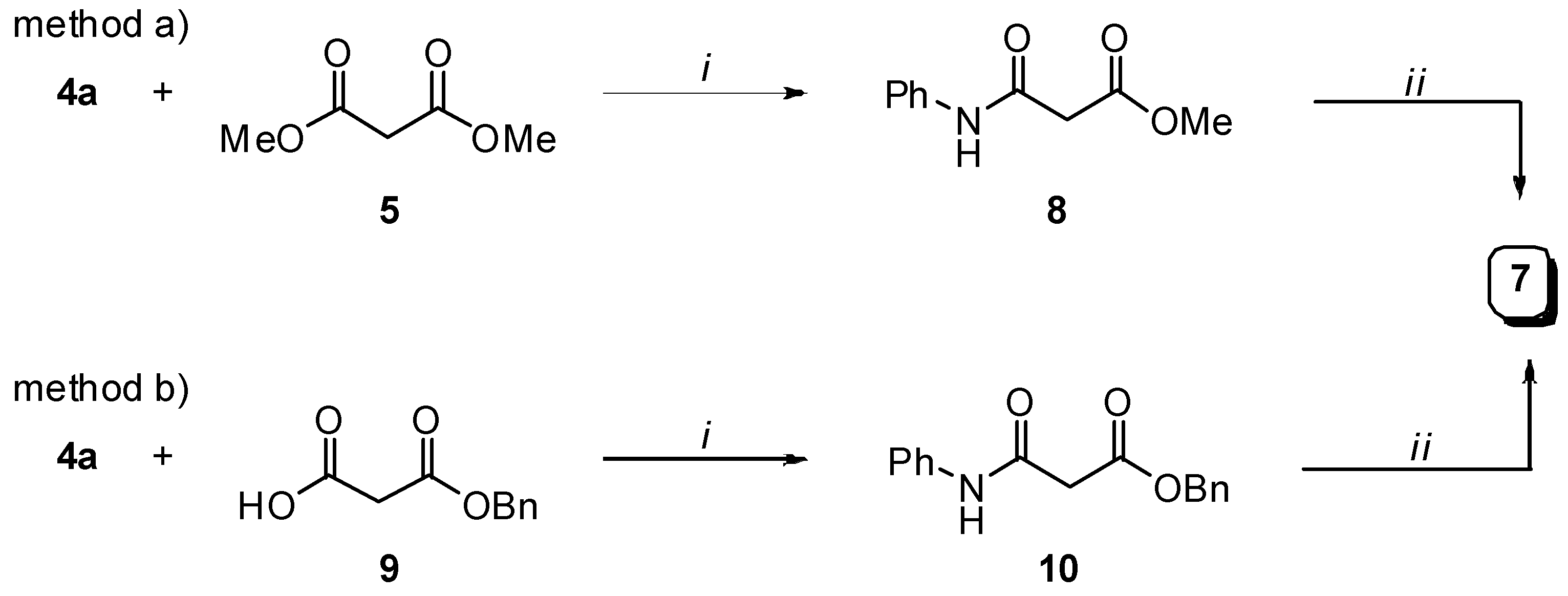
Preparation of bis-amides.
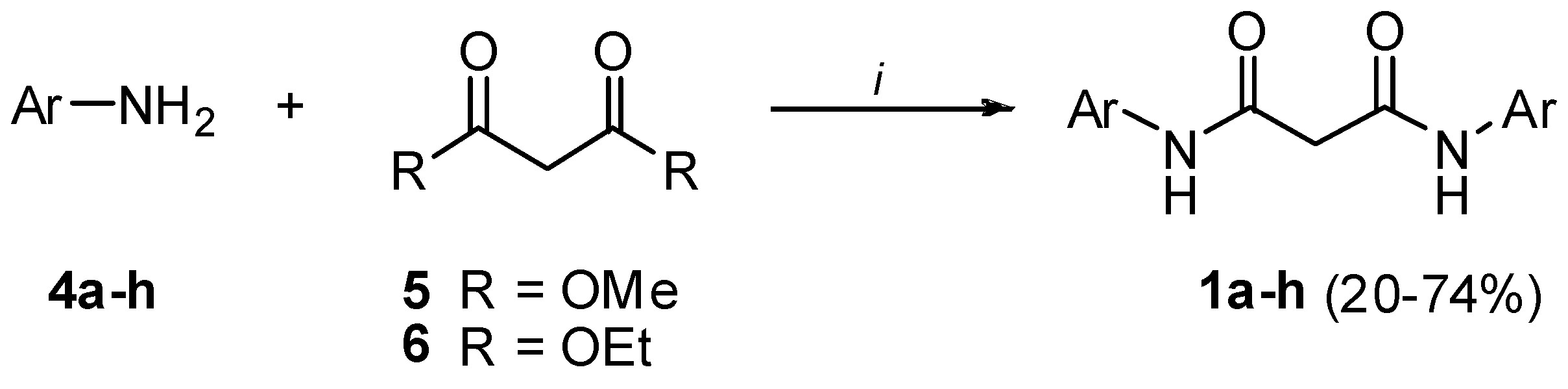
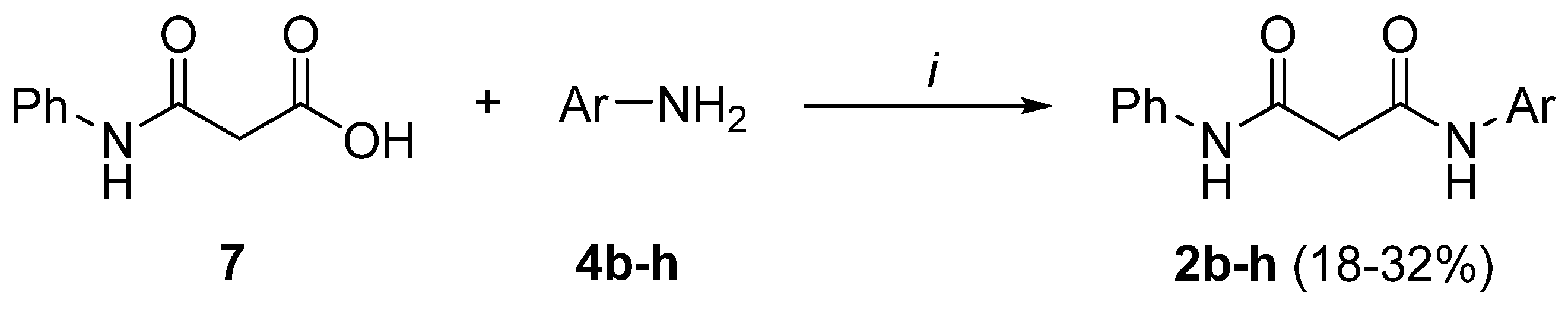
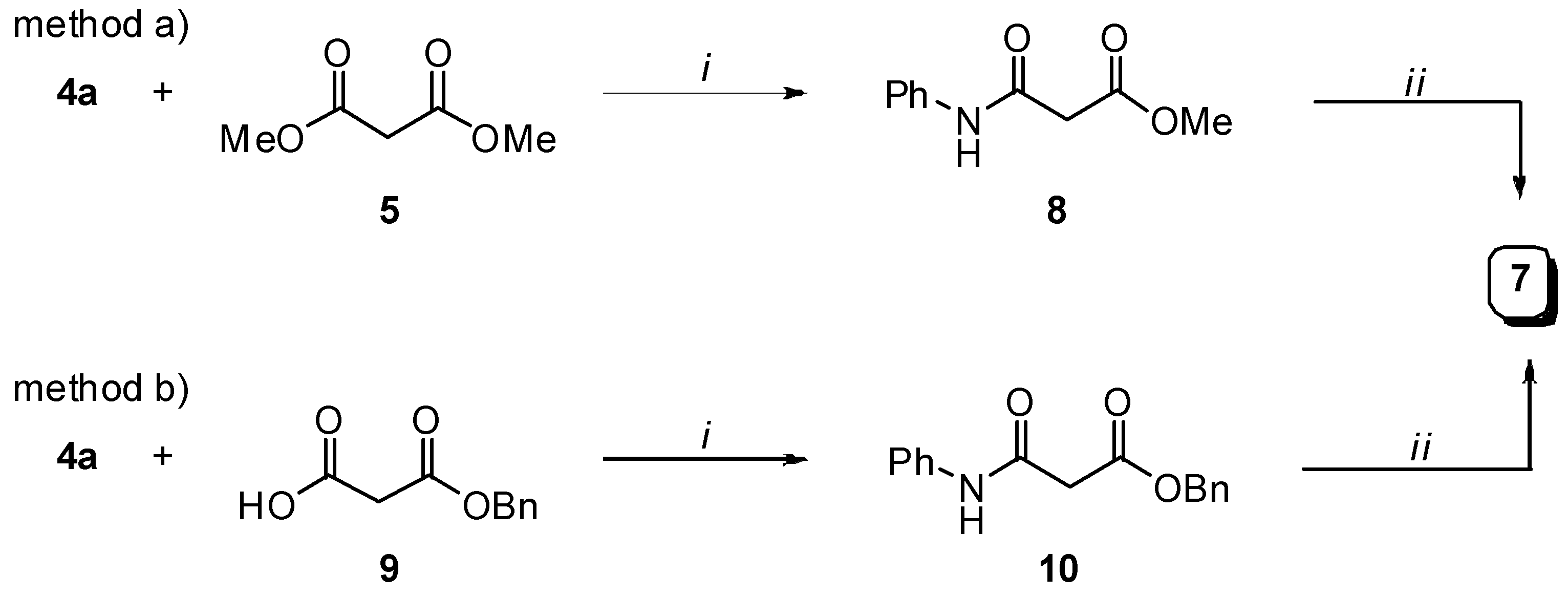
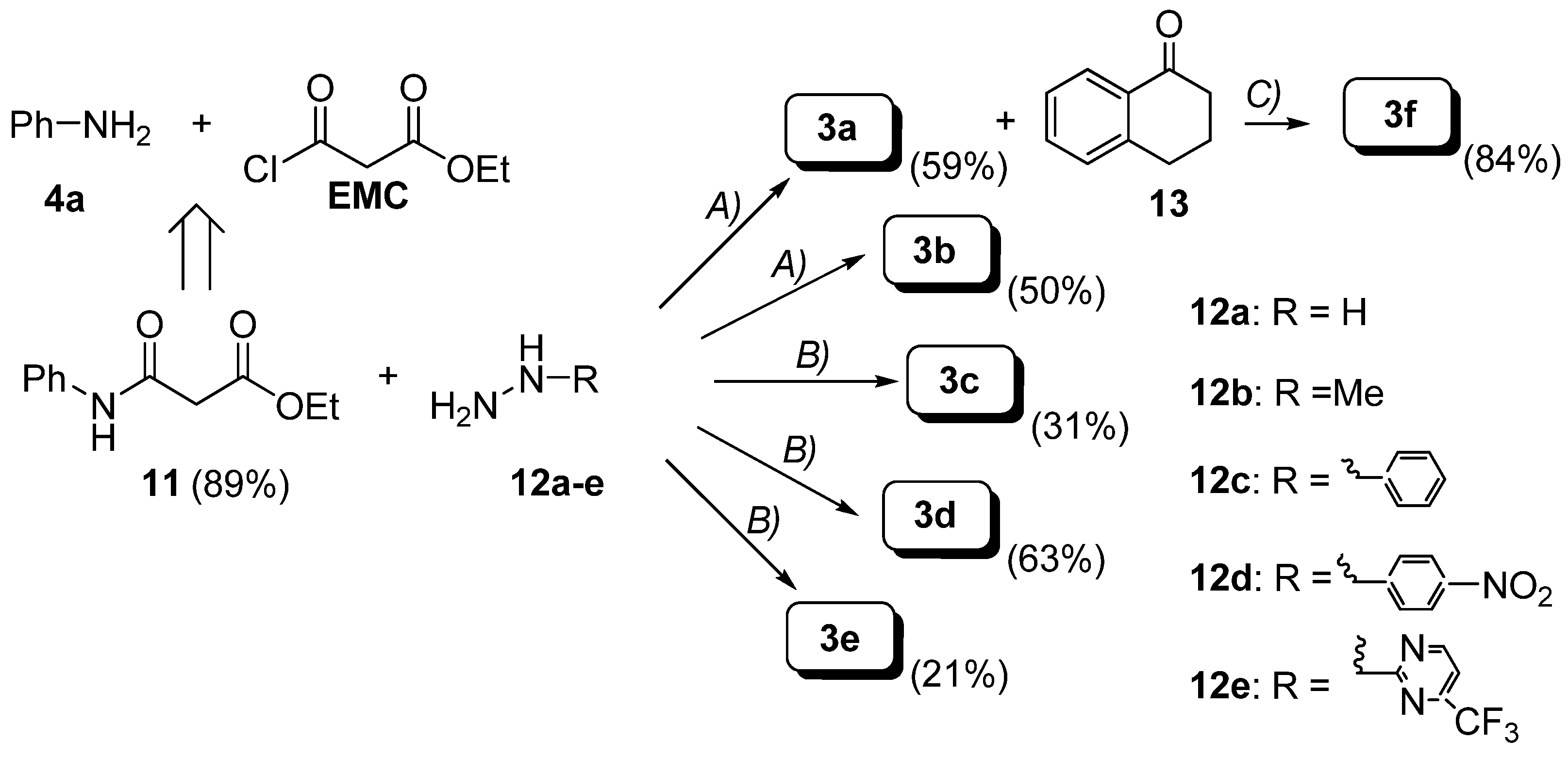
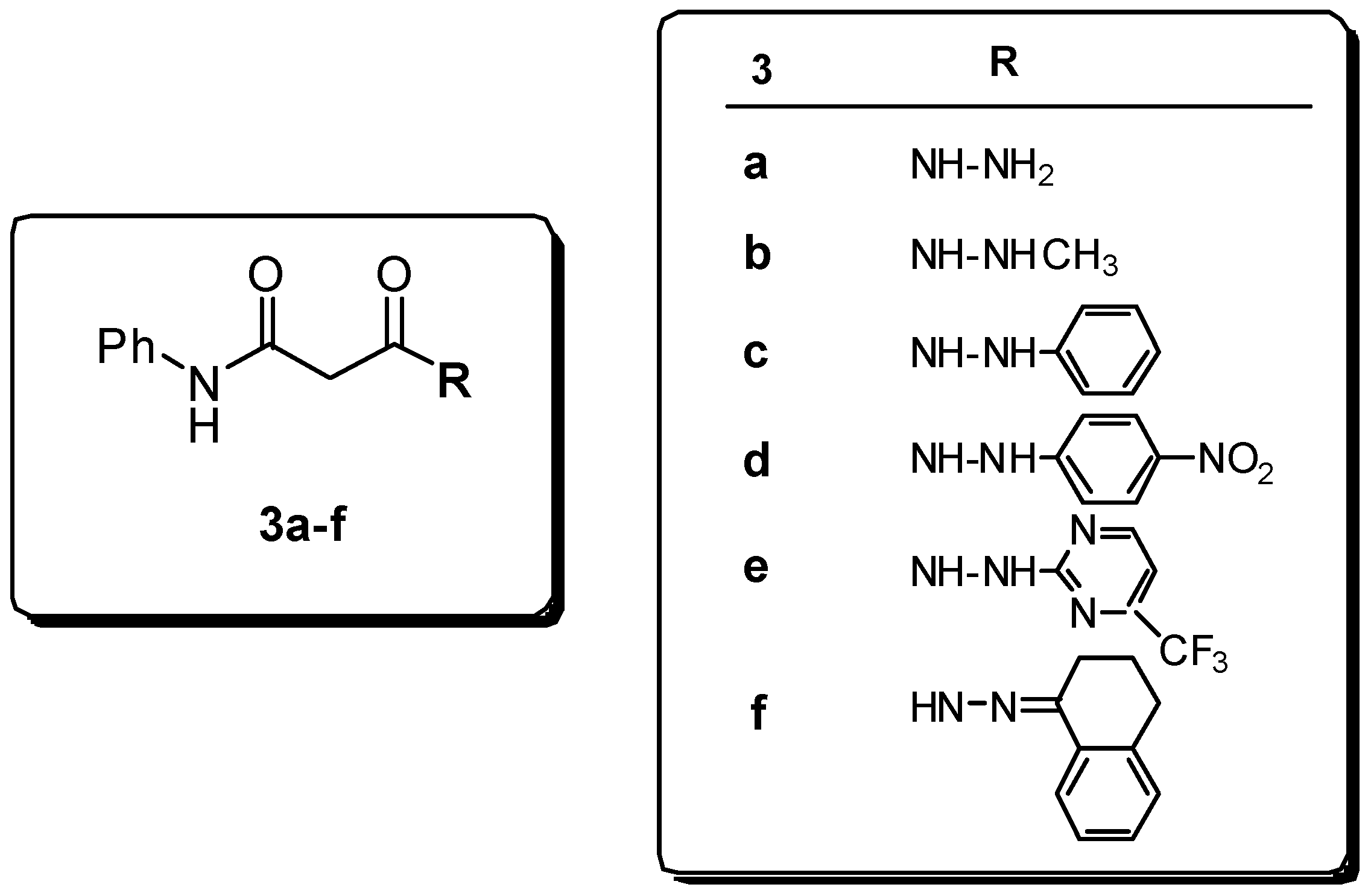
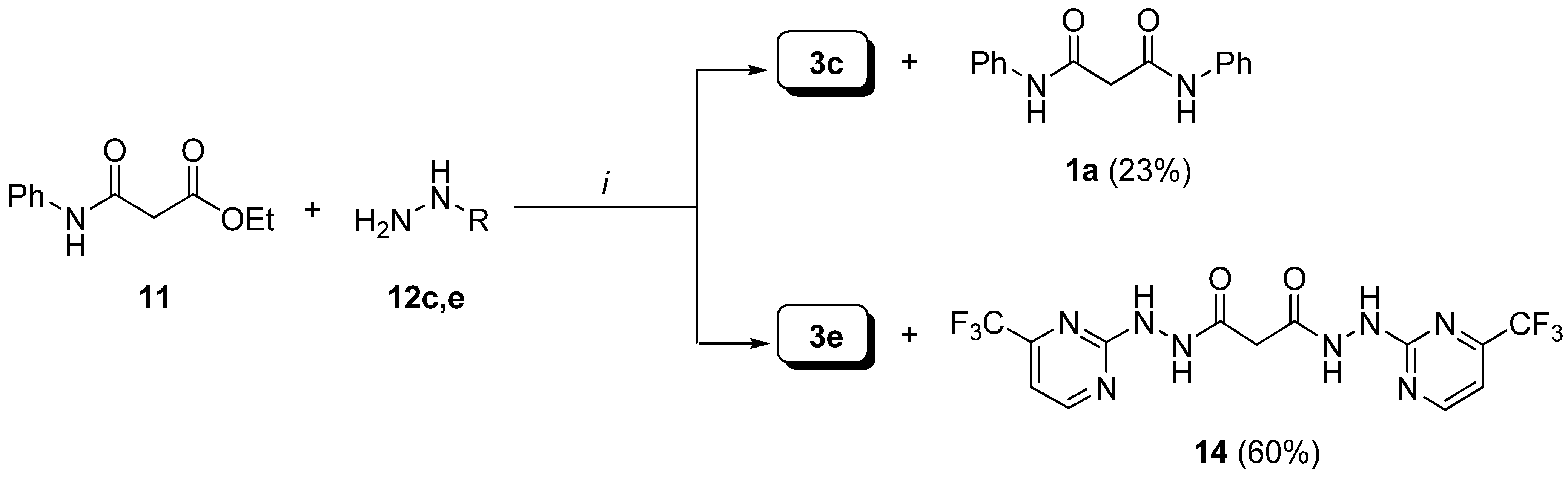
Preparation of hydrazides.
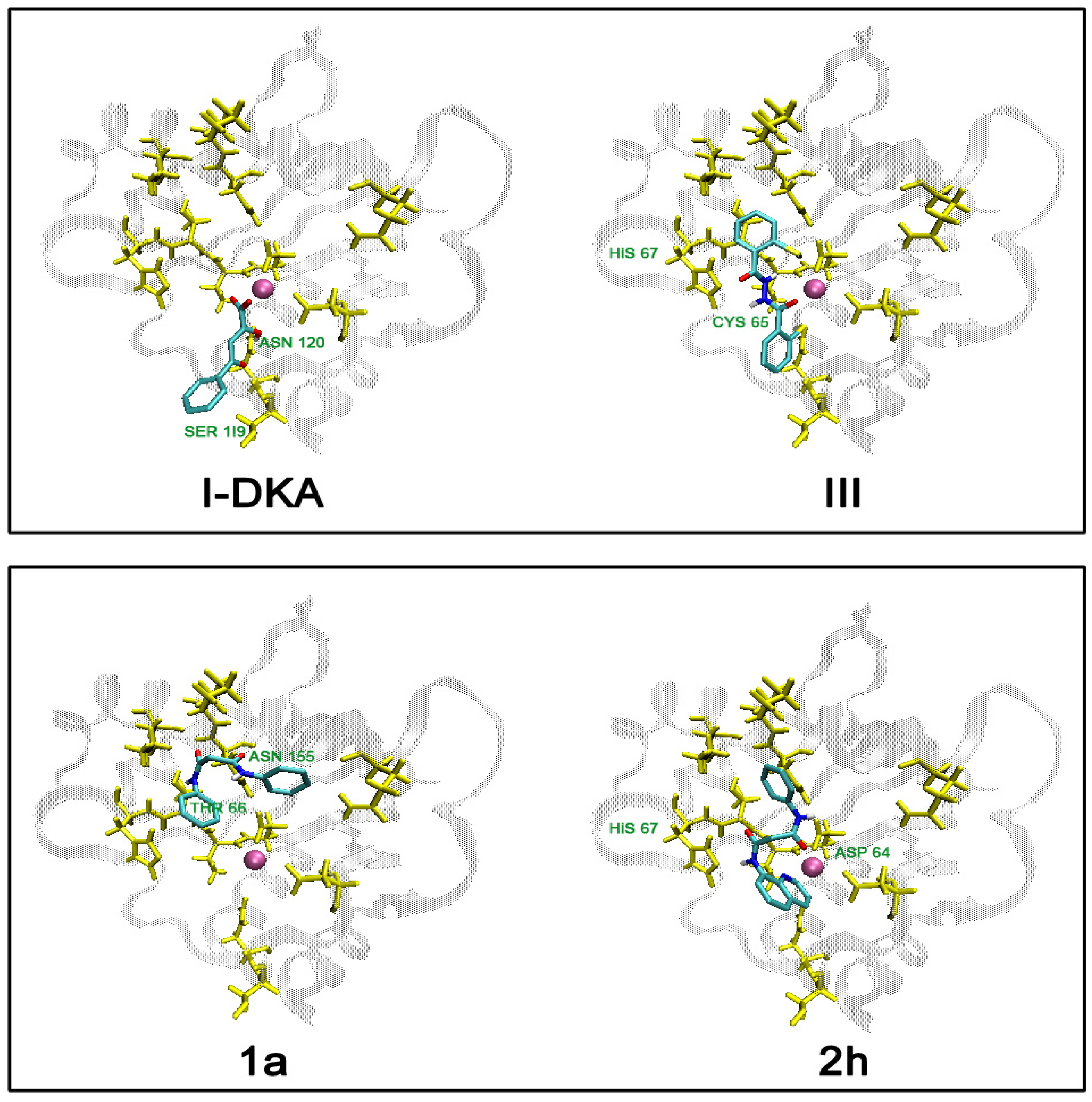
Molecular Modeling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
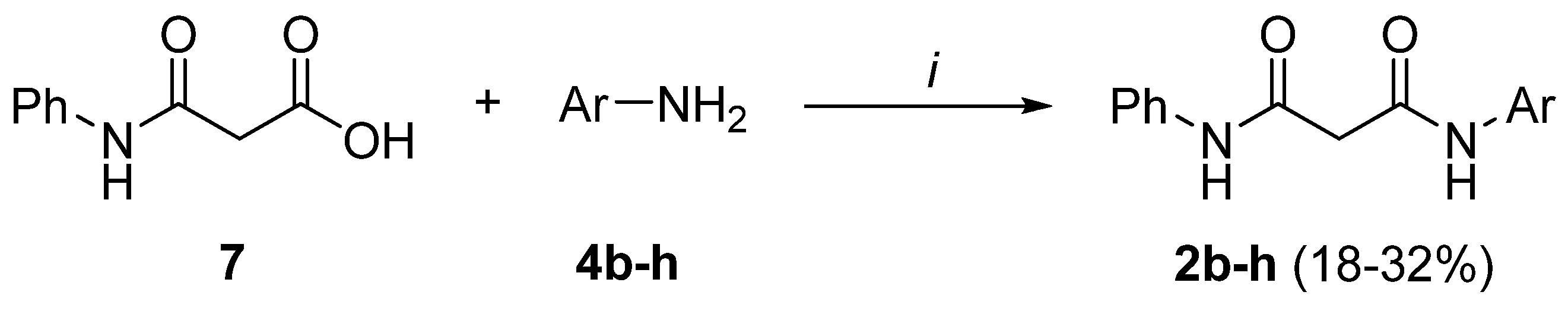{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | *Ntot | †focc | °F.D.E. | ‡∆Gbind | #Ki | H-bonds |
| I-Phe-DKA | 35 | 5/4 | -8.94 | -7,78 | 2.00E-06 | Ser119, Asn120 |
| III | 51 | 8/8 | -8.76 | -6,78 | 1.07E-05 | Cys65, His67, Glu92 |
| 1a | 39 | 10/7 | -6.65 | -5,49 | 9.38E-05 | Thr66, Asn155 |
| 2h | 25 | 19/9 | -8.19 | -6,86 | 9.29E-06 | Asp64, His67 |
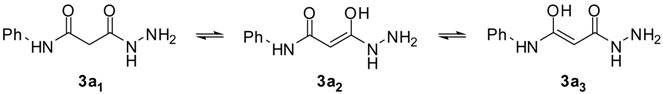
| 3a1 | 21 | 20/14 | -5.79 | -4,41 | 5.81E-04 | Cys65, Asp116, Asn155 |
| 3a2 | 50 | 13/7 | -6.62 | -5,56 | 8.34E-05 | Cys65, Asn120 |
| 3a3 | 31 | 10/7 | -6.88 | -5,61 | 7.66E-05 | Asp64, Cys65 |
| 3c | 28 | 14/10 | -8.21 | -6,55 | 1.57E-05 | Asp64, His67 |
| 3d | 17 | 26/13 | -8.26 | -6,56 | 1.55E-05 | Asp64, Cys65, His67, Ly156 |
| 3e | 21 | 27/12 | -7.42 | -5,61 | 7.68E-05 | Cys65, Asn155 |
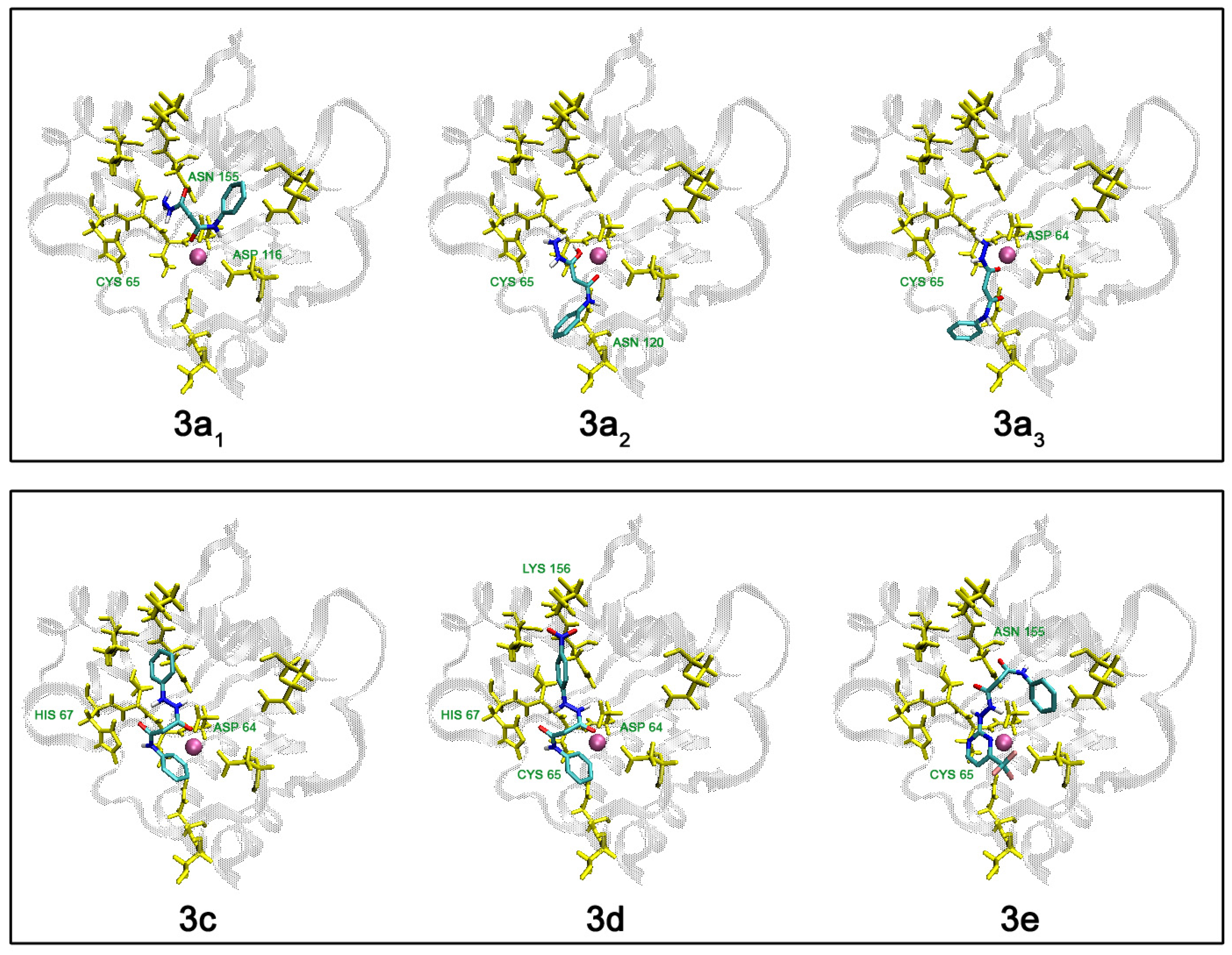
Conclusions
Experimental
General
General procedure for the synthesis of symmetric bis-amides (1a-h)
Preparation of N-phenylmalonamic acid (7).
Molecular Modeling
Acknowledgements
References and Notes
- Pommier, Y.; Johnson, A. A.; Marchand, C. Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug Discov. 2005, 4, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Neamati, N. Patented small molecule inhibitors of HIV-1 integrase: a ten-year saga. Expert Opin. Ther. Pat. 2002, 12, 709–724. [Google Scholar] [CrossRef]
- Cotelle, P. Patented HIV-1 integrase inhibitors (1998-2005). Recent Patents Anti-Infect. Drug Disc. 2006, 1, 1–15. [Google Scholar] [CrossRef]
- Wang, Y.; Serradell, N.; Bolos, J.; Rosa, E. MK-0518, HIV integrase inhibitor. Drugs Fut. 2007, 32, 118–122. [Google Scholar] [CrossRef]
- Pais, G. C. G.; Burke, T. R. Novel aryl diketo-containing inhibitors of HIV-1 integrase. Drugs Fut. 2002, 27, 1101–1111. [Google Scholar]
- Neamati, N.; Lin, Z.; Karki, R. G.; Orr, A.; Cowansage, K.; Strumberg, D.; Pais, G. C. G.; Voigt, J. H.; Nicklaus, M. C.; Winslow, H. E.; Zhao, H.; Turpin, J. A.; Yi, J.; Skalka, A. M.; Burke, T.R., Jr.; Pommier, Y. Metal-dependent inhibition of HIV-1 integrase. J. Med. Chem. 2002, 45, 5661–5670. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P. E.; Duus, F.; Bolvig, S.; Jagodzinski, T. S. Intramolecular hydrogen bonding of the enol forms of β-ketoamides and β-ketothioamides. Deuterium isotope effects on 13C chemical shifts. J. Mol. Struct. 1996, 378, 45–59. [Google Scholar] [CrossRef]
- Hanson, J. C. Structure of a Copper-Isoniazid Complex. J. Med. Chem. 1981, 24, 1369–1371. [Google Scholar] [CrossRef] [PubMed]
- Jahagirdar, J. A.; Patil, B. G.; Havinale, B. R. Cobalt(II), nickel(II), and copper(II) complexes of malonoanilic acidhydrazones. Ind. J. Chem., A: Inorg., Phys., Theor. Anal. 1990, 29A, 924–926. [Google Scholar]
- David, L.; Rusu, M.; Cozar, O.; Rusu, D.; Todica, M.; Balan, C. Spectroscopic and magnetic investigations of some transition metal complexes with N-4-methoxyphenyl-N-4-chlorobenzoyl hydrazide as ligand. J. Mol. Struct. 1999, 482-483, 149–152. [Google Scholar] [CrossRef]
- Shulgin, V. F.; Pevzner, N. S.; Zub, V. Y.; Strizhakova, N. G.; Maletin, Y. A. The synthesis and structure of nickel(II) and copper(II) complexes with N,N-dimethyl-N′-(2,4-dichlorophenoxy)acetyl hydrazine. Inorg. Chem. Comm. 2001, 3, 134–137. [Google Scholar] [CrossRef]
- Vennerstrom, J. L., Jr.; Holmes, T. J., Jr. Prostaglandin-H synthase inhibition by malonamides. Ring-opened analogues of phenylbutazone. J. Med. Chem. 1987, 30, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Kunishima, M.; Kawachi, C.; Morita, J.; Terao, K.; Iwasaki, F.; Tani, S. 4-(4,6-Dimethoxy- 1,3,5-triazin-2-yl)-4-methyl-morpholinum chloride: an efficient condensing agent leading to formation of amides and esters. Tetrahedron 1999, 55, 13159–13170. [Google Scholar] [CrossRef]
- Falchi, A.; Porcheddu, A.; Taddei, M. 4-(4,6-Dimethoxy[1,3,4]triazin-2-yl)-4-methyl-morpholinum chloride (DMTMM): a valuable activating agent for carboxylic acids in solution, liquid and solid phase synthesis. Acros Org. Acta 2001, 8, 8–10. [Google Scholar]
- Chattaway, F. D.; Olmsted, J. M. D. The action of aromatic amines on ethyl malonate. J. Chem. Soc. 1910, 938–941. [Google Scholar] [CrossRef]
- Kaminski, Z. J. 2-Chloro-4,6-disubstituted-1,3,5-triazines: a novel group of condensing reagents. Tetrahedron Lett. 1985, 26, 2901–2904. [Google Scholar] [CrossRef]
- Dhapalapur, M. G.; Sabnis, S. S.; Deliwala, C. V. Potential Anticancer Agents. I. Synthesis of Some Nitrogen Mustard Containing Benzylidenehydrazides. J. Med. Chem. 1968, 11, 154–156. [Google Scholar] [CrossRef]
- Kutyrev, A.; Kappe, T. Methanetricarboxylates as Key Reagents for the Simple Preparation of Heteroarylcarboxamides with Potential Biological Activity. Part 1. Reaction of Methanetricarboxylates with Indoline and 1,2,3,4-Tetrahydroquinoline. J. Heter. Chem. 1997, 34, 969–972. [Google Scholar] [CrossRef]
- Ukrainets, I. V.; Bezugly, P. A.; Treskach, V. I.; Taran, S. G.; Gorokhova, O. V. Ethyl Esters of Malonanilic Acids. Synthesis and Pyrolysis. Tetrahedron 1994, 50, 10331–10338. [Google Scholar] [CrossRef]
- Nishino, H.; Ishida, K.; Hashimoto, H.; Kurosawa, K. Manganese(III)-Mediated Oxidative Radical Cyclization 3. Synthesis of 3-Azabicyclo[3,3,0]-octan-2-ones and Related Compounds in the Reaction of 1,1,6,6-Tetraarylhexa-1,5-dienes with N,N’-Disubstituted Malonamides. Synthesis 1996, 888–896. [Google Scholar] [CrossRef]
- Sotriffer, C. A.; Ni, H.; McCammon, A. J. HIV-1 Integrase inhibitor interactions at the active site: prediction of binding modes unaffected by crystal packing. J. Am. Chem. Soc. 2000, 122, 6136–6137. [Google Scholar] [CrossRef]
- Sechi, M.; Angotzi, G.; Dallocchio, R.; Dessì, A.; Carta, F.; Sannia, L.; Mariani, A.; Fiori, S.; Sanchez, T.; Movsessian, L.; Plasencia, C.; Neamati, N. Design and synthesis of novel dihydroxyindole-2-carboxylic acids as HIV-1 integrase inhibitors. Antiv. Chem Chemother. 2004, 15, 67–81. [Google Scholar] [CrossRef]
- Sotriffer, C.A.; Ni, H.; McCammon, A.J. Active site binding modes of HIV-1 Integrase inhibitors. J. Med. Chem. 2000, 43, 4109–4117. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Sotriffer, C. A.; McCammon, J. A. Ordered water and ligand mobility in the HIV-1 integrase5CITEP complex: a molecular dynamics study. J. Med. Chem. 2001, 44, 3043–3047. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M. L.; Lee, K. W.; Chimirri, A.; Briggs, J. M. Molecular dynamics studies of the wild-type and double mutant HIV-1 integrase complexed with the 5CITEP inhibitor: Mechanism for inhibition and drug resistance. Biophys. J. 2003, 84, 1450–1463. [Google Scholar] [CrossRef]
- Zeinalipour-Loizidou, E.; Nicolaou, C.; Nicolaides, A.; Kostrikis, L. G. HIV-1 Integrase: From Biology to Chemotherapeutics. Curr. HIV Res. 2007, 5, 365–388. [Google Scholar] [CrossRef] [PubMed]
- Goldgur, Y.; Craigie, R.; Cohen, G. H.; Fujiwara, T.; Yoshinaga, T.; Fujishita, T.; Sugimoto, H.; Endo, T.; Murai, H.; Davies, D. R. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc. Natl. Acad. Sci. USA 1999, 96, 13040–13043. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D. S.; Huey, R.; Olson, A. Distributed automated docking of flexible ligands to proteins: Parallel application of AutoDock 2.4*. J. Comp. Aid. Mol. Des. 1996, 10, 293–304. [Google Scholar] [CrossRef]
- Neamati, N.; Marchand, C.; Pommier, Y. HIV-1 integrase inhibitors: past, present, and future. In Advances in Pharmacology; Academic Press: San Diego, USA, 2000; volume 49, pp. 147–165. [Google Scholar]
- Semenova, E. A.; Marchand, C.; Pommier, Y. HIV-1 integrase inhibitors: update and perspectives. Adv. Pharmacol. 2008, 56, 199–228. [Google Scholar] [PubMed]
- Sechi, M.; Bacchi, A.; Carcelli, M.; Compari, C.; Duce, E.; Fisicaro, E.; Rogolino, D.; Gates, P.; Derudas, M.; Al-Mawsawi, L. Q.; Neamati, N. From Ligand to Complexes: Inhibition of Human Immunodeficiency Virus Type 1 Integrase by β-Diketo Acid Metal Complexes. J. Med. Chem. 2006, 49, 4248–4260. [Google Scholar] [CrossRef] [PubMed]
- Shobana, N.; Yeshoda, P.; Shanmugam, P. A convenient approach to the synthesis of prenyl-, furo- and pyranoquinoline alkaloids of the Rutaceae. Tetrahedron 1989, 45, 757–762. [Google Scholar] [CrossRef]
- Price, C. C.; Velzen, B. H. Some derivatives of oxanilide. J. Org. Chem. 1947, 12, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xu, J.; Li, Z.; Huang, B. An 8-Aminoquinoline-based Fluorescent Sensor of Transition Metal Ions. J. Chem. Res., Syn. 1998, 8, 444–445. [Google Scholar] [CrossRef]
- Grimm, J.; Harrington, P.; Heidebrecht, R., Jr.; Miller, T.; Otte, K.; Siliphaivanh, P.; Sloman, D.; Stanton, M.; Wilson, K.; Witter, D.; Kattar, S.; Tempest, P. Preparation of modified malonates, particularly 2-[4-[[(2-aminophenyl)amino]carbonyl]phenyl]-N,N'-disubstituted malonamides and related derivatives, as histone deacetylase inhibitors useful for treating neoplasm and other proliferative diseases. [PCT Int. Appl., WO 2007002248 2007].
- Bakulev, V. A.; Morzherin, Y. Y.; Lebedev, A. T.; Dankova, E. F.; Kolobov, M. Y.; Shafran, Y.M. Study of polyfunctional diazo compound reactivity in heterocyclization by the method of intramolecular competitive reactions. Bull. Soc. Chim. Belg. 1993, 102, 493–502. [Google Scholar] [CrossRef]
- Ziegler, E.; Brus, G. Synthesis of functionally mixed malonic acid derivatives. Monatsh. Chem. 1967, 98, 1100–1113. [Google Scholar] [CrossRef]
- Macromodel, version 6.0; Columbia University: New York, USA, 1997.
- SYBYL, version 6.2; Tripos Inc.: St. Louis, MO., USA, 2001.
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity - a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Chang, G.; Guida, W. C.; Still, W. C. An internal coordinate Monte Carlo method for searching conformational space. J. Am. Chem. Soc. 1989, 111, 4379–4386. [Google Scholar] [CrossRef]
- Weiner, S. J.; Kollman, P. A.; Case, D. A.; Singh, U. C.; Ghio, C.; Alagona, G. S.; Profeta, P.; Weiner, P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984, 106, 765–784. [Google Scholar] [CrossRef]
- Morris, G. M.; Goodsell, D. S.; Halliday, R. S.; Huey, R.; Hart, W. E.; Belew, R. K.; Olson, A. J. Automated docking using a Lamarckian genetic algorithm an empirical binding free energy function. J. Comp. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Sanner, M. F. Python: A Programming Language for Software Integration and Development. J. Mol. Graphics Mod. 1999, 17, 57–61. [Google Scholar]
- Sample Availability: Samples of the compounds 1a-h, 2a-h, 3a-f are available from the authors.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sechi, M.; Azzena, U.; Delussu, M.P.; Dallocchio, R.; Dessì, A.; Cosseddu, A.; Pala, N.; Neamati, N. Design and Synthesis of Bis-amide and Hydrazide-containing Derivatives of Malonic Acid as Potential HIV-1 Integrase Inhibitors. Molecules 2008, 13, 2442-2461. https://doi.org/10.3390/molecules13102442
Sechi M, Azzena U, Delussu MP, Dallocchio R, Dessì A, Cosseddu A, Pala N, Neamati N. Design and Synthesis of Bis-amide and Hydrazide-containing Derivatives of Malonic Acid as Potential HIV-1 Integrase Inhibitors. Molecules. 2008; 13(10):2442-2461. https://doi.org/10.3390/molecules13102442
Chicago/Turabian StyleSechi, Mario, Ugo Azzena, Maria Paola Delussu, Roberto Dallocchio, Alessandro Dessì, Alessia Cosseddu, Nicolino Pala, and Nouri Neamati. 2008. "Design and Synthesis of Bis-amide and Hydrazide-containing Derivatives of Malonic Acid as Potential HIV-1 Integrase Inhibitors" Molecules 13, no. 10: 2442-2461. https://doi.org/10.3390/molecules13102442
APA StyleSechi, M., Azzena, U., Delussu, M. P., Dallocchio, R., Dessì, A., Cosseddu, A., Pala, N., & Neamati, N. (2008). Design and Synthesis of Bis-amide and Hydrazide-containing Derivatives of Malonic Acid as Potential HIV-1 Integrase Inhibitors. Molecules, 13(10), 2442-2461. https://doi.org/10.3390/molecules13102442