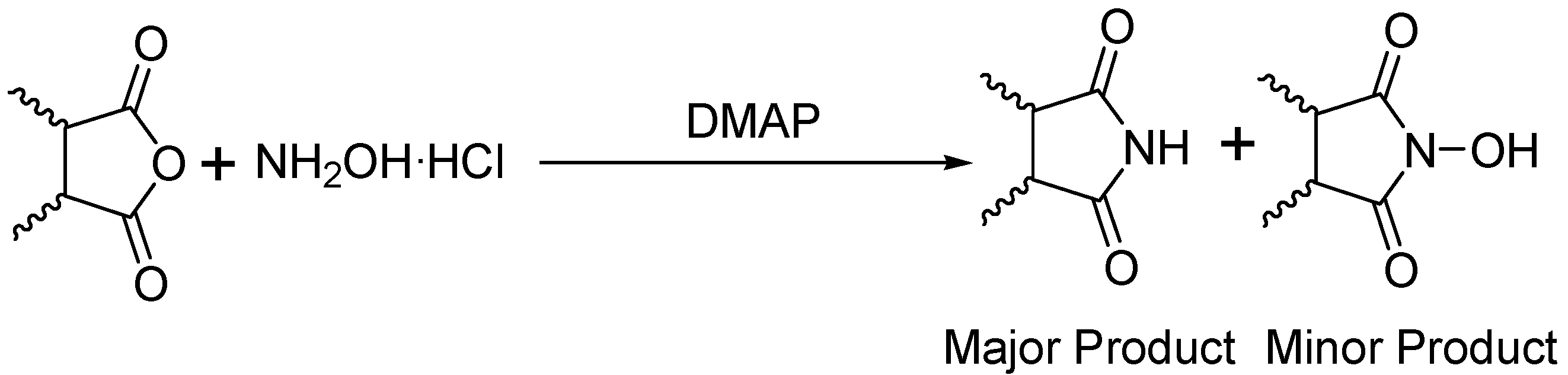
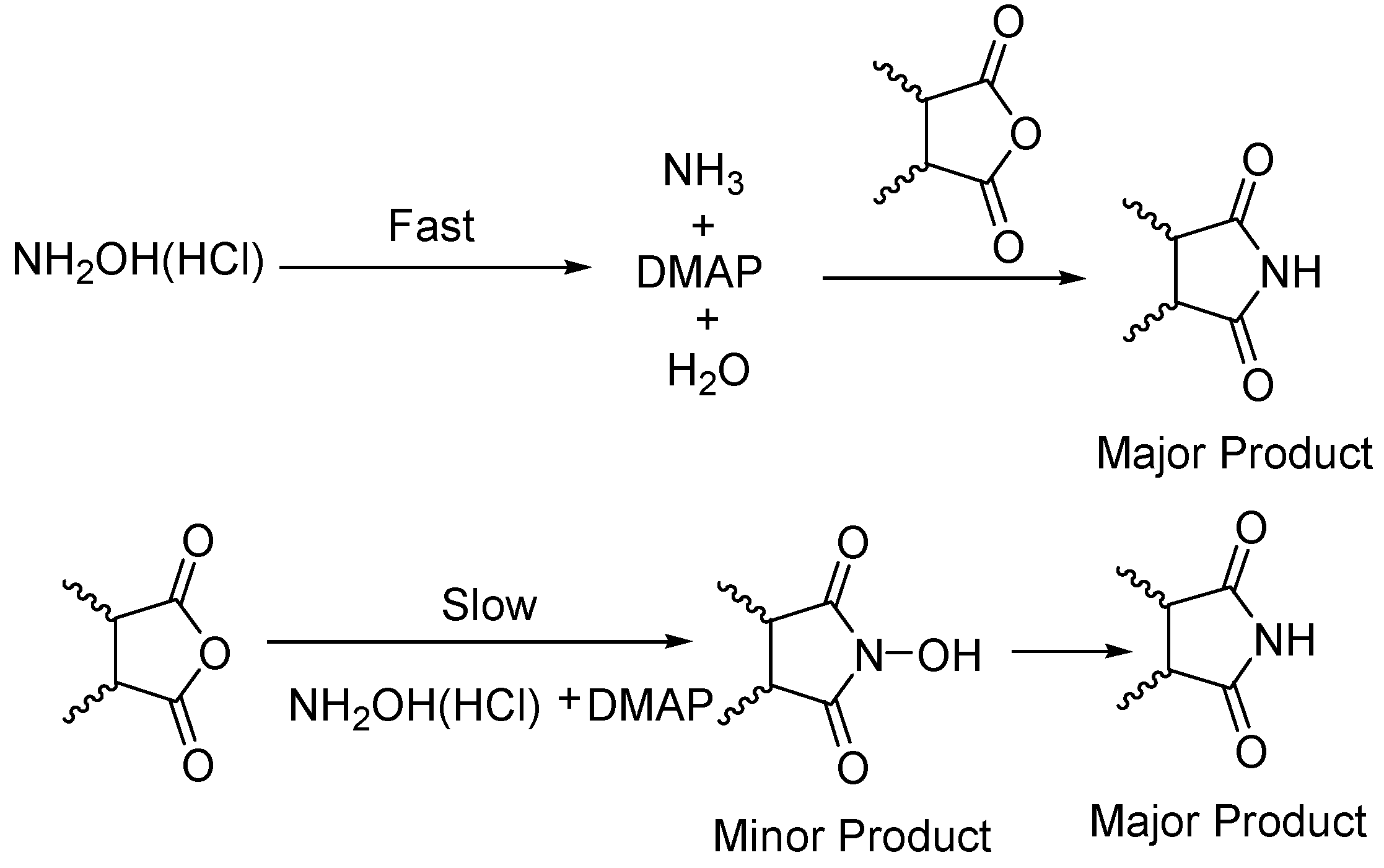
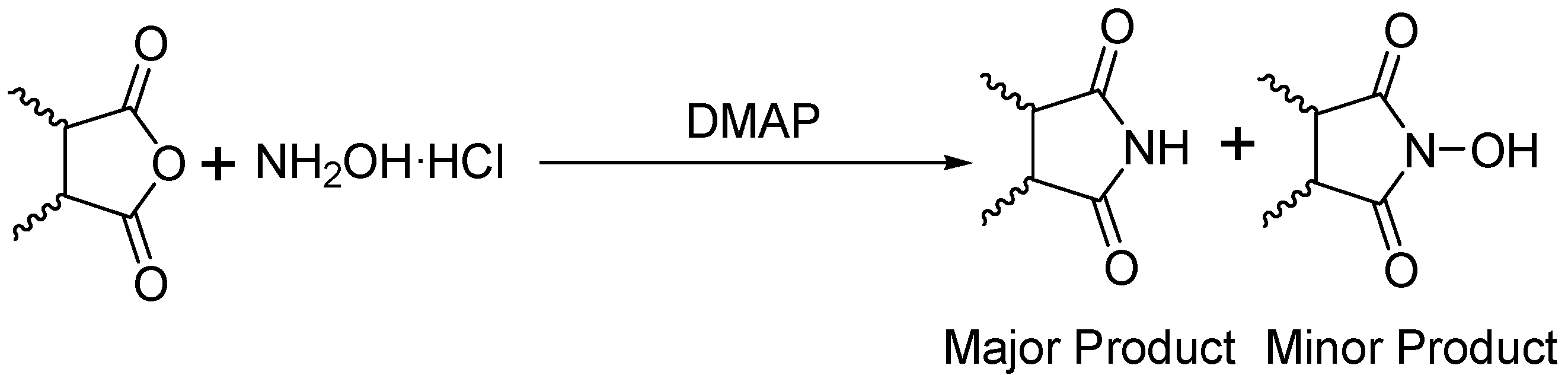
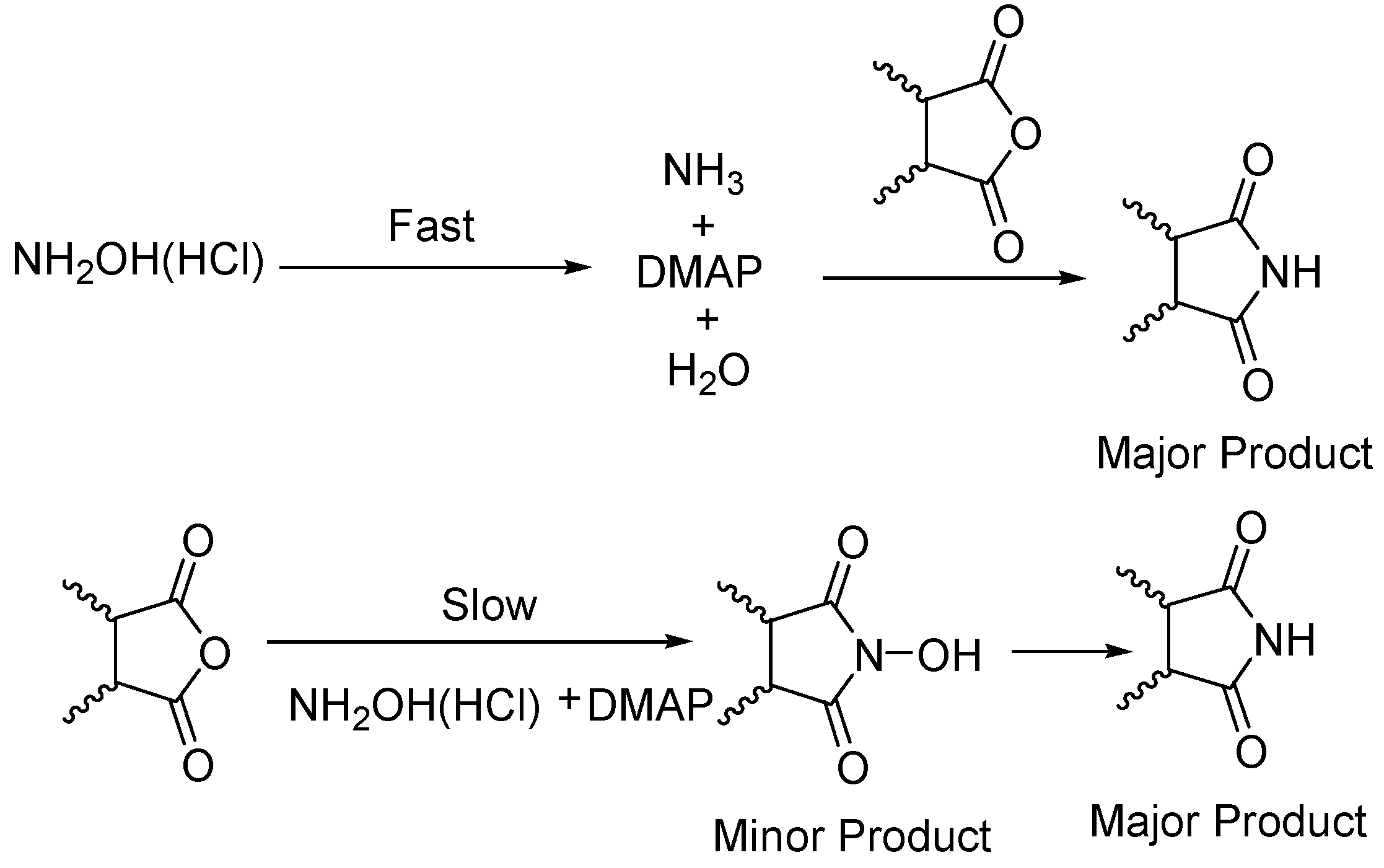
General
The monomode microwave reactions were carried out in a CEM Discover Microwave. Multimode microwave reactions were done in a Kennmore Microwave Oven (Household) Output: 1100 Watts (Frequency: 2450 MHz). All Gas Chromatograph Mass Spectrometry (GC-MS) was performed using a Shimadzu GC-17A and GCMS-QP5050A Labsolutions system. All reagents were purchased from Aldrich Chemical Company and were used without further purification. Since two different methods are described in this article
Method 1 will refer to synthesis carried out under monomode conditions, while,
Method 2 will refer to synthesis under multimode conditions. Only the GCMS data is given, however unsubstituted cyclic imides data matched that reported earlier [
15].
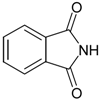
Phthalimide (1): Method 1. Phthalic anhydride (0.20 g, 1.35 mmol), NH2OH·HCl (0.09 g, 1.30 mmol), and DMAP (0.04 g, 0.33 mmol) were thoroughly mixed in a CEM vial with a stirrer. This was capped and heated in a CEM Discover microwave for 5 minutes at 150 oC. This was rapidly cooled to room temperature yielding a dark brown solid. The reaction mixture was dissolved in AcOEt (4 mL) and was washed with distilled water (2 x 2 mL). The organic layer was concentrated to obtain a white solid (0.14 g, 70%); MS m/z 147 (M+) 104, 76, 50.
Phthalimide (1): Method 2. Phthalic anhydride (1.0 g, 6.75 mmol), NH2OH·HCl (0.54 g, 7.7 mmol), and DMAP (0.08 g, 0.65 mmol) were mixed in an 8 mL Teflon capped vial. The mixture was allowed to heat for 4 minutes and 11 seconds at 30 percent power in the multimode microwave and then cooled to room temperature. The sample was dissolved in acetone and flash chromatographed using silica (~30 g) with pure acetone as the mobile phase to obtain a yellow solid. Yield: 0.96 g (97%); MS m/z 147 (M+) 104, 76, 50.
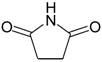
Succinimide (2): Method 1. Succinic anhydride (0.20 g, 2.00 mmol), NH2OH·HCl (0.14 g, 2.0 mmol), and DMAP (0.04 g, 0.33 mmol) were thoroughly mixed in a CEM vial with a stirrer. This was capped and heated in a CEM Discover microwave for 5 minutes at 150 °C. This was rapidly cooled to room temperature yielding a dark brown solid. The reaction mixture was dissolved in AcOEt (4 mL) and was washed with distilled water (2 x 2 mL). The organic layer was concentrated to obtain a white solid (0.14 g, 71%); MS m/z 99 (M+) 56.
Succinimide (2): Method 2. Succinic anhydride (1.0 g, 10 mmol), NH2OH·HCl (0.80 g, 11 mmol), and DMAP (0.12 g, 0.98 mmol) were mixed in an 8 mL Teflon capped vial. The mixture was allowed to heat for 1 minute 49 seconds at full power in the multimode microwave and then cooled to room temperature. The sample was dissolved in acetone and flash chromatographed using silica (~30 g) with pure acetone as the mobile phase to obtain a yellow solid (0.95 g, 96%); MS m/z 99 (M+) 56.
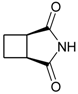
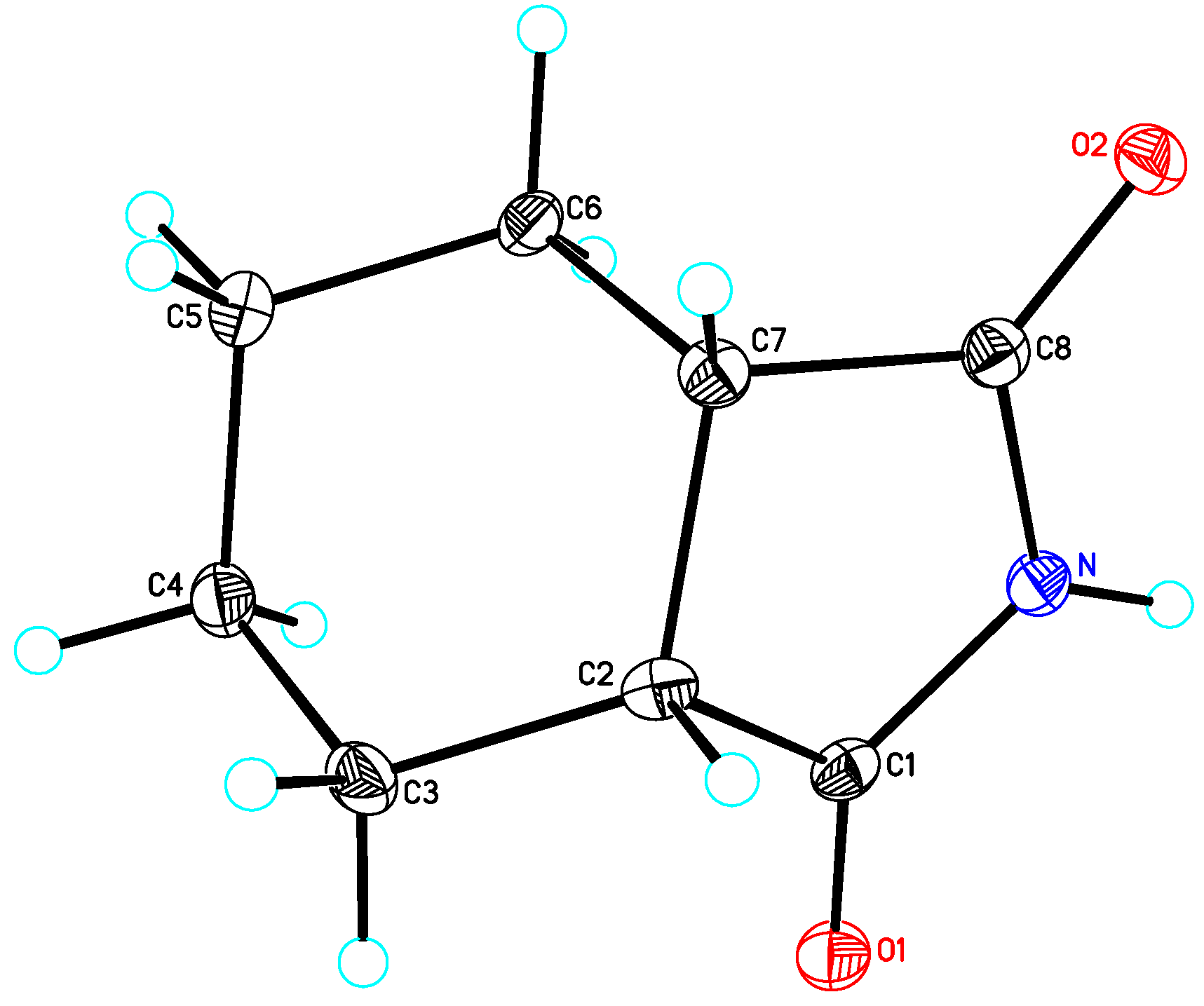
cis-1,2-Cyclobutanedicarboximide (3): Method 1. cis-1,2-Cyclobutanedicarboxylic acid anhydride (0.20 g, 1.59 mmol), NH2OH∙HCl) (0.11 g, 1.58 mmol), and DMAP (0.04 g, 0.33 mmol) were thoroughly mixed in a CEM vial with a stirrer. This was capped and heated in a CEM Discover microwave for 5 minutes at 150 °C. This was rapidly cooled to room temperature yielding a white solid. The reaction mixture was dissolved in AcOEt (4 mL) and was washed with distilled water (2 x 2 mL). The organic layer was concentrated to obtain a white solid (0.12 g, 61%); MS m/z 125 (M+) 82, 54.
cis-1,2-Cyclobutanedicarboximide (3): Method 2. cis-1,2-Cyclobutanedicarboxylic acid anhydride (1.0 g, 7.9 mmol), NH2OH∙HCl (0.63 g, 9.1 mmol), and DMAP (0.10 g, 0.82 mmol) were mixed in an 8 mL Teflon capped vial. The mixture was allowed to heat for 1 minute 12 seconds at full power in the multimode microwave and then cooled to room temperature. The sample was dissolved in acetone and flash chromatographed using silica (~30 g) with pure acetone as the mobile phase to obtain a light brown solid, (0.95 g, 96%); MS m/z 125 (M+) 82, 54.
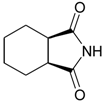
3a,4,5,6,7,7a-Hexahydro-1H-isoindole-1,3(2H)-dione (4): Method 1. cis-1,2-Cyclohexanedicarbo-xylic acid anhydride (0.20 g, 1.30 mmol), NH2OH∙HCl (0.09 g, 1.29 mmol), and DMAP (0.04 g, 0.33 mmol) were thoroughly mixed in a CEM vial with a stirrer. This was capped and heated in a CEM Discover microwave for 5 minutes at 150 °C. This was rapidly cooled to room temperature yielding a white solid. The reaction mixture was dissolved in AcOEt (4 mL) and was washed with distilled water (2 x 2 mL). The organic layer was concentrated to obtain a white solid (0.12 g, 61%); MS m/z 153 (M+) 125, 99, 82, 67, 54, 41.
3a,4,5,6,7,7a-Hexahydro-1H-isoindole-1,3(2H)-dione (4): Method 2. cis-1,2-Cyclohexanedicarboxylic acid anhydride (1.0 g, 6.5 mmol), NH2OH∙HCl (0.51 g, 7.3 mmol), and DMAP (0.08 g, 0.65 mmol) were mixed in an 8 mL Teflon capped vial. The mixture was allowed to heat for 1 minute 49 seconds at full power in the multimode microwave and then cooled to room temperature. The sample was dissolved in acetone and flash chromatographed using silica (~30 g) with pure acetone as the mobile phase to obtain a white solid (0.83 g, 84%); MS m/z 153 (M+) 125, 99, 82, 67, 54, 41.
Glutarimide (5): Method 1. Glutaric anhydride (0.20 g, 1.75 mmol), NH2OH∙HCl (0.12 g, 1.73 mmol), and DMAP (0.04 g, 0.33 mmol) were thoroughly mixed in a CEM vial with a stirrer. This was capped and heated in a CEM Discover microwave for 5 minutes at 150 °C. This was rapidly cooled to room temperature yielding a dark brown solid. The reaction mixture was dissolved in AcOEt (4 mL) and was washed with distilled water (2 x 2 mL). The organic layer was concentrated to obtain a light brown solid (0.12 g, 61%); MS m/z 113 (M+) 70, 42.
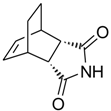
3a,4,7,7a-Tetrahydro-4,7-ethano-1H-isoindole-1,3(2H)-dione (6): Method 1. cis-Bicyclo[2.2.2]oct-5-ene-2,3-dicarboxylic acid anhydride (0.10 g, 0.56 mmol), NH2OH∙HCl (0.05 g, 0.72 mmol) and DMAP (0.02 g, 0.16 mmol) were mixed thoroughly in a CEM-sealed vial with a magnetic stirrer. The mixture was heated for 5 min at 150 °C in a CEM Discover microwave powered at 150 W. The sample was then cooled rapidly to 40 °C. The reaction mixture was dissolved in AcOEt (4 mL) and was washed with distilled water (2 x 2 mL). The organic layer was concentrated to obtain a light brown solid (0.08 g, 81 %); MS m/z 177 (M+) 149, 99, 78, 51.


{kind=link}
{kind=link}
{kind=link}