Synthesis of Novel Steroid-Peptoid Hybrid Macrocycles by Multiple Multicomponent Macrocyclizations Including Bifunctional Building Blocks (MiBs)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
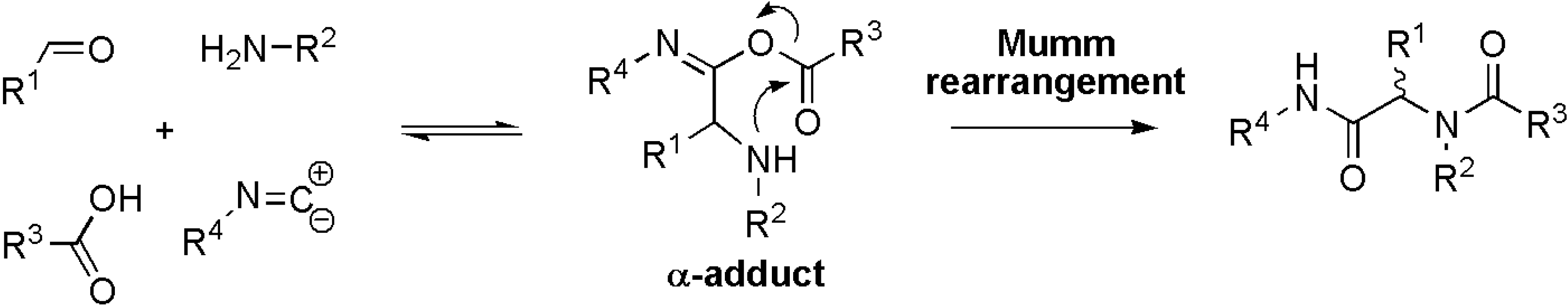
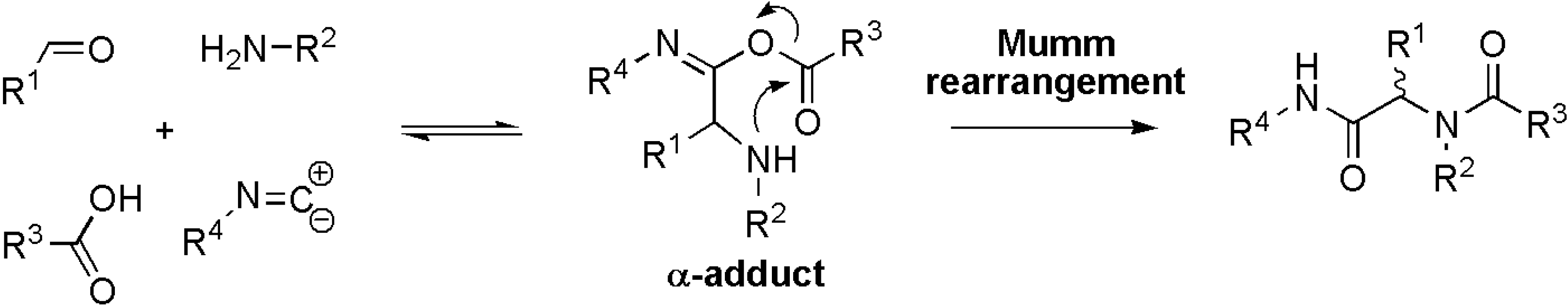
:Introduction
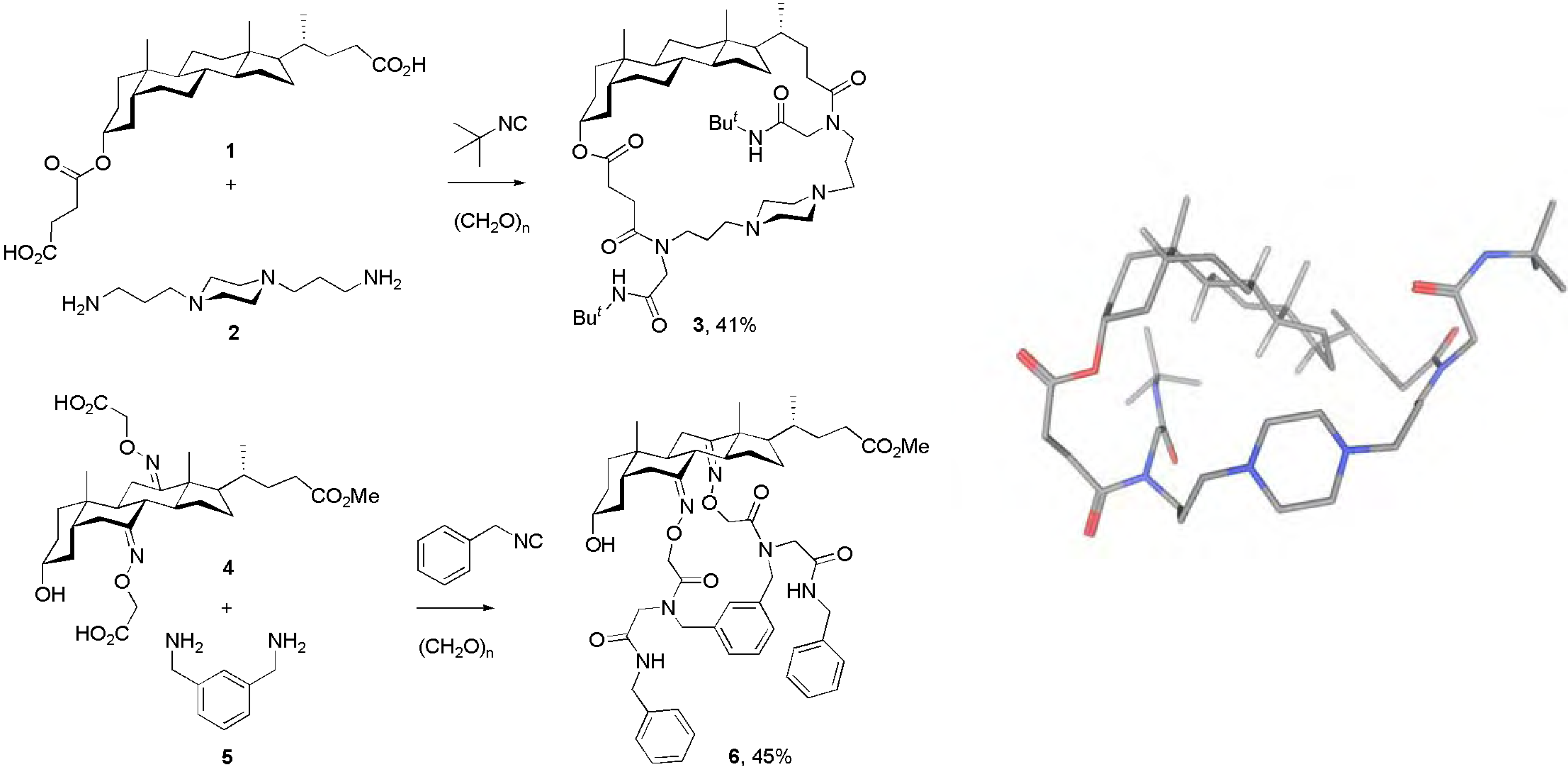
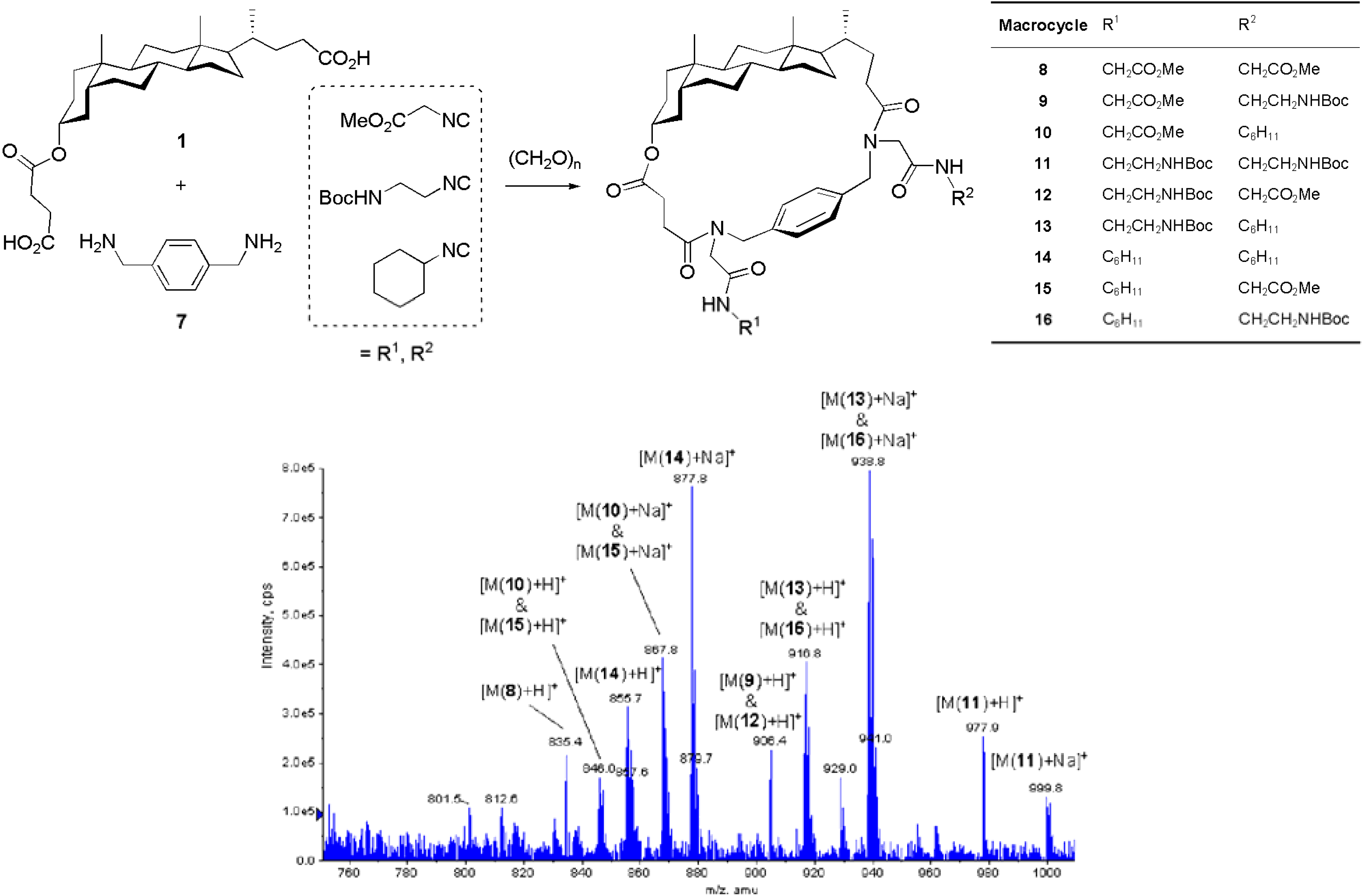
Results and Discussion
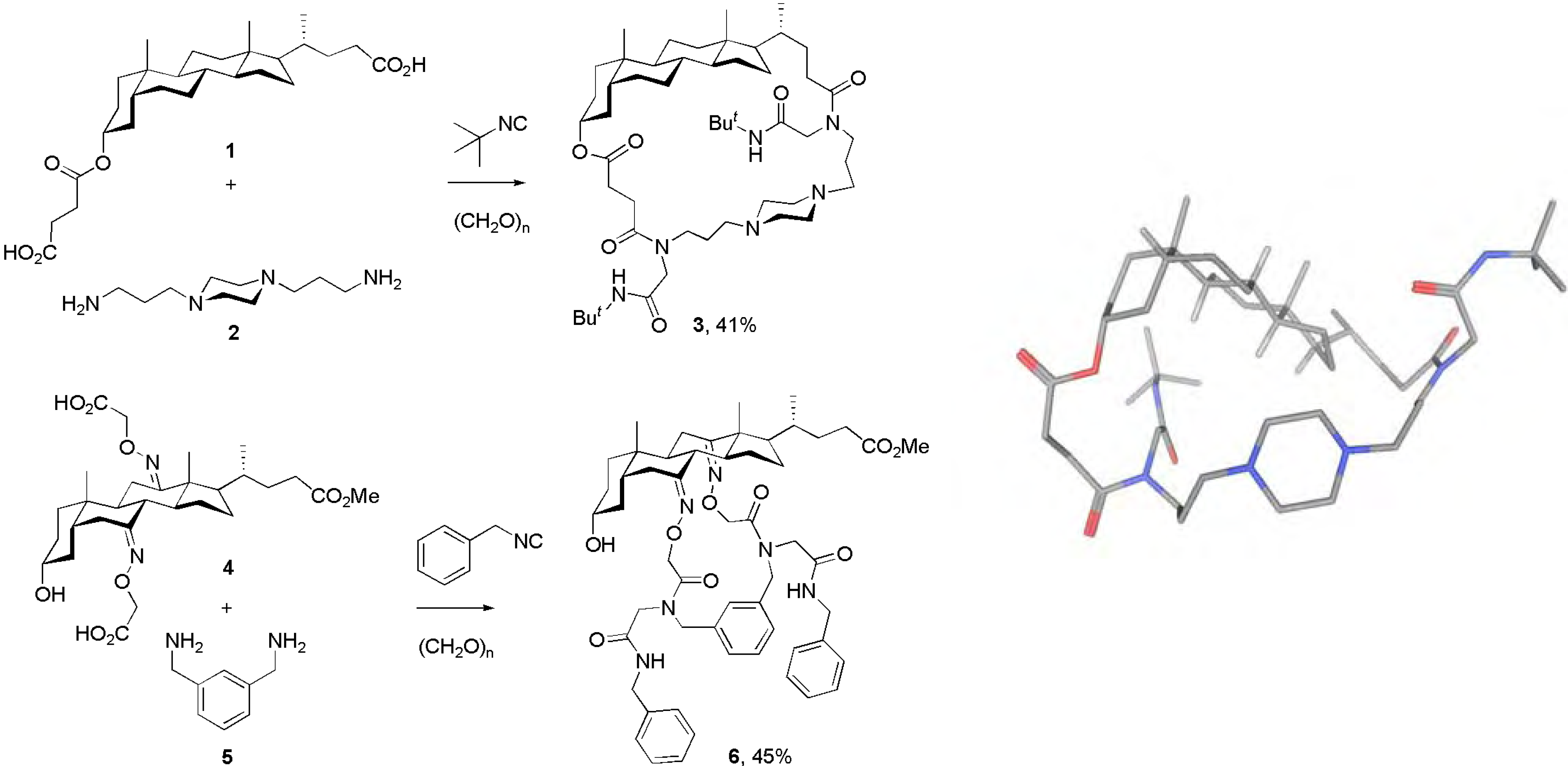
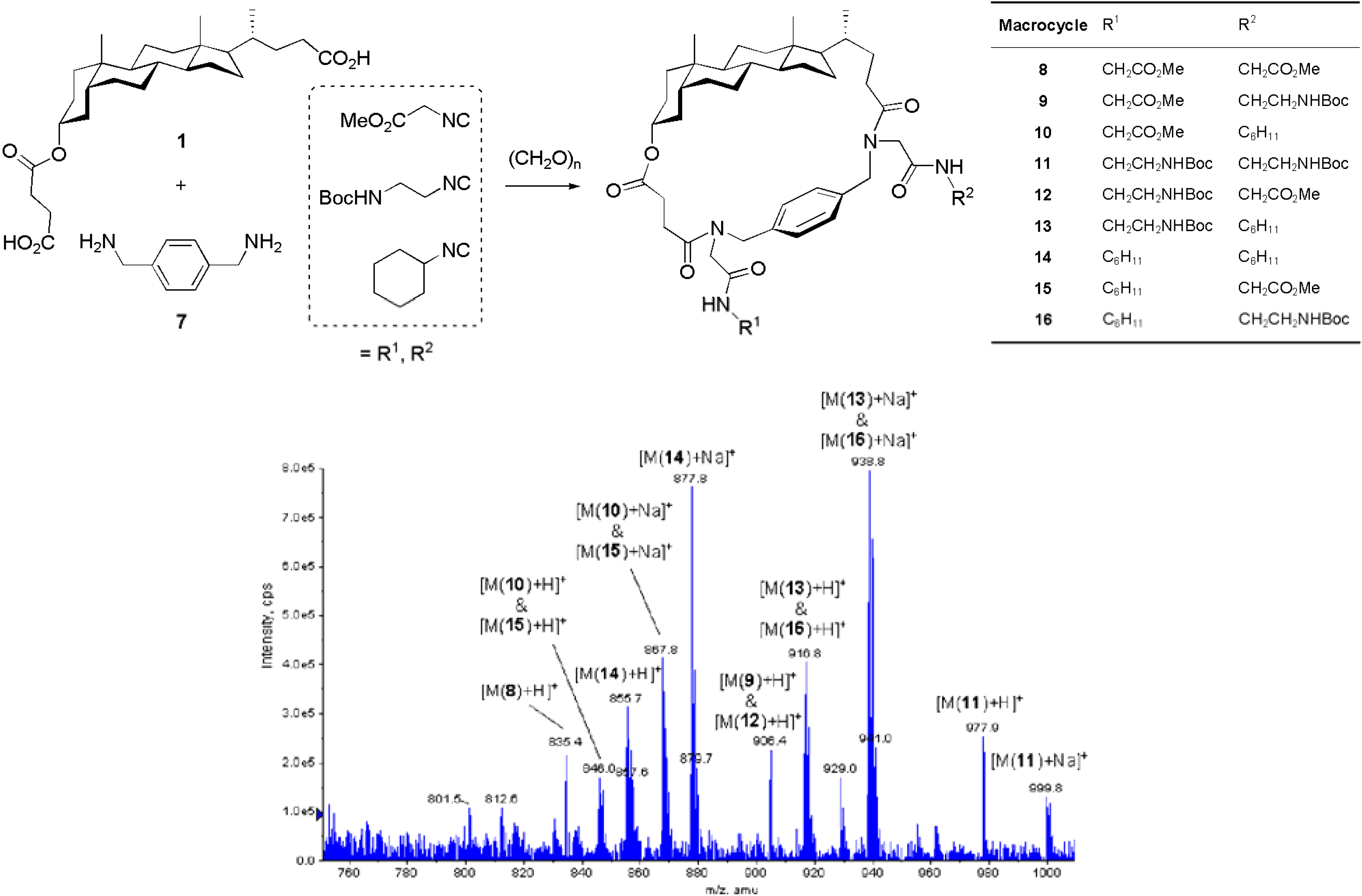
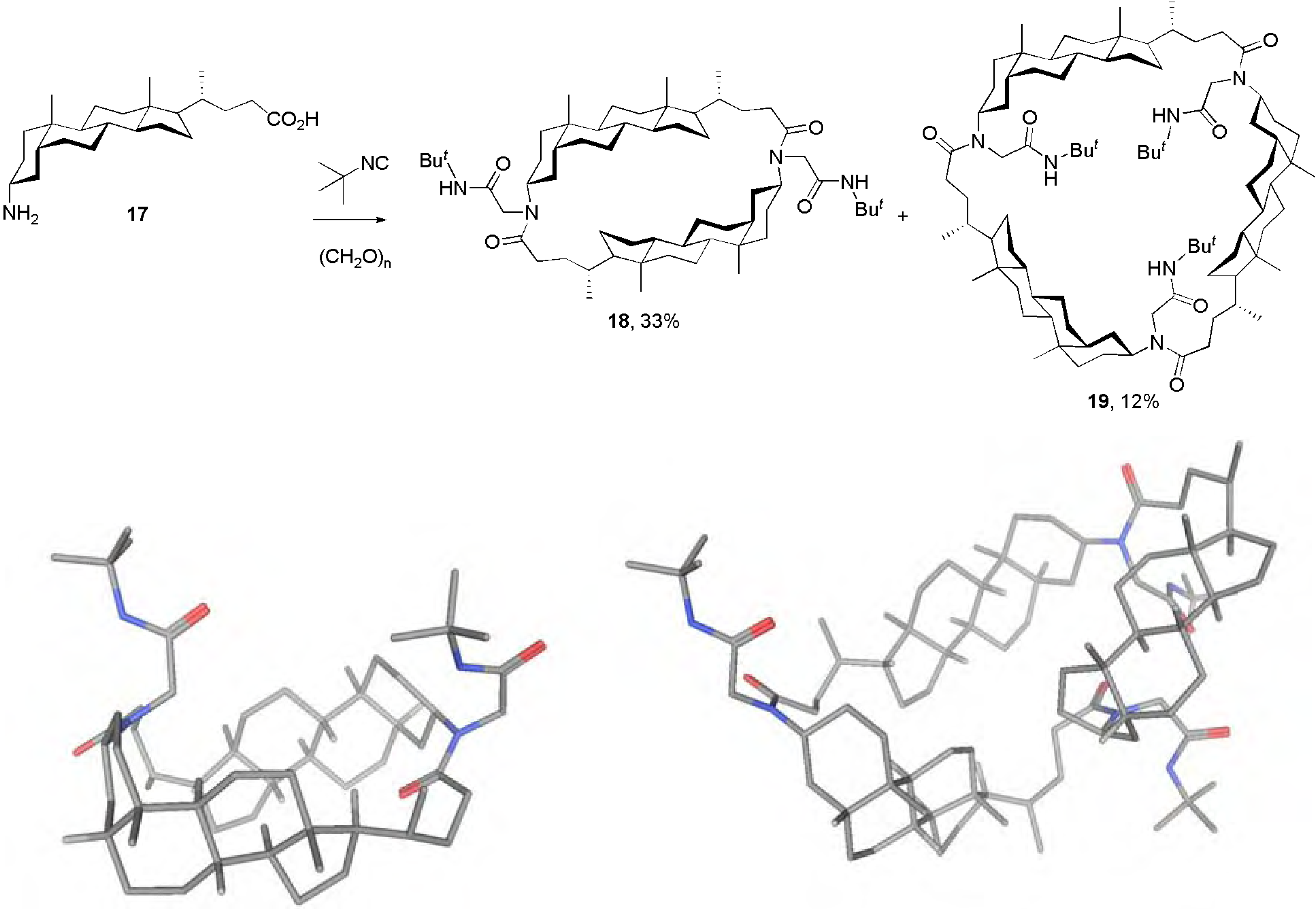
Conclusions
Experimental
General
General procedure for the double Ugi-4CR-based macrocyclizations of the diacid/diamine combination
Combinatorial double Ugi-4CR-based macrocyclization
Procedure for the unidirectional double Ugi-4CR-based macrocyclization
Acknowledgements
References
- Davis, A.P. Cholaphanes et al.; Steroids as Structural Components in Molecular Engineering. Chem. Soc. Rev. 1993, 22, 243–253. [Google Scholar] [CrossRef]
- Walliman, P.; Marti, T.; Fürer, A.; Diederich, F. Steroids in Molecular Recognition. Chem. Rev. 1997, 97, 1567–1608. [Google Scholar] [CrossRef]
- Tamminen, J.; Kolehmainen, E. Bile Acids as Building Blocks of Supramolecular Hosts. Molecules 2001, 6, 21–46. [Google Scholar] [CrossRef]
- Virtanen, E.; Kolehmainen, E. Use of Bile Acids in Pharmacological and Supramolecular Applications. Eur. J. Org. Chem. 2004, 3385–3399. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular Chemistry: Concepts and Perspectives; Wiley-VCH: Weinheim, 1995. [Google Scholar]
- Dias, J.R.; Paskal, R.A.; Morrill, J.; Holder, A.J.; Gao, H.; Barnes, C. Remarkable Structures of Cyclotri(deoxycholate) and Cyclotetra(24-norcholate) Acetate Esters. J. Am. Chem. Soc. 2002, 124, 4647–4652. [Google Scholar] [CrossRef]
- Gao, H.; Dias, J.R. Cyclocholates with 12-Oxo and 7,12-Oxo Groups. Eur. J. Org. Chem. 1998, 719–724. [Google Scholar] [CrossRef]
- Brady, P.A.; Bonar-Law, R.P.; Rowan, S.J.; Suckling, C.J.; Sanders, J.K.M. ‘Living’ macrolactonization: Thermodynamically-controlled cyclization and interconversion of oligocholates. Chem. Commun. 1996, 319–320. [Google Scholar]
- Bonar-Law, R.P.; Sanders, J.K.M. Cyclocholates: Synthesis and Ion Binding. Tetrahedron Lett. 1992, 33, 2071–2074. [Google Scholar] [CrossRef]
- Feigel, M.; Ladberg, R.; Winter, M.; Bläaser, D.; Boese, R. Synthesis and Structure of Macrolactams of 3α-Aminodeoxycholanic Acid. Eur. J. Org. Chem. 2006, 371–377. [Google Scholar]
- Davis, A.P.; Walsh, J.J. Steroid-based receptors with tunable cavities; stepwise and direct synthesis of a C3-symmetrical prototype. Chem. Commun. 1996, 449–451. [Google Scholar] [CrossRef]
- Sisson, A.L.; Clare, J.P.; Davis, A.P. Contra-Hofmeister anion extraction by cyclosteroidal receptors. Chem. Commun. 2005, 5263–5265. [Google Scholar] [CrossRef]
- Albert, D.; Feigel, M.A. Steroidal Cyclopeptide, Synthesis and Shape of the Cavity. Tetrahedron Lett. 1994, 35, 565–568. [Google Scholar] [CrossRef]
- Albert, D.; Feigel, M. β-Loop, γ-Loop, and Helical Peptide Conformations in Cyclopeptides Containing a Steroidal Pseudo-Amino Acid. Helv. Chim. Acta 1997, 80, 2168–2181. [Google Scholar] [CrossRef]
- Maitra, U.; Bag, B.G. Synthesis and Cation Binding Properties of a Novel “Chola-Crown”. J. Org. Chem. 1994, 59, 6114–6115. [Google Scholar] [CrossRef]
- Voigt, B.; Rivera, D.G. Diversity Oriented One-pot Synthesis of Complex Macrocycles: Very Large Steroid-Peptoid Hybrids via Multiple Multicomponent Reactions Including Bifunctional Building Blocks (MiBs). Angew. Chem. Int. Ed. 2005, 44, 4785–4790. [Google Scholar] [CrossRef]
- Rivera, D.G.; Wessjohann, L.A. Supramolecular Compounds by Multiple Multicomponent Macrocyclizations: Peptoid-based Cryptands, Cages and Cryptophanes. J. Am. Chem. Soc. 2006, 128, 7122–7123. [Google Scholar] [CrossRef]
- Wessjohann, L.A.; Ruijter, E. Macrocycles rapidly produced by multiple multicomponent reactions including bifunctional building blocks (MiBs). Mol. Diversity 2005, 9, 159–169. [Google Scholar] [CrossRef]
- Dömling, A.; Ugi, I. Multicomponent Reactions with Isocyanides. Angew. Chem. Int. Ed. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Wessjohann, L.A.; Rivera, D.G.; Coll, F. Steroid-Biarylether Hybrid Macrocycles with High Skeletal and Side Chain Variability by Multiple Multicomponent Macrocyclization including Bifunctional Building Blocks (MiBs). J. Org. Chem. 2006, 71, 7521–7526. [Google Scholar] [CrossRef]
- Rivera, D.G.; Vercillo, O.E.; Wessjohann, L.A. Combinatorial Synthesis of Macrocycles by Multiple Multicomponent Macrocyclization including Bifunctional Building Blocks (MiB). Synlett 2007, 308–312. [Google Scholar]
- Bon, R.S.; van Vliet, B.; Sprenkels, N.E.; Schmitz, R.F.; de Kanter, F.J.J.; Stevens, C.; Swart, M.; Bickelhaupt, F.M.; Groen, M.B.; Orru, R.V.A. Multicomponent Synthesis of 2-Imidazolines. J. Org. Chem. 2005, 70, 3542–3553. [Google Scholar] [CrossRef]
- Failli, A.; Immer, H.; Götz, M. The synthesis of cyclic peptides by the four component condensation (4CC). Can. J. Chem. 1979, 57, 3257–3261. [Google Scholar] [CrossRef]
- Davis, A.P.; Dresen, S.; Lawless, L.J. Mitsunobu Reaction with Methanesulfoic Acid; The Replacement of Equatorial Hydroxyl Groups by Azide with Net Retention of Configuration. Tetrahedron Lett. 1997, 38, 4305–4308. [Google Scholar] [CrossRef]
- Davis, A.P.; Wareham, R.S. Carbohydrate Recognition through Noncovalent Interactions: A Challenge for Biomimetic and Supramolecular Chemistry. Angew. Chem. Int. Ed. 1999, 38, 2978–2996. [Google Scholar] [CrossRef]
- Batta, A.K.; Aggarwal, S.K.; Salen, G.; Shefer, S. Selective reduction of oxo bile acids: 3β-, 7β-, and 12β-hydroxy bile acids. J. Lipid Res. 1991, 32, 977–983. [Google Scholar]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] Halgren, T.A. MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J. Comput. Chem. 1999, 20, 730–748. [Google Scholar]
- Sample Availability: Contact the author.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Rivera, D.G.; Wessjohann, L.A. Synthesis of Novel Steroid-Peptoid Hybrid Macrocycles by Multiple Multicomponent Macrocyclizations Including Bifunctional Building Blocks (MiBs). Molecules 2007, 12, 1890-1899. https://doi.org/10.3390/12081890
Rivera DG, Wessjohann LA. Synthesis of Novel Steroid-Peptoid Hybrid Macrocycles by Multiple Multicomponent Macrocyclizations Including Bifunctional Building Blocks (MiBs). Molecules. 2007; 12(8):1890-1899. https://doi.org/10.3390/12081890
Chicago/Turabian StyleRivera, Daniel G., and Ludger A. Wessjohann. 2007. "Synthesis of Novel Steroid-Peptoid Hybrid Macrocycles by Multiple Multicomponent Macrocyclizations Including Bifunctional Building Blocks (MiBs)" Molecules 12, no. 8: 1890-1899. https://doi.org/10.3390/12081890
APA StyleRivera, D. G., & Wessjohann, L. A. (2007). Synthesis of Novel Steroid-Peptoid Hybrid Macrocycles by Multiple Multicomponent Macrocyclizations Including Bifunctional Building Blocks (MiBs). Molecules, 12(8), 1890-1899. https://doi.org/10.3390/12081890