(-)-Epicatechin-3-gallate, a Green Tea Polyphenol Is a Potent Agent Against UVB-induced Damage in HaCaT Keratinocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
Results and Discussion
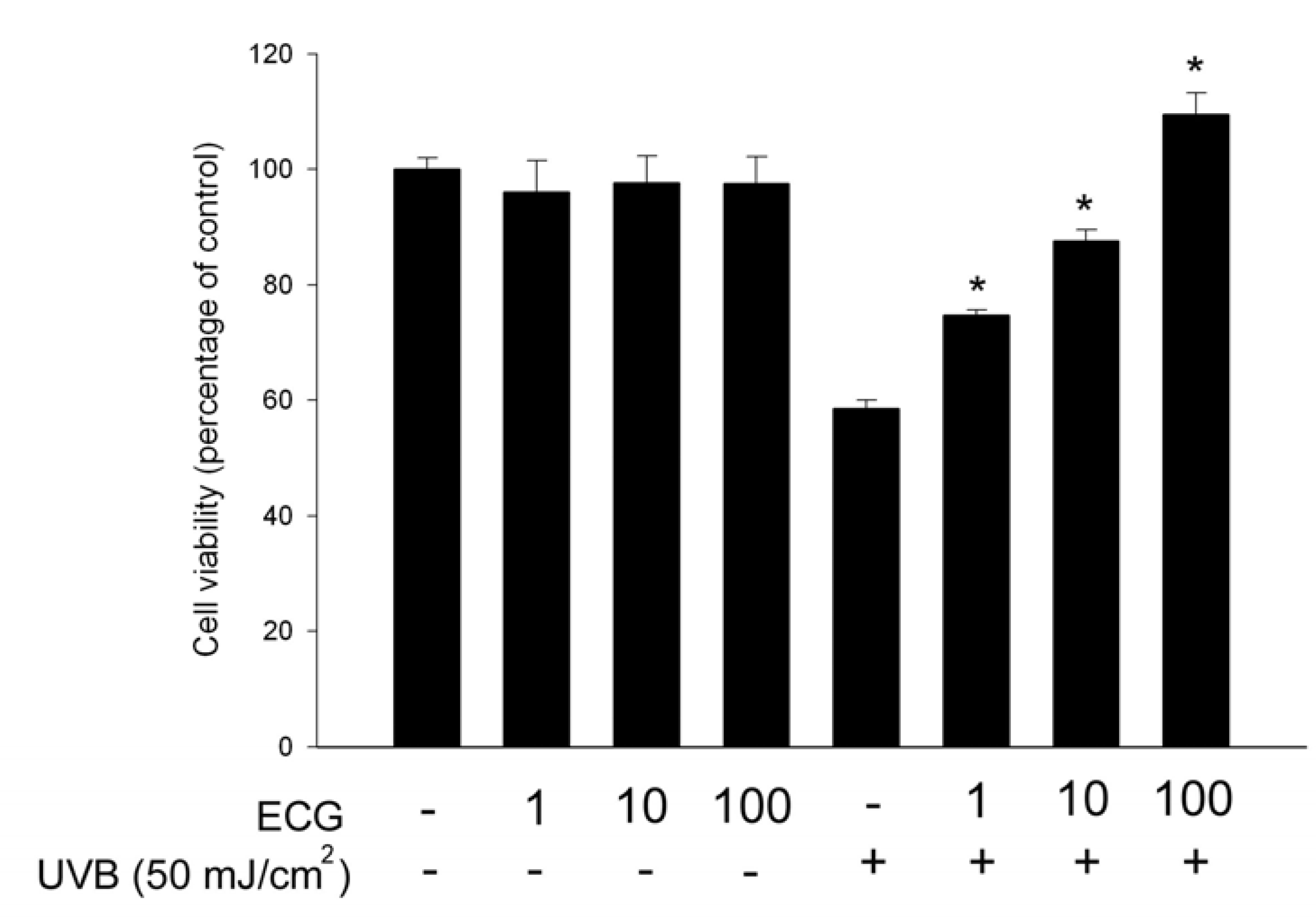
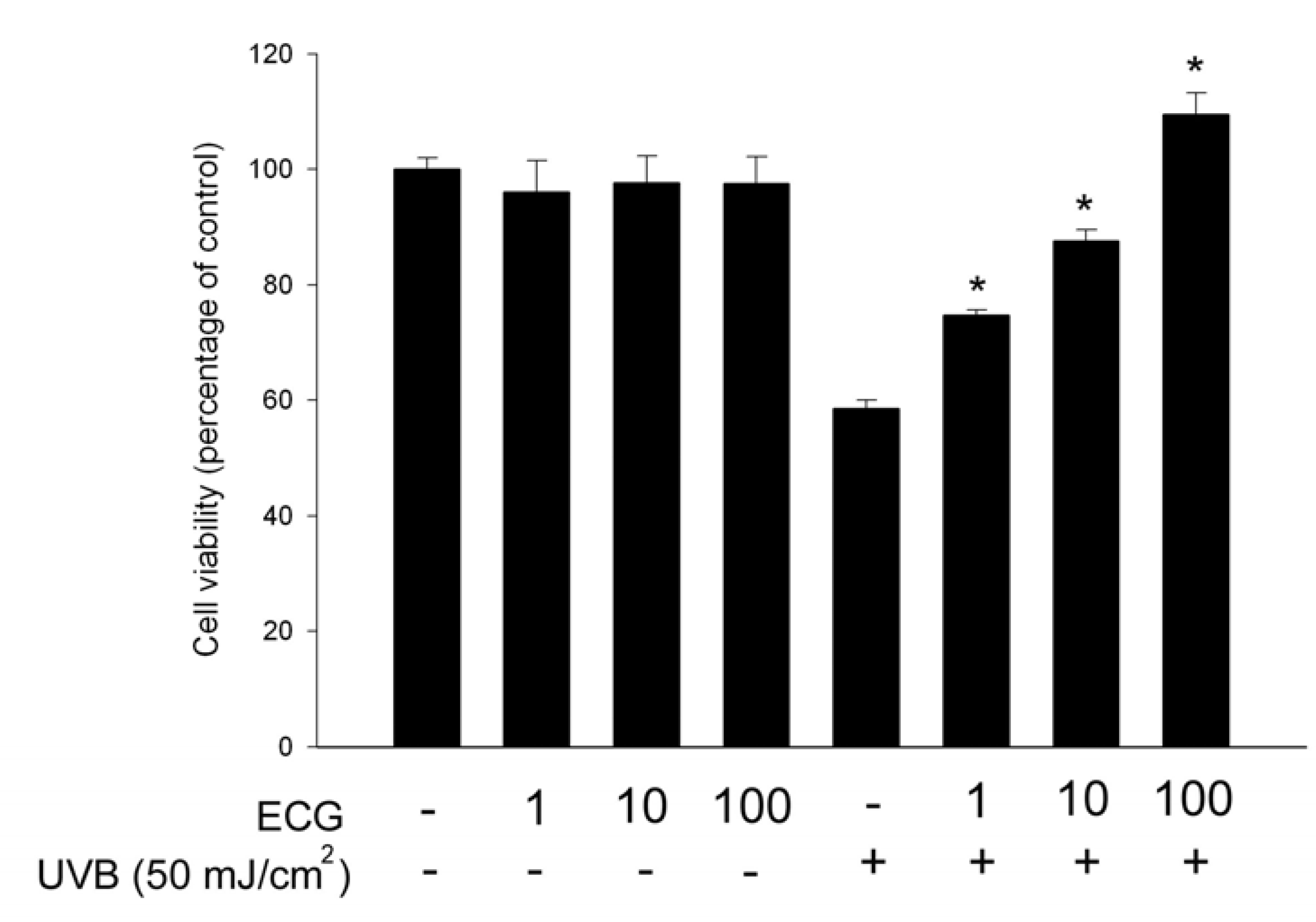
ECG inhibits UVB-induced keratinocyte death
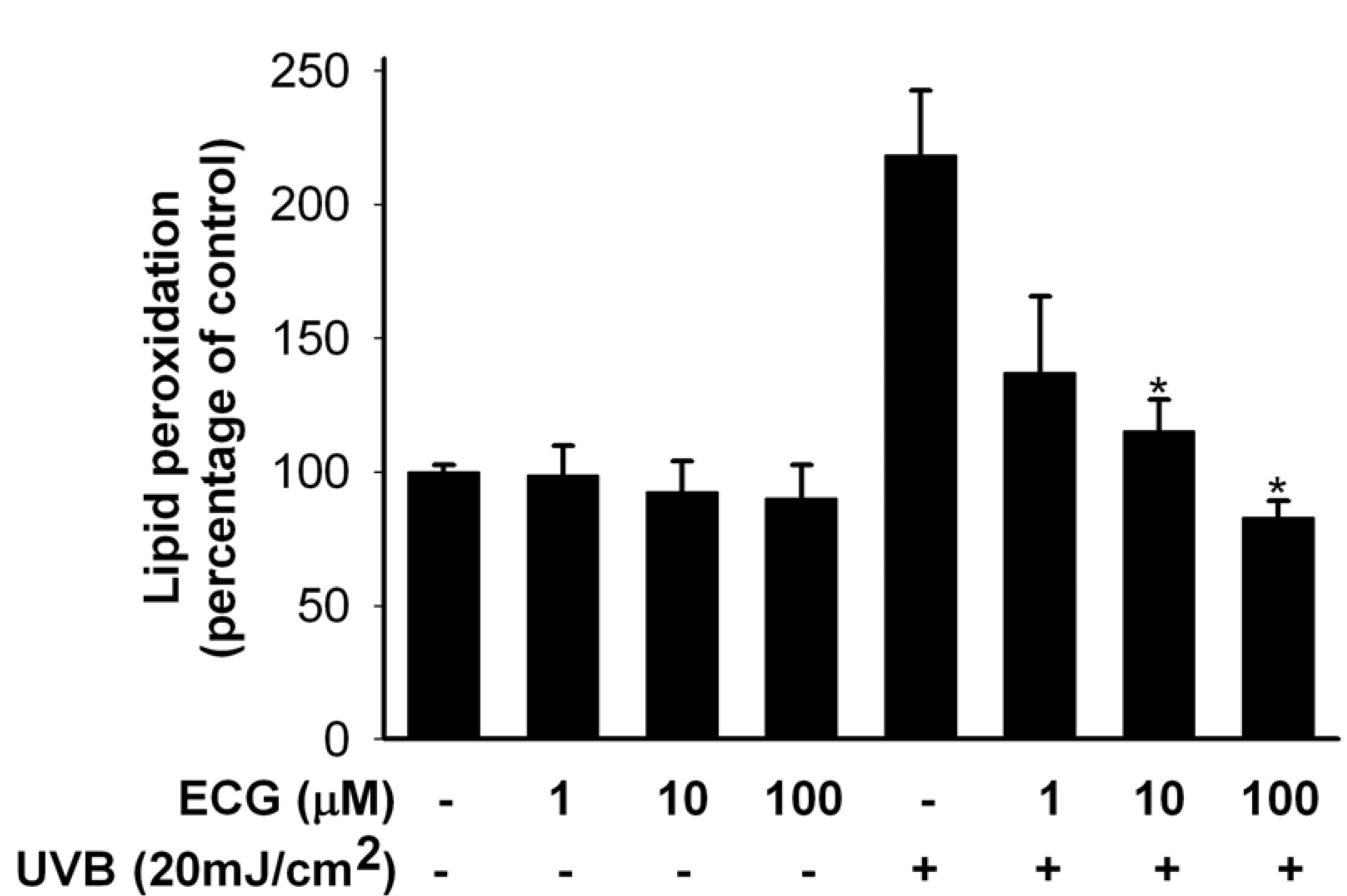
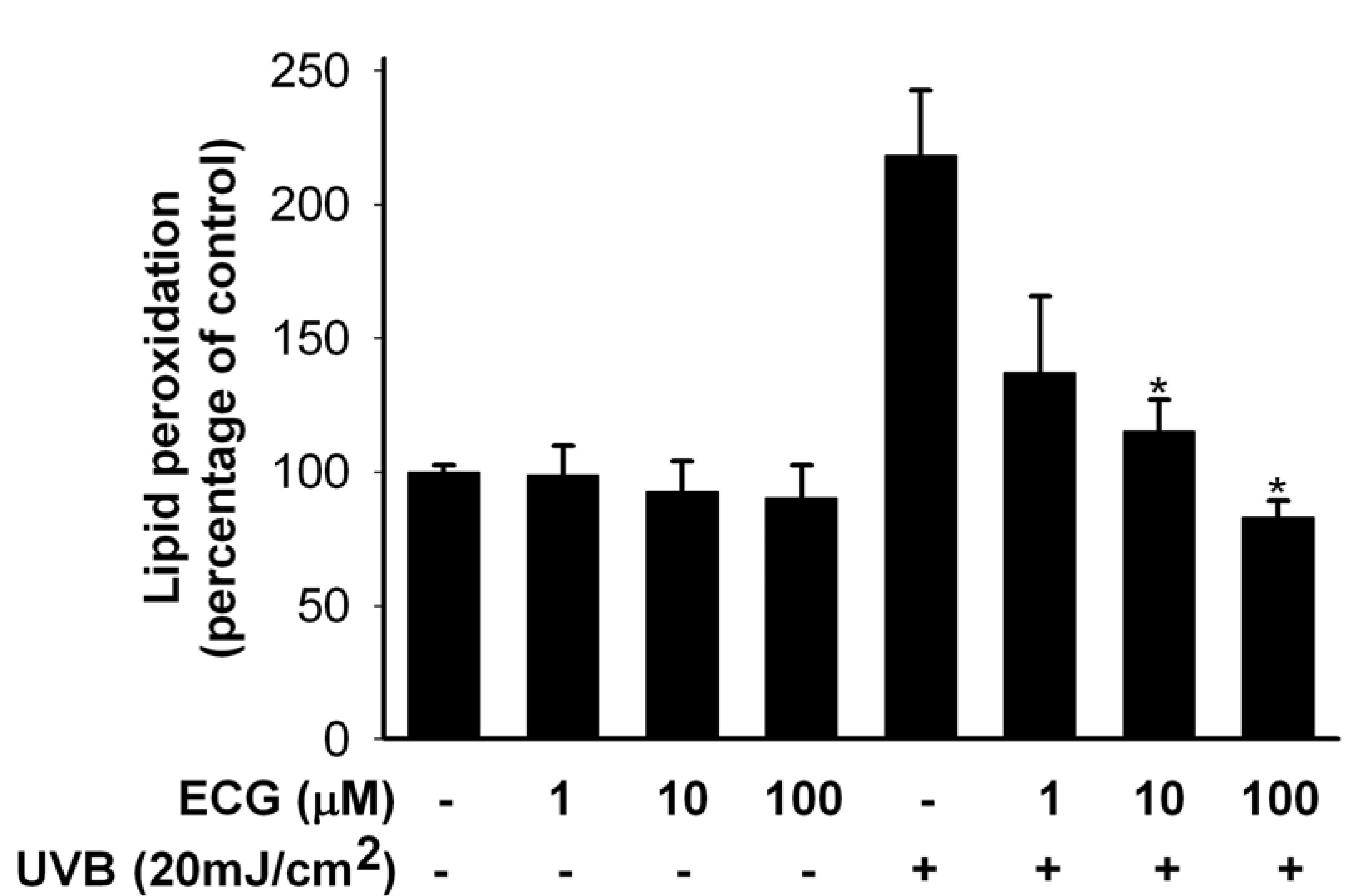
ECG inhibits lipid peroxidation (LPO) by UVB irradiation
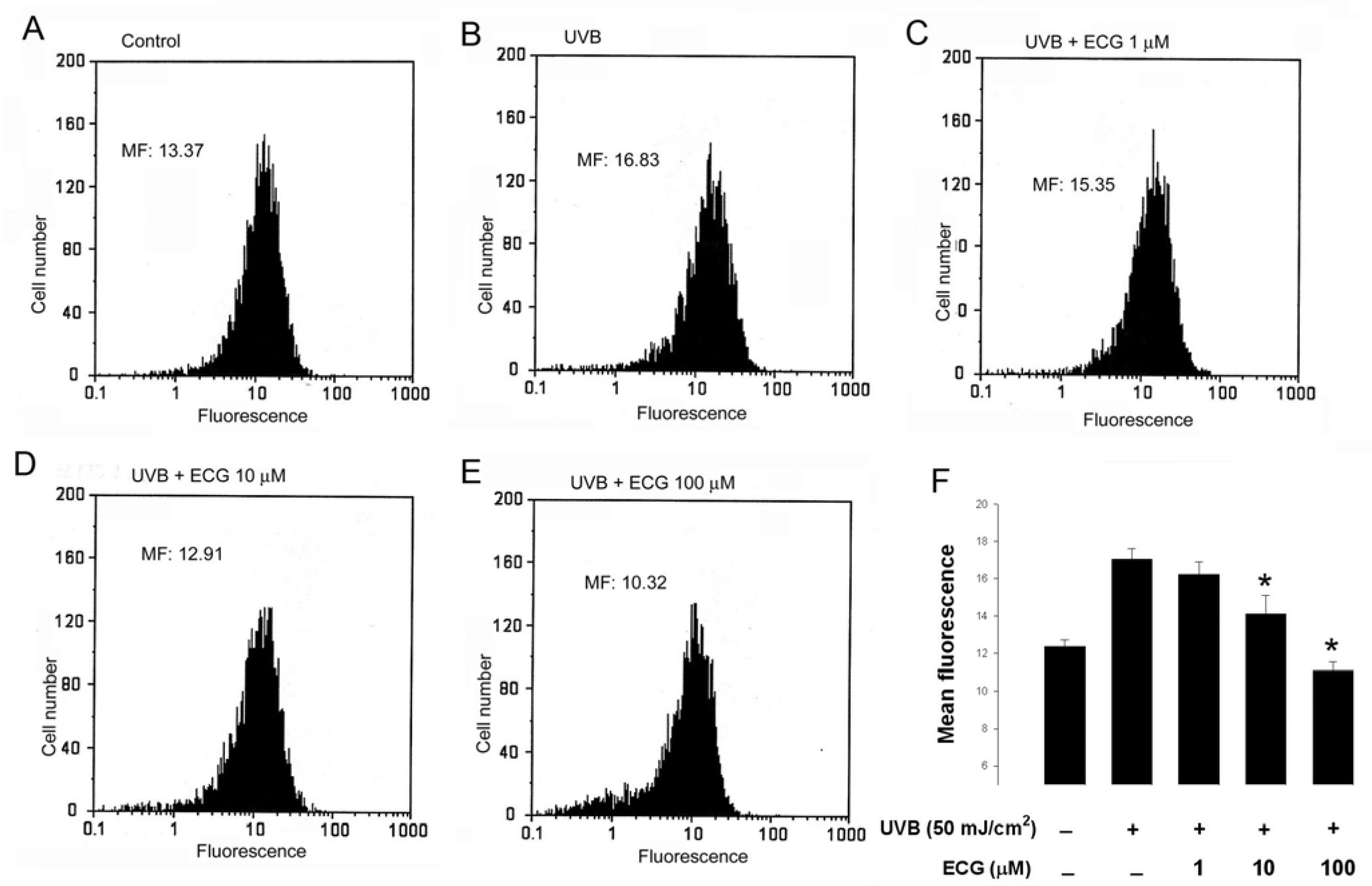
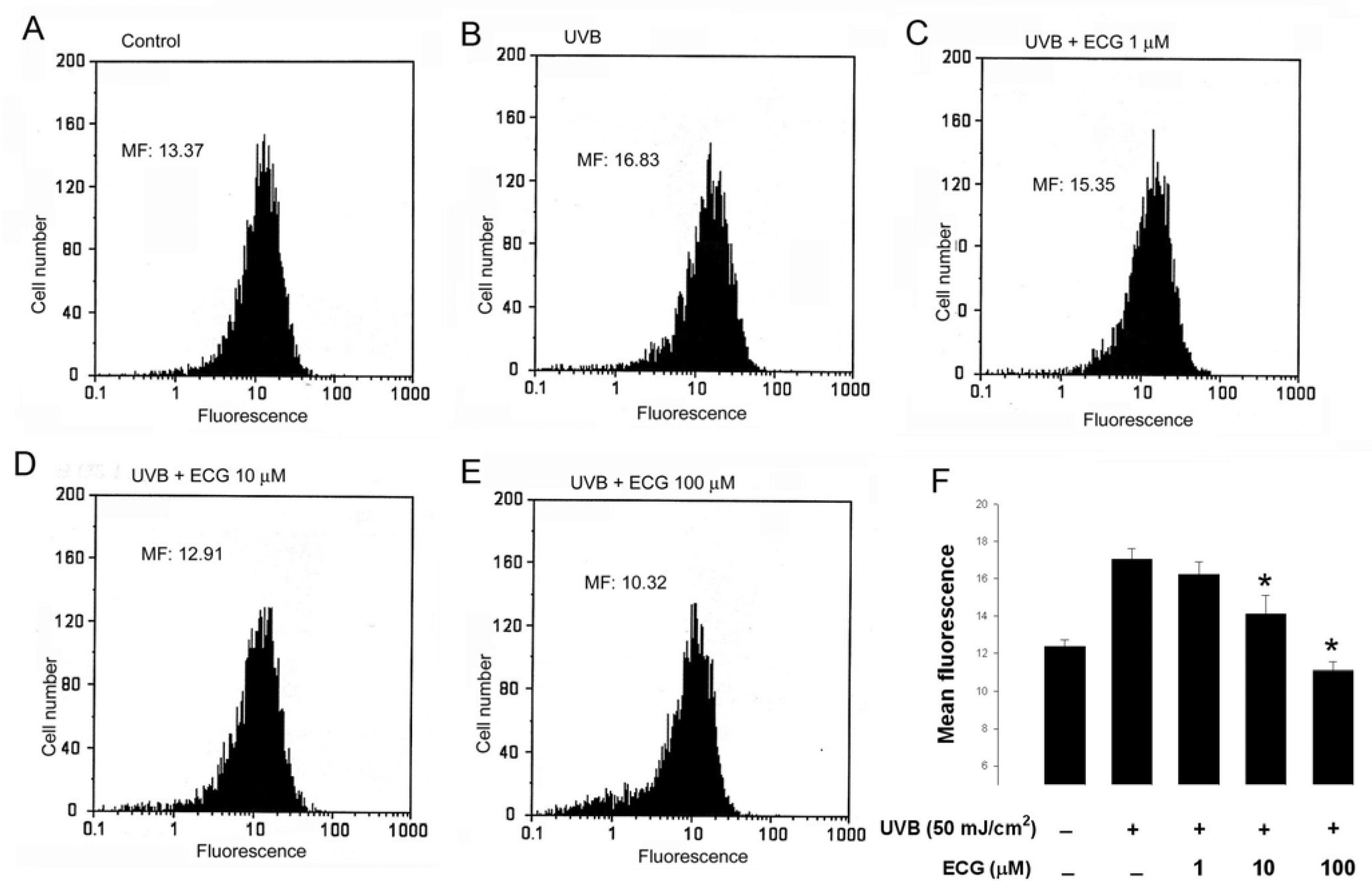
ECG inhibits UVB-induced H2O2 generation in keratinocytes
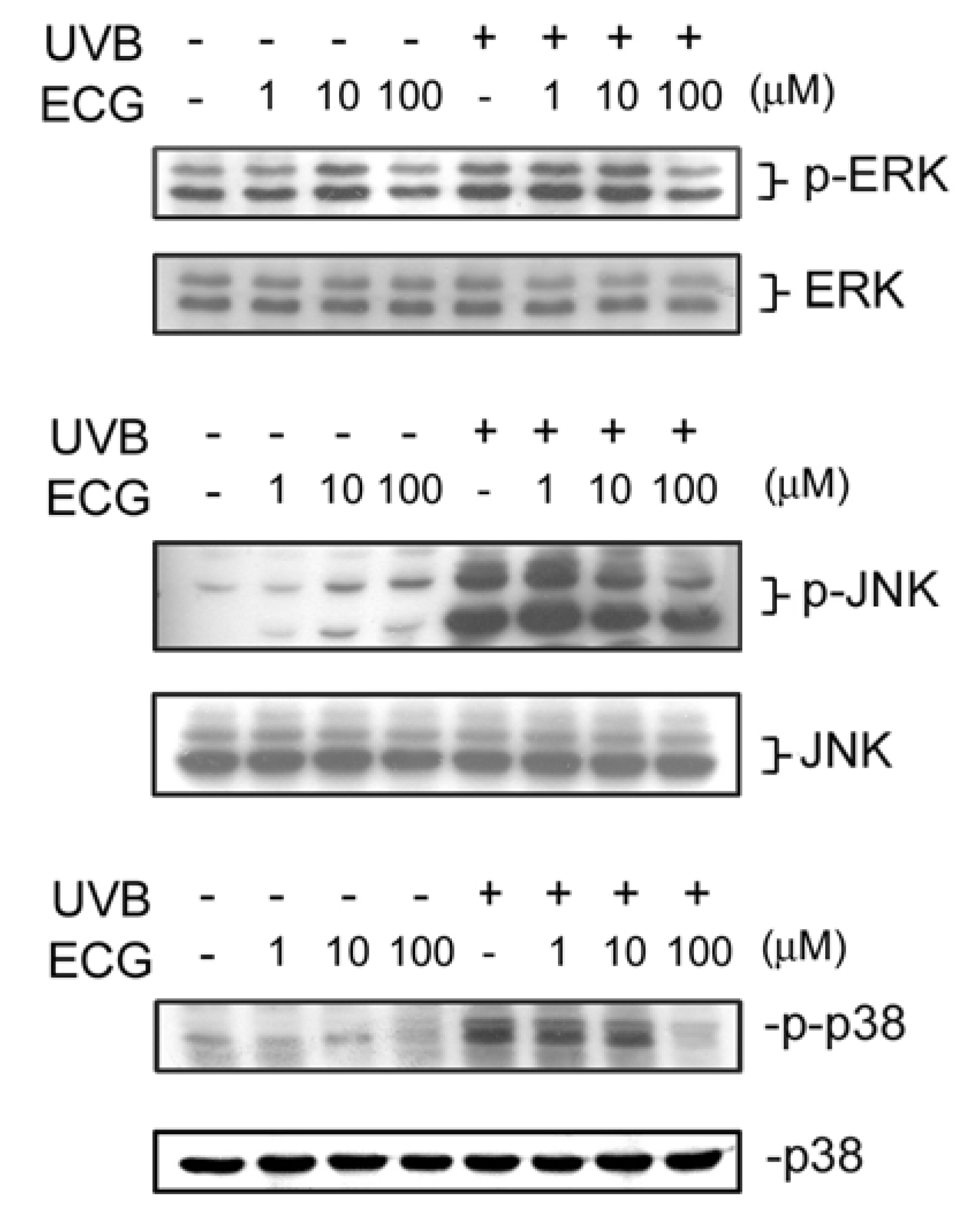
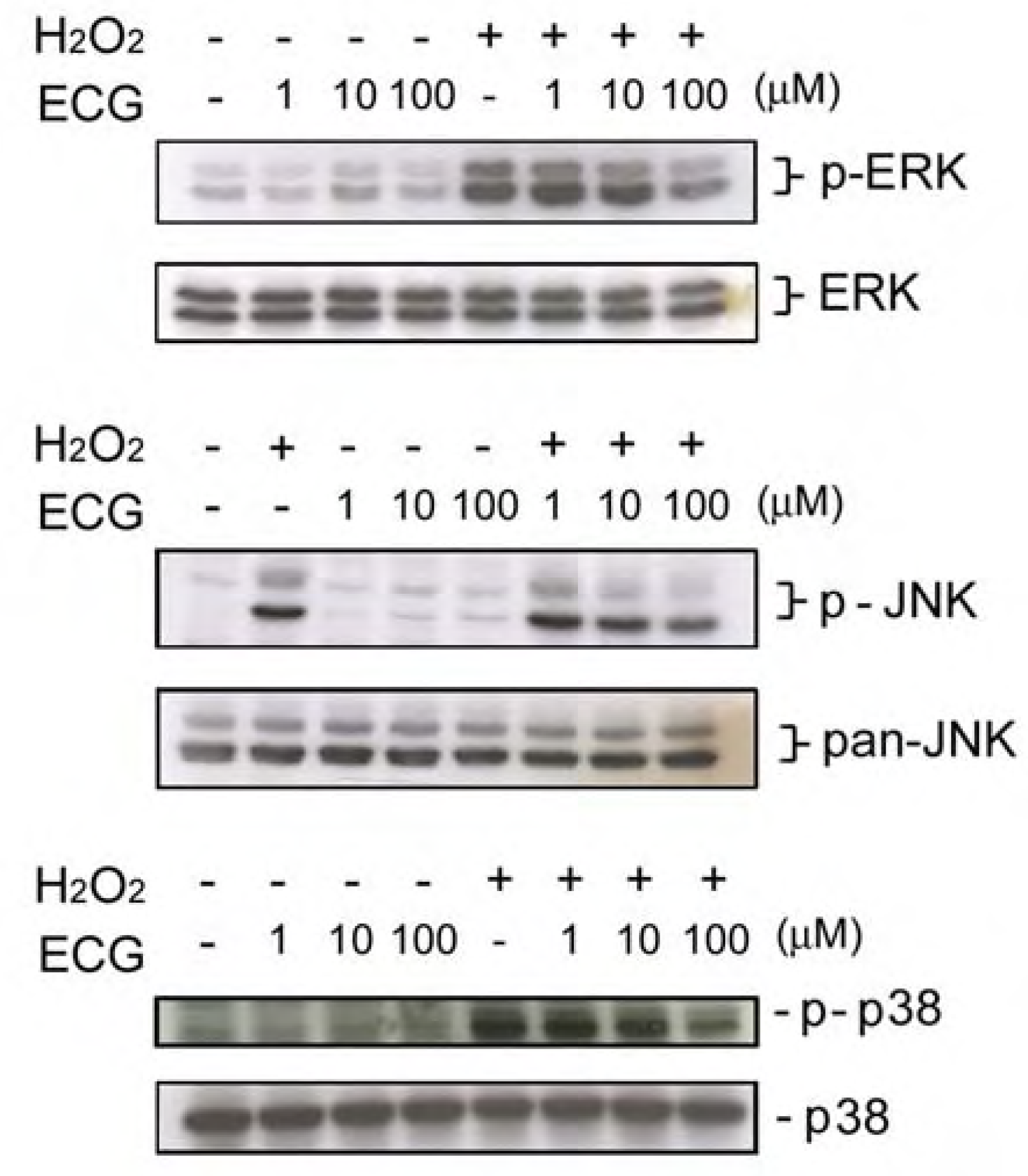
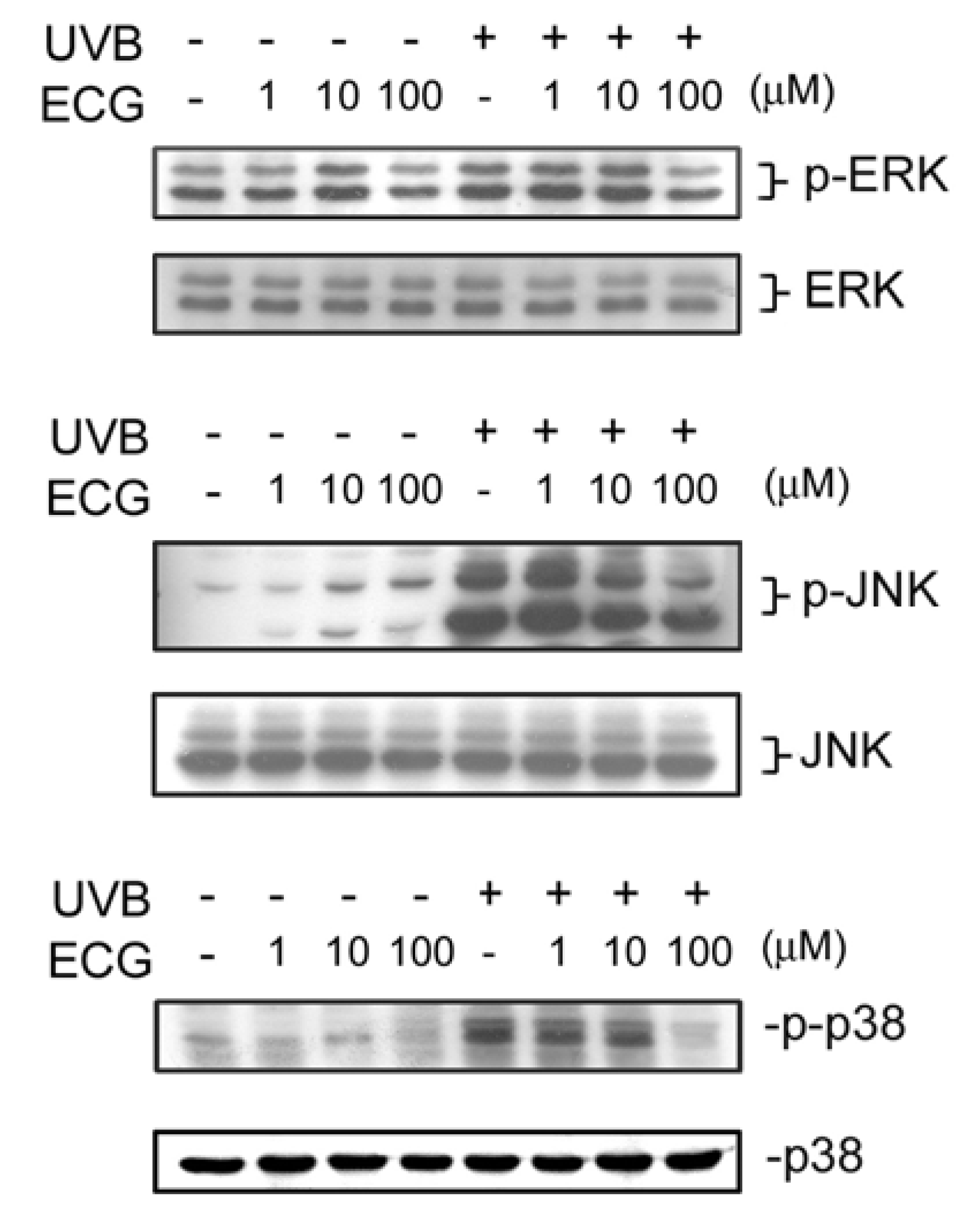
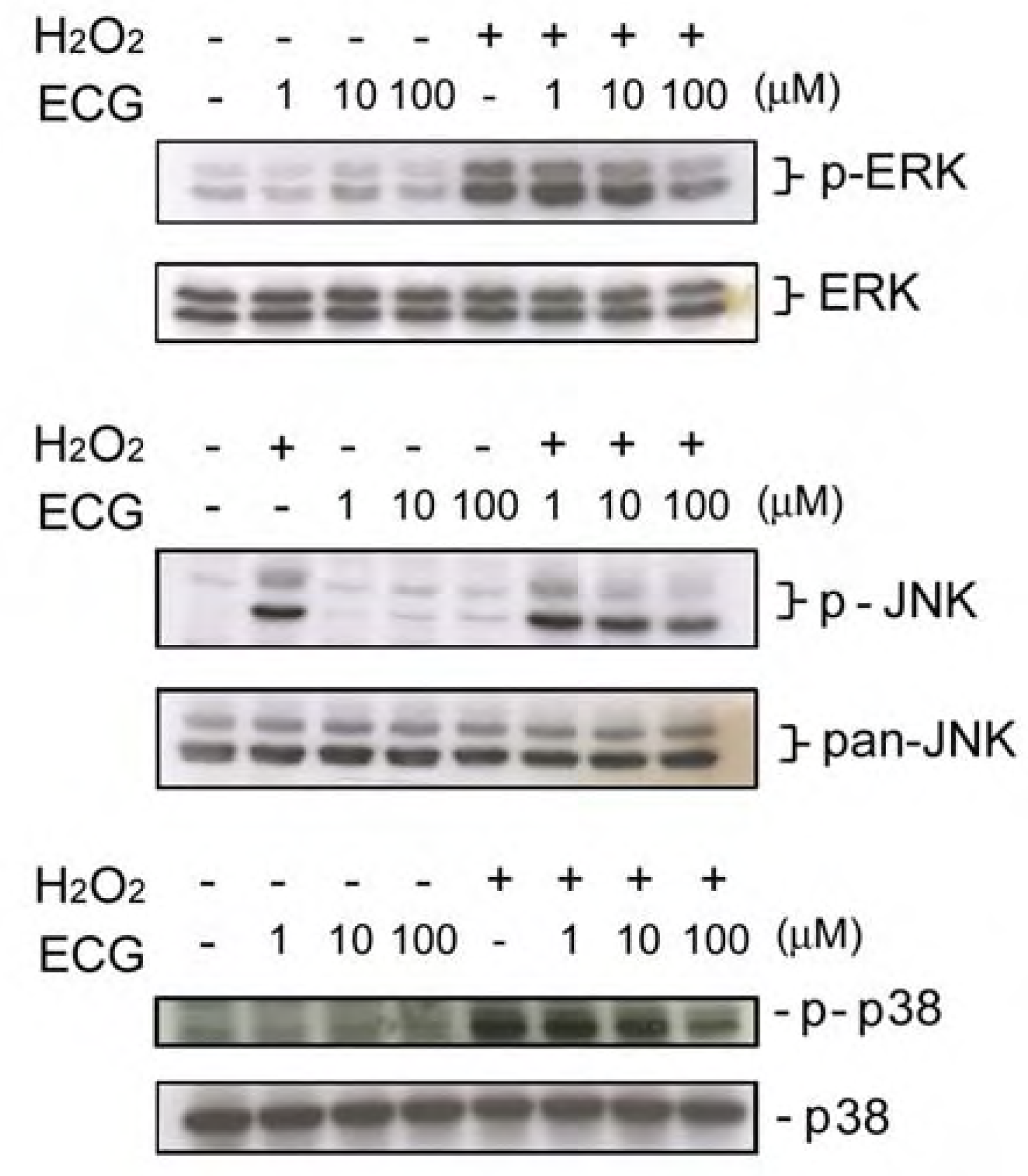
ECG inhibits UVB-induced ERK1/2, JNK andp38 activation
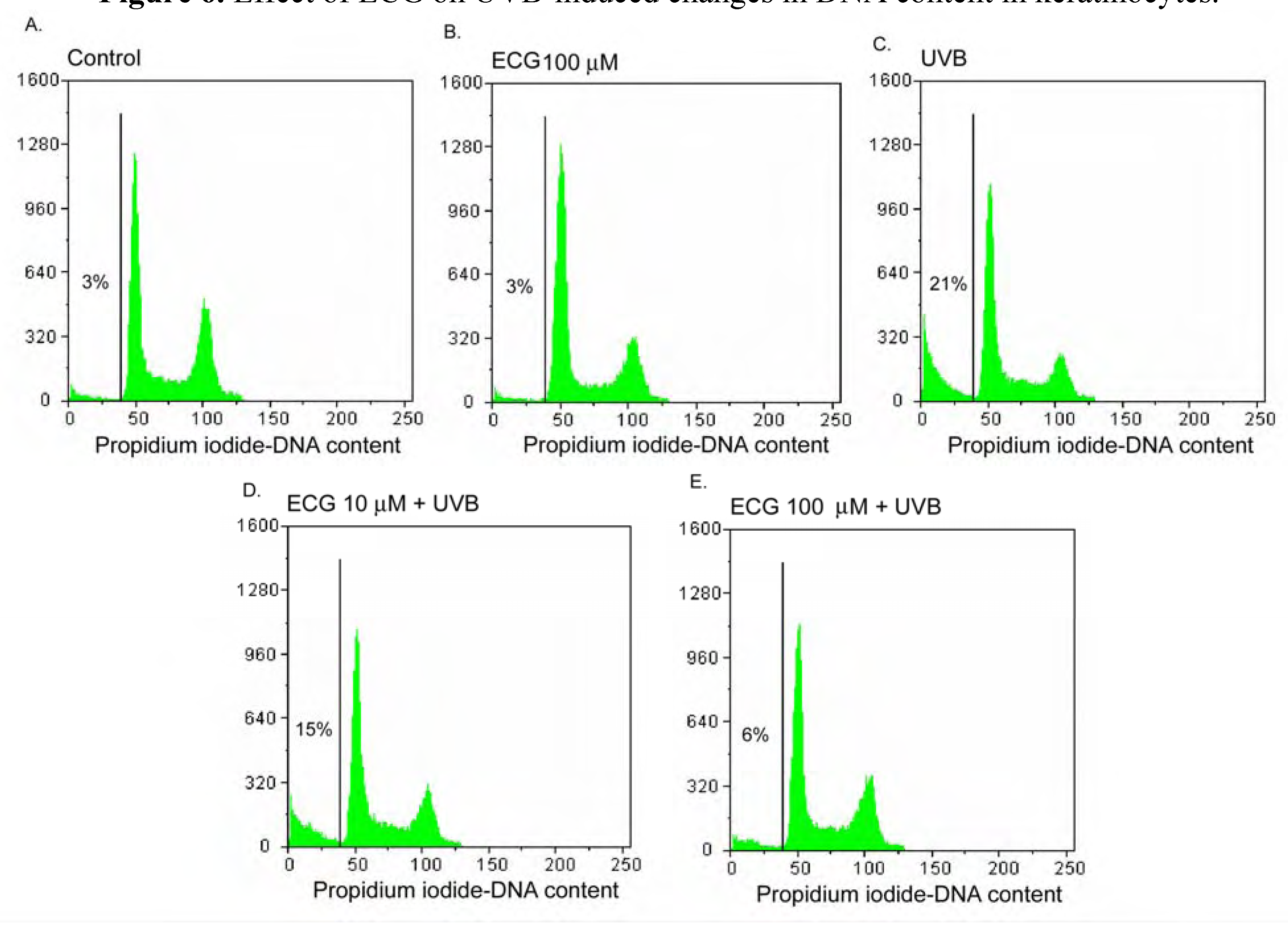
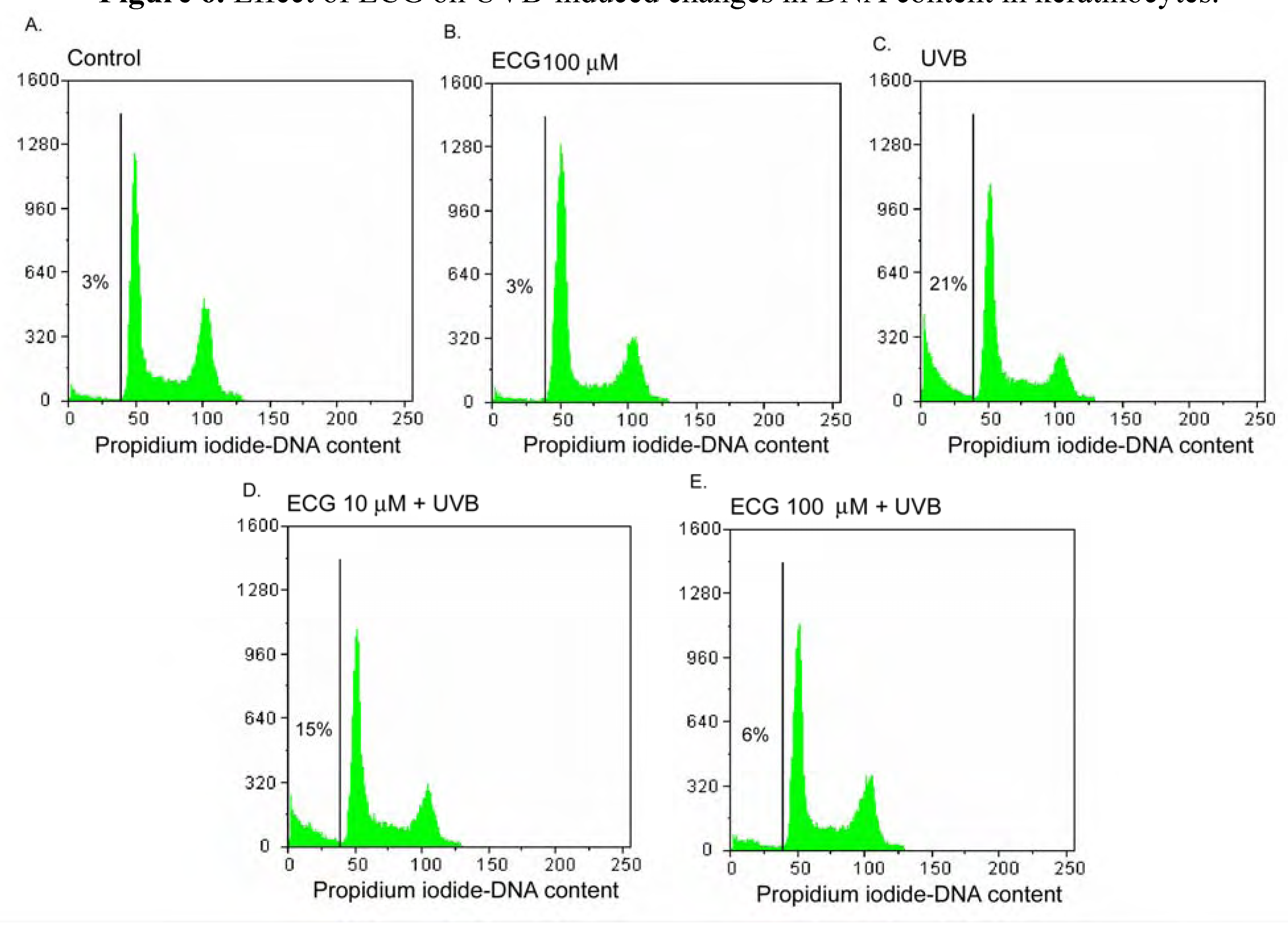
Effect of ECG on DNA content in human keratinocytes
Conclusions
Experimental
Materials
Cell Culture
Drug treatment and UVB irradiation
Cell viability assay (MTT assay)
Lipid peroxidation assay
Flow cytometric analysis of intracellular H2O2
Cell lysate preparation and Western blot analysis of ERK1/2, JNK and p38
Flow cytometric analysis of cell cycle
Statistical analysis
Acknowledgements
References
- Lyons, N.M.; O'Brien, N.M. Modulatory effects of an algal extract containing astaxanthin on UVA-irradiated cells in culture. J. Dermatol. Sci. 2002, 30, 73–84. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Rae, V.; Bruins-Slot, W.; Van den Berg, J.W.; Taylor, J.R.; Streilein, J.W. Susceptibility to effects of UVB radiation on induction of contact hypersensitivity as a risk factor for skin cancer in humans. J. Invest. Dermatol. 1990, 95, 530–536. [Google Scholar]
- Donawho, C.K.; Muller, H.K.; Bucana, C.D.; Kripke, M.L. Enhanced growth of murine melanoma in ultraviolet-irradiated skin is associated with local inhibition of immune effector mechanisms. J. Immunol. 1996, 157, 781–786. [Google Scholar]
- Goihman-Yahr, M. Skin aging and photoaging: an outlook. Clin. Dermatol. 1996, 14, 153–160. [Google Scholar] [CrossRef]
- Miyachi, Y. Photoaging from an oxidative standpoint. J. Dermatol. Sci. 1995, 9, 79–86. [Google Scholar] [CrossRef]
- Dunham, W.B.; Zuckerkandl, E.; Reynolds, R.; Willoughby, R.; Marcuson, R.; Barth, R.; Pauling, L. Effects of intake of L-ascorbic acid on the incidence of dermal neoplasms induced in mice by ultraviolet light. Proc. Natl. Acad. Sci. U S A 1982, 79, 7532–7536. [Google Scholar] [CrossRef]
- Gensler, H.L.; Magdaleno, M. Topical vitamin E inhibition of immunosuppression and tumorigenesis induced by ultraviolet irradiation. Nutr. Cancer 1991, 15, 97–106. [Google Scholar] [CrossRef]
- Black, H.S.; Chan, J.T. Suppression of ultraviolet light-induced tumor formation by dietary antioxidants. J. Invest. Dermatol. 1975, 65, 412–414. [Google Scholar]
- Graham, H.N. Green tea composition, consumption, and polyphenol chemistry. Prev. Med. 1992, 21, 334–350. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Afaq, F.; Adhami, V.M.; Mukhtar, H. Photochemoprevention of ultraviolet B signaling and photocarcinogenesis. Mutat. Res. 2005, 571, 153–173. [Google Scholar] [CrossRef]
- Afaq, F.; Mukhtar, H. Photochemoprevention by botanical antioxidants. Skin Pharmacol. Appl. Skin Physiol. 2002, 15, 297–306. [Google Scholar] [CrossRef]
- F'guyer, S.; Afaq, F.; Mukhtar, H. Photochemoprevention of skin cancer by botanical agents. Photodermatol. Photoimmunol. Photomed. 2003, 19, 56–72. [Google Scholar] [CrossRef]
- Katiyar, S.K.; Elmets, C.A. Green tea polyphenolic antioxidants and skin photoprotection (Review). Int. J. Oncol. 2001, 18, 1307–1313. [Google Scholar]
- Katiyar, S.K.; Afaq, F.; Perez, A.; Mukhtar, H. Green tea polyphenol (-)-epigallocatechin-3-gallate treatment of human skin inhibits ultraviolet radiation-induced oxidative stress. Carcinogenesis 2001, 22, 287–294. [Google Scholar] [CrossRef]
- Elmets, C.A.; Singh, D.; Tubesing, K.; Matsui, M.; Katiyar, S.; Mukhtar, H. Cutaneous photoprotection from ultraviolet injury by green tea polyphenols. J. Am. Acad. Dermatol. 2001, 44, 425–432. [Google Scholar] [CrossRef]
- Katiyar, S.K.; Afaq, F.; Azizuddin, K.; Mukhtar, H. Inhibition of UVB-induced oxidative stress-mediated phosphorylation of mitogen-activated protein kinase signaling pathways in cultured human epidermal keratinocytes by green tea polyphenol (-)-epigallocatechin-3-gallate. Toxicol. Appl. Pharmacol. 2001, 176, 110–117. [Google Scholar] [CrossRef]
- Katiyar, S.K.; Mukhtar, H. Green tea polyphenol (-)-epigallocatechin-3-gallate treatment to mouse skin prevents UVB-induced infiltration of leukocytes, depletion of antigen-presenting cells, and oxidative stress. J. Leukoc. Biol. 2001, 69, 719–726. [Google Scholar]
- Hursting, S.D.; Slaga, T.J.; Fischer, S.M.; DiGiovanni, J.; Phang, J.M. Mechanism-based cancer prevention approaches: targets, examples, and the use of transgenic mice. J. Natl. Cancer Inst. 1999, 91, 215–225. [Google Scholar] [CrossRef]
- Katiyar, S.K.; Elmets, C.A.; Agarwal, R.; Mukhtar, H. Protection against ultraviolet-B radiation-induced local and systemic suppression of contact hypersensitivity and edema responses in C3H/HeN mice by green tea polyphenols. Photochem. Photobiol. 1995, 62, 855–861. [Google Scholar]
- Agarwal, R.; Katiyar, S.K.; Khan, S.G.; Mukhtar, H. Protection against ultraviolet B radiation-induced effects in the skin of SKH-1 hairless mice by a polyphenolic fraction isolated from green tea. Photochem. Photobiol. 1993, 58, 695–700. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Agarwal, R.; Bickers, D.R.; Mukhtar, H. Protection against ultraviolet B radiation-induced photocarcinogenesis in hairless mice by green tea polyphenols. Carcinogenesis 1991, 12, 1527–1530. [Google Scholar] [CrossRef]
- Huang, C.C.; Fang, J.Y.; Wu, W.B.; Chiang, H.S.; Wei, Y.J.; Hung, C.F. Protective effects of (-)-epicatechin-3-gallate on UVA-induced damage in HaCaT keratinocytes. Arch. Dermatol. Res. 2005, 296, 473–481. [Google Scholar] [CrossRef]
- Lim, Y.C.; Lee, S.H.; Song, M.H.; Yamaguchi, K.; Yoon, J.H.; Choi, E.C.; Baek, S.J. Growth inhibition and apoptosis by (−)-epicatechin gallate are mediated by cyclin D1 suppression in head and neck squamous carcinoma cells. Eur. J. Cancer 2006, 42, 3260–3266. [Google Scholar] [CrossRef]
- Huang, C.C.; Fang, J.Y.; Wu, W.B.; Chiang, H.S.; Wei, Y.J.; Hung, C.F. Protective effects of (-)-epicatechin-3-gallate on UVA-induced damage in HaCaT keratinocytes. Arch. Dermatol. Res. 2005, 296, 473–481. [Google Scholar] [CrossRef]
- Brunet, S.; Thibault, L.; Lepage, G.; Seidman, E.G.; Dube, N.; Levy, E. Modulation of endoplasmic reticulum-bound cholesterol regulatory enzymes by iron/ascorbate-mediated lipid peroxidation. Free Radic. Biol. Med. 2000, 28, 46–54. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Riley, D.P.; Caputi, A.P.; Salvemini, D. Antioxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol. Rev. 2001, 53, 135–159. [Google Scholar]
- Afaq, F.; Syed, D.N.; Malik, A.; Hadi, N.; Sarfaraz, S.; Kweon, M.H.; Khan, N.; Zaid, M.A.; Mukhtar, H. Delphinidin, an anthocyanidin in pigmented fruits and vegetables, protects human HaCaT keratinocytes and mouse skin against UVB-mediated oxidative stress and apoptosis. J. Invest. Dermatol. 2007, 127, 222–232. [Google Scholar] [CrossRef]
- Peus, D.; Meves, A.; Vasa, R.A.; Beyerle, A.; O'Brien, T.; Pittelkow, M.R. H2O2 is required for UVB-induced EGF receptor and downstream signaling pathway activation. Free Radic. Biol. Med. 1999, 27, 1197–1202. [Google Scholar] [CrossRef]
- Peus, D.; Vasa, R.A.; Beyerle, A.; Meves, A.; Krautmacher, C.; Pittelkow, M.R. UVB activates ERK1/2 and p38 signaling pathways via reactive oxygen species in cultured keratinocytes. J. Invest. Dermatol. 1999, 112, 751–756. [Google Scholar] [CrossRef]
- Maziere, C.; Conte, M.A.; Leborgn, L.; Levade, T.; Hornebeck, W.; Santus, R.; Maziere, J.C. UVA radiation stimulates ceramide production: relationship to oxidative stress and potential role in ERK, JNK, and p38 activation. Biochem. Biophys. Res. Commun. 2001, 281, 289–294. [Google Scholar] [CrossRef]
- Darr, D.; Fridovich, I. Free radicals in cutaneous biology. J. Invest. Dermatol. 1994, 102, 671–675. [Google Scholar]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995, 270, 16483–16486. [Google Scholar] [CrossRef]
- Peus, D.; Vasa, R.A.; Meves, A.; Beyerle, A.; Pittelkow, M.R. UVB-induced epidermal growth factor receptor phosphorylation is critical for downstream signaling and keratinocyte survival. Photochem. Photobiol. 2000, 72, 135–140. [Google Scholar] [CrossRef]
- Cobb, M.H.; Goldsmith, E.J. How MAP kinases are regulated. J. Biol. Chem. 1995, 270, 14843–14846. [Google Scholar] [CrossRef]
- Robinson, M.J.; Cobb, M.H. Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 1997, 9, 180–186. [Google Scholar] [CrossRef]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)-from inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef]
- Chen, W.; Borchers, A.H.; Dong, Z.; Powell, M.B.; Bowden, G.T. UVB irradiation-induced activator protein-1 activation correlates with increased c-fos gene expression in a human keratinocyte cell line. J. Biol. Chem. 1998, 273, 32176–32181. [Google Scholar]
- Guyton, K.Z.; Gorospe, M.; Kensler, T.W.; Holbrook, N.J. Mitogen-activated protein kinase (MAPK) activation by butylated hydroxytoluene hydroperoxide: implications for cellular survival and tumor promotion. Cancer Res. 1996, 56, 3480–3485. [Google Scholar]
- Wang, X.; Martindale, J.L.; Liu, Y.; Holbrook, N.J. The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem. J. 1998, 333, 291–300. [Google Scholar]
- Wilmer, W.A.; Tan, L.C.; Dickerson, J.A.; Danne, M.; Rovin, B.H. Interleukin-1beta induction of mitogen-activated protein kinases in human mesangial cells. Role of oxidation. J. Biol. Chem. 1997, 272, 10877–10881. [Google Scholar]
- Sample availability: Contact the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Huang, C.-C.; Wu, W.-B.; Fang, J.-Y.; Chiang, H.-S.; Chen, S.-K.; Chen, B.-H.; Chen, Y.-T.; Hung, C.-F. (-)-Epicatechin-3-gallate, a Green Tea Polyphenol Is a Potent Agent Against UVB-induced Damage in HaCaT Keratinocytes. Molecules 2007, 12, 1845-1858. https://doi.org/10.3390/12081845
Huang C-C, Wu W-B, Fang J-Y, Chiang H-S, Chen S-K, Chen B-H, Chen Y-T, Hung C-F. (-)-Epicatechin-3-gallate, a Green Tea Polyphenol Is a Potent Agent Against UVB-induced Damage in HaCaT Keratinocytes. Molecules. 2007; 12(8):1845-1858. https://doi.org/10.3390/12081845
Chicago/Turabian StyleHuang, Chieh-Chen, Wen-Bin Wu, Jia-You Fang, Han-Sun Chiang, Shao-Kuan Chen, Bing-Huei Chen, Ying-Ting Chen, and Chi-Feng Hung. 2007. "(-)-Epicatechin-3-gallate, a Green Tea Polyphenol Is a Potent Agent Against UVB-induced Damage in HaCaT Keratinocytes" Molecules 12, no. 8: 1845-1858. https://doi.org/10.3390/12081845
APA StyleHuang, C.-C., Wu, W.-B., Fang, J.-Y., Chiang, H.-S., Chen, S.-K., Chen, B.-H., Chen, Y.-T., & Hung, C.-F. (2007). (-)-Epicatechin-3-gallate, a Green Tea Polyphenol Is a Potent Agent Against UVB-induced Damage in HaCaT Keratinocytes. Molecules, 12(8), 1845-1858. https://doi.org/10.3390/12081845