Ultrasound Assisted Synthesis of 5,9-Dimethylpentadecane and 5,9-Dimethylhexadecane – the Sex Pheromones of Leucoptera coffeella


Abstract
:Introduction
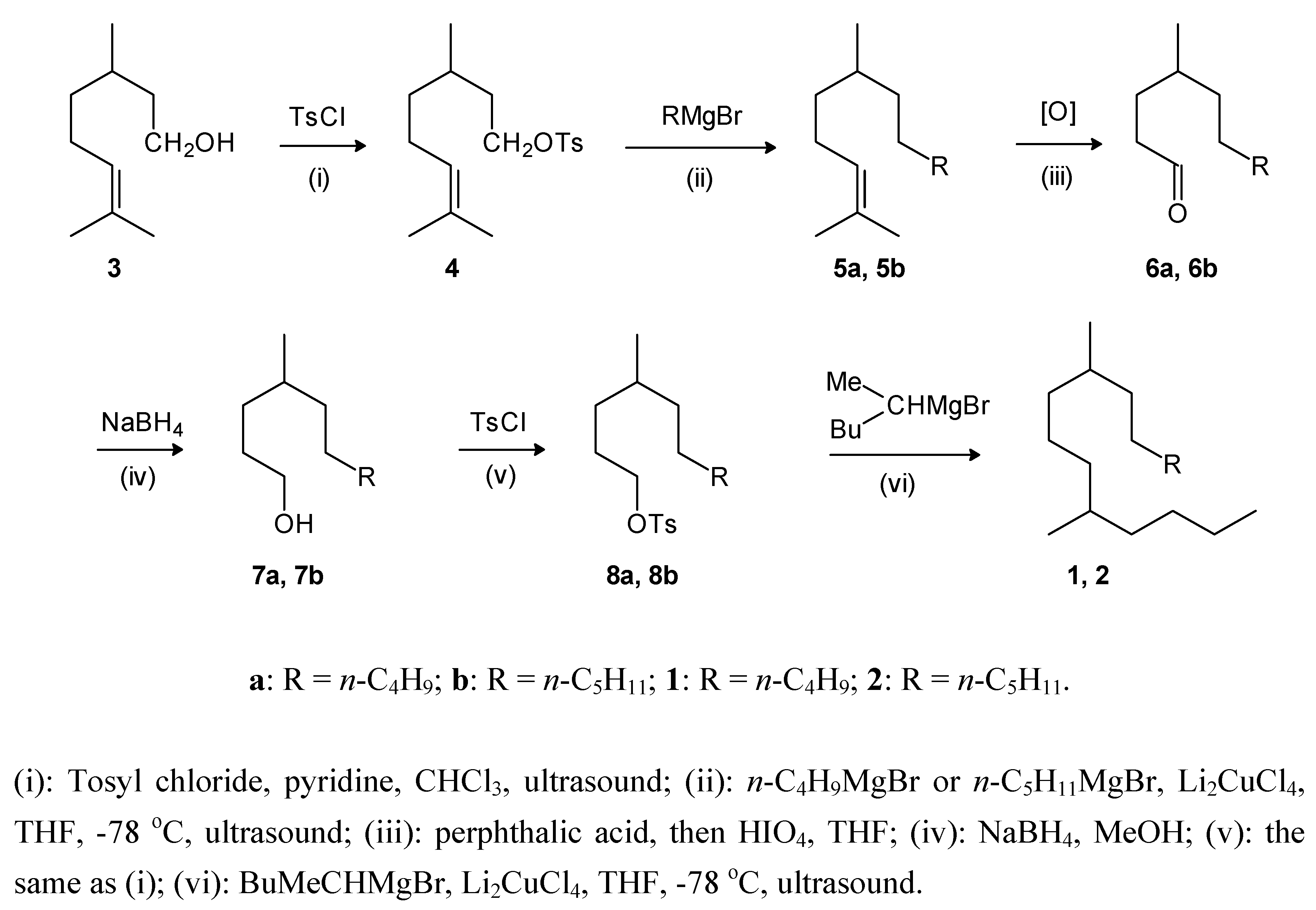
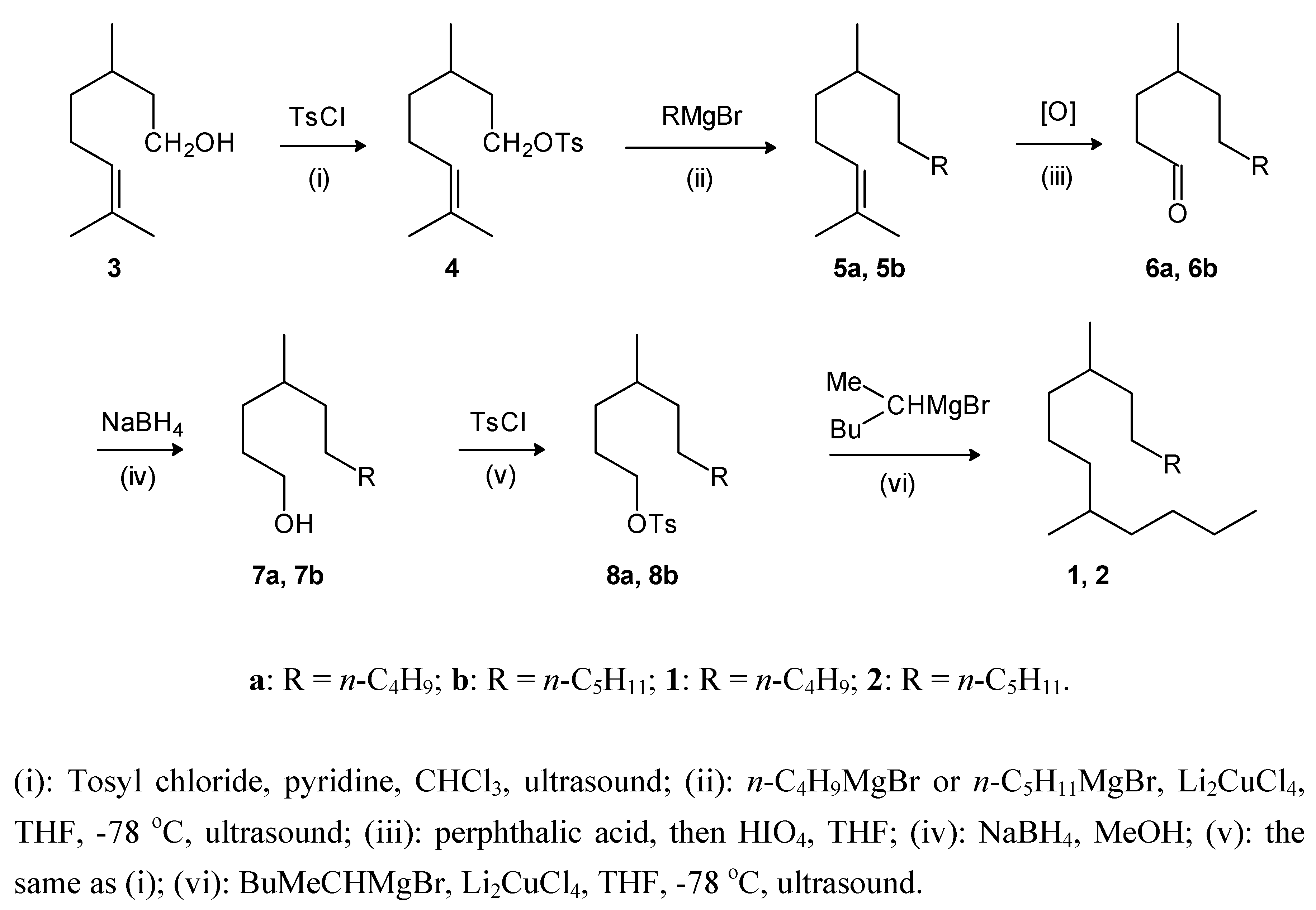
Results and Discussion
Conclusions
Acknowledgement
Experimental
General
General procedure for the tosylation of alcohols: 3,7-dimethyl-6-octenyl 4-methylbenzenesulfonate (4)
General procedure for the cross coupling reactions: 2,6-dimethyl-2-dodecene (5a)
General procedure for oxidation: 4-methyldecanal (6a)
General procedure for reduction: 4-methyl-1-decanol (7a)
References and Notes
- Mori, K. Synthesis of optically active pheromones. Tetrahedron 1989, 45, 3233–3298. [Google Scholar] [CrossRef]Maier, C. T.; Gries, R.; Gries, G. Sex pheromone components of pitch pine looper, Lambdina pellucidaria. J. Chem. Ecol. 1998, 24, 491–500. [Google Scholar] [CrossRef]Kimura, T.; Carlson, D. A.; Mori, K. Synthesis of all the stereoisomers of 13,17-dimethyl-1-tritriacontene and 13,17-dimethyl-1-pentatriacontene, the contact sex pheromone components of the female tsetse fly, Glossina austeni. Eur. J. Org. Chem. 2001, 3385–3390. [Google Scholar] [CrossRef]
- Francke, W.; Toth, M.; Szocs, G.; Krieg, W.; Ernst, H.; Buschmann, E. Identification and synthesis of dimethylalkanes as sex attractants of female leaf miner moths (Lyonetiidae). Z. Naturforsch. 1988, 43C, 787–789. [Google Scholar]
- Moreira, J. A.; Correa, A. G. Enantioselective synthesis of three stereoisomers of 5,9-dimethylpentadecane, sex pheromone component of Leucoptera coffeella, from (-)-isopulegol. Tetrahedron: Asym. 2003, 14, 3787–3795. [Google Scholar] [CrossRef]
- Kuwahara, S.; Liang, T.; Leal, W. S.; Ishikawa, J.; Kodama, O. Synthesis of All Four Possible Stereoisomers of 5,9-Dimethyldecane, the Major Sex Pheromones Component of the Coffee Leaf Minor Moth, Perileucoptera coffeella. Biosci. Biotechnol. Biochem. 2000, 64, 2723–2726. [Google Scholar] [CrossRef]
- Zarbin, P. H. G.; Princival, J. L.; Lima, E. R.; Santos, A. A.; Ambrogio, B. G.; Oliveira, A. R. M. Unsymmetrical double Wittig olefination on the syntheses of insect pheromones. Part 1: Synthesis of 5,9-dimethylpentadecane, the sexual pheromone of Leucoptera coffeella. Tetrahedron Lett. 2004, 45, 239–241. [Google Scholar]
- Liang, T.; Kuwahara, S.; Hasegawa, M.; Kodama, O. Simple Synthesis of 5,9-Dimethylated Long-Chain Alkanes, the Sex Pheromones of Leaf Minor Moths. Biosci. Biotechnol. Biochem. 2000, 64, 2474–2477. [Google Scholar] [CrossRef] [PubMed]
- Poppe, L.; Novák, L.; Dévényi, J.; Szántay, Cs. Baker's yeast mediated synthesis of (5SR,9S)-5,9-dimethylheptadecane and (5SR,9S)-5,9-dimethylpentadecane; the main sex-pheromone components of Leocoptera scitella and Perileucoptera coffeella enriched in 9S-isomers. Terahedron Lett. 1991, 32, 2643–2646. [Google Scholar] [CrossRef]
- Luche, J. L. Synthetic Organic Sonochemistry; Plenum Press: NewYork, 1998. [Google Scholar]
- Bauer, K.; Garbe, D.; Surburg, H. Common Fragrance and Flavor Materials, 2nd ed.; VCH: Weinheim, 1990. [Google Scholar]
- Kabalka, G. W.; Varma, M.; Varma, R. S.; Srivastava, P. C.; Knapp, F. F., Jr. Tosylation of alcohols. J. Org. Chem. 1986, 51, 2386–2388. [Google Scholar] [CrossRef]
- Fouquet, C.; Schlosser, M. Improved carbon-carbon linking by controlled copper catalysis. Angew. Chem. Int. Ed. Engl. 1974, 13, 83–85. [Google Scholar] [CrossRef]
- Zarbin, P. H. G.; Cruz, W. O.; Ferreira, J. T. B. Stereospecific synthesis of two isomers of (4,8) - dimethyldecanal: the aggregation pheromone of Tribolium spp. J. Braz. Chem. Soc. 1998, 9, 511–513. [Google Scholar] [CrossRef]
- Furniss, B. S.; Tatchell, A. R.; Hannaford, A. J.; Smith, P. W. G. Vogel's Textbook of Practical Organic Chemistry, 5th ed.; Longman: London, 1989; pp. 454–457. [Google Scholar]
- Sufficiently pure 2-bromohexane could not be procured commercially, minor contents of the isomeric 3-bromohexane being an apparently unavoidable impurity. Therefore, 2-bromohexane was prepared with the purity of 98% (GC) from 2-hexanol and PBr3 at a temperature below 5 oC ([13]; pp. 559-564).
- The signals are separated from each other by Δδ = 0.002 ppm.
- Resolved resonances for the same carbon atom of two diastereomerically different forms.
- Sample Availability: Available from the authors.

{kind=link}
| Entry | 3 : TsCl : pyridine (molar ratios) | Time (minutes) | Yield of 4 (%) |
|---|---|---|---|
| 1 | 1:1:1 | 30 | 82 |
| 2 | 1:1:1 | 60 | 83 |
| 3 | 1:1:2 | 30 | 86 |
| 4 | 1:1:2 | 60 | 84 |
| 5 | 1:1.5:1 | 60 | 80 |
| 6 | 1:1.5:2 | 30 | 91 |
| 7 | 1:1.5:2 | 20 | 84 |
| 8 | 1:1.5:2 | 40 | 92 |
| 9 | 1:1.5:3 | 30 | 92 |
| 10 | 1:2:2 | 20 | 86 |
| 11 | 1:2:2 | 30 | 92 |
| Entry | Tosylate | Time (minutes) | Yield (%)a |
|---|---|---|---|
| 1 | 8a | 30 | 74 |
| 2 | 8a | 45 | 92 |
| 3 | 8a | 60 | 93 |
| 4 | 8b | 30 | 78 |
| 5 | 8b | 45 | 91 |
| 6 | 8b | 60 | 91 |
| Entry | Product | Time (minutes) | Yield (%)a |
|---|---|---|---|
| 1 | 5a | 20 | 80 |
| 2 | 5a | 30 | 95 |
| 3 | 5a | 45 | 95 |
| 4 | 5b | 30 | 94 |
| 5 | 1 | 45 | 93 |
| 6 | 2 | 45 | 91 |
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Doan, N.N.; Le, T.N.; Nguyen, H.C.; Hansen, P.E.; Duus, F. Ultrasound Assisted Synthesis of 5,9-Dimethylpentadecane and 5,9-Dimethylhexadecane – the Sex Pheromones of Leucoptera coffeella. Molecules 2007, 12, 2080-2088. https://doi.org/10.3390/12082080
Doan NN, Le TN, Nguyen HC, Hansen PE, Duus F. Ultrasound Assisted Synthesis of 5,9-Dimethylpentadecane and 5,9-Dimethylhexadecane – the Sex Pheromones of Leucoptera coffeella. Molecules. 2007; 12(8):2080-2088. https://doi.org/10.3390/12082080
Chicago/Turabian StyleDoan, Nhuan Ngoc, Thach Ngoc Le, Hao Cong Nguyen, Poul Erik Hansen, and Fritz Duus. 2007. "Ultrasound Assisted Synthesis of 5,9-Dimethylpentadecane and 5,9-Dimethylhexadecane – the Sex Pheromones of Leucoptera coffeella" Molecules 12, no. 8: 2080-2088. https://doi.org/10.3390/12082080
APA StyleDoan, N. N., Le, T. N., Nguyen, H. C., Hansen, P. E., & Duus, F. (2007). Ultrasound Assisted Synthesis of 5,9-Dimethylpentadecane and 5,9-Dimethylhexadecane – the Sex Pheromones of Leucoptera coffeella. Molecules, 12(8), 2080-2088. https://doi.org/10.3390/12082080