Facile Synthesis of Optically Active Imidazole Derivatives

Abstract
:Introduction
Results and Discussion
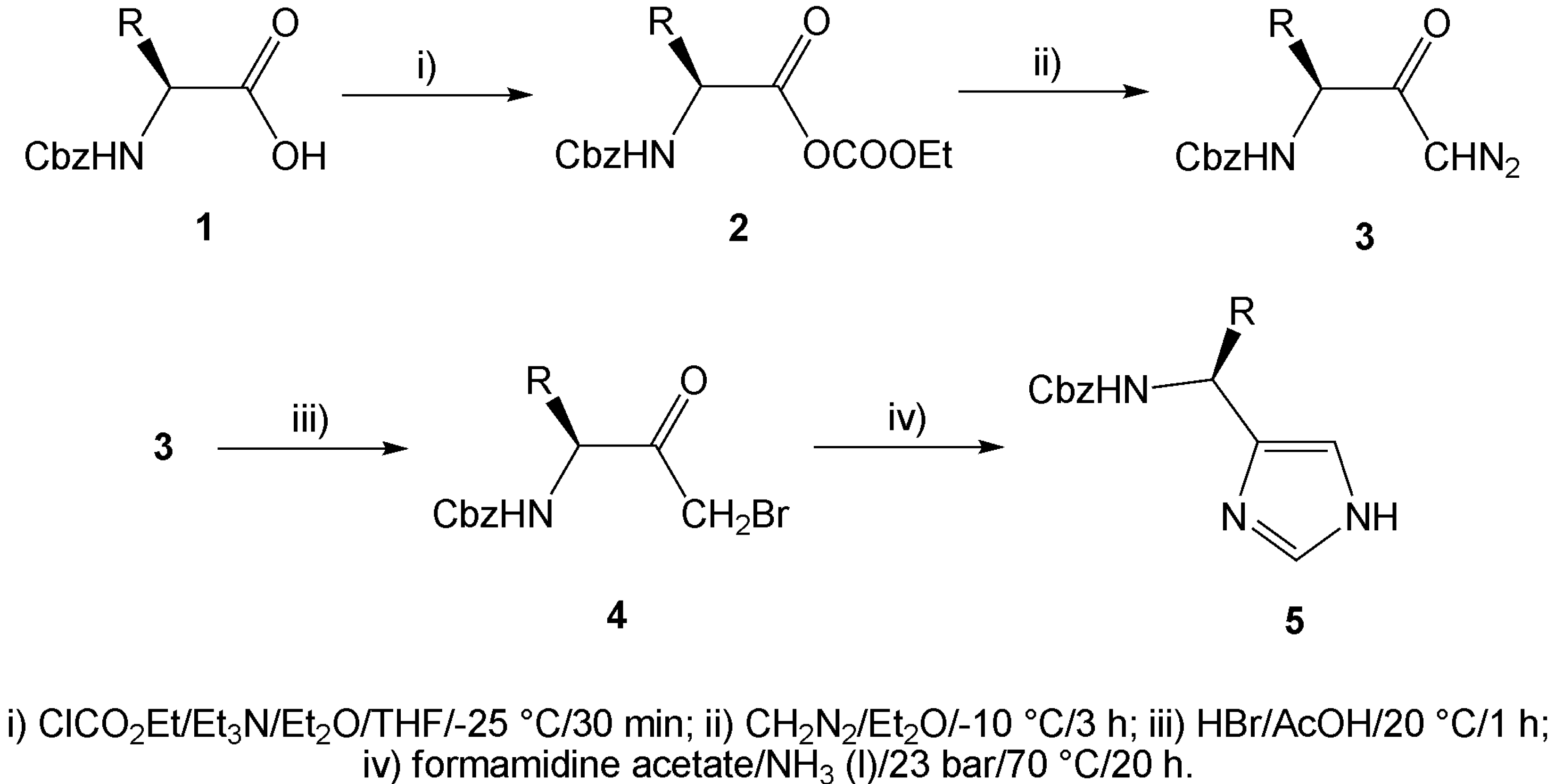

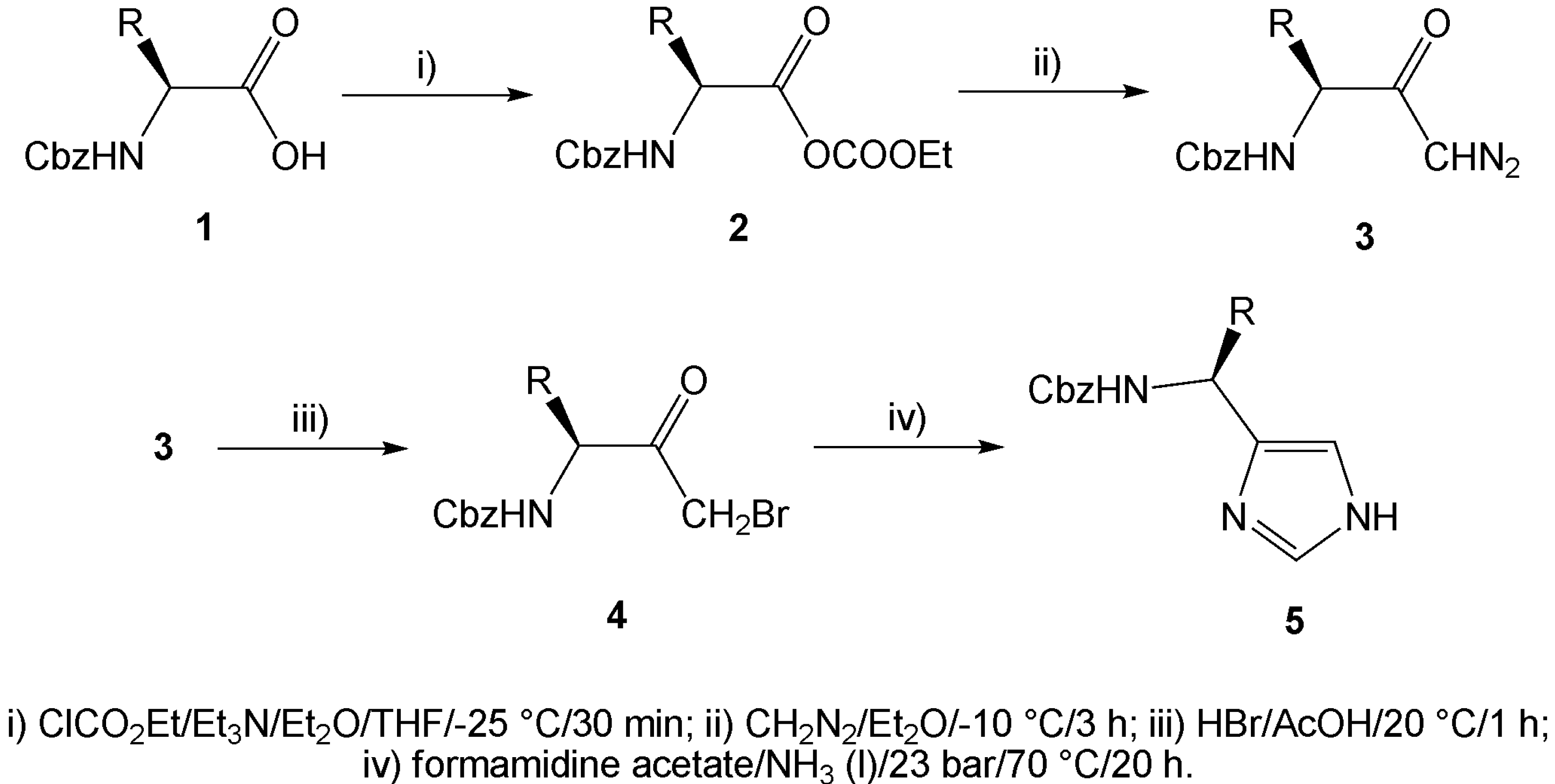{kind=link}
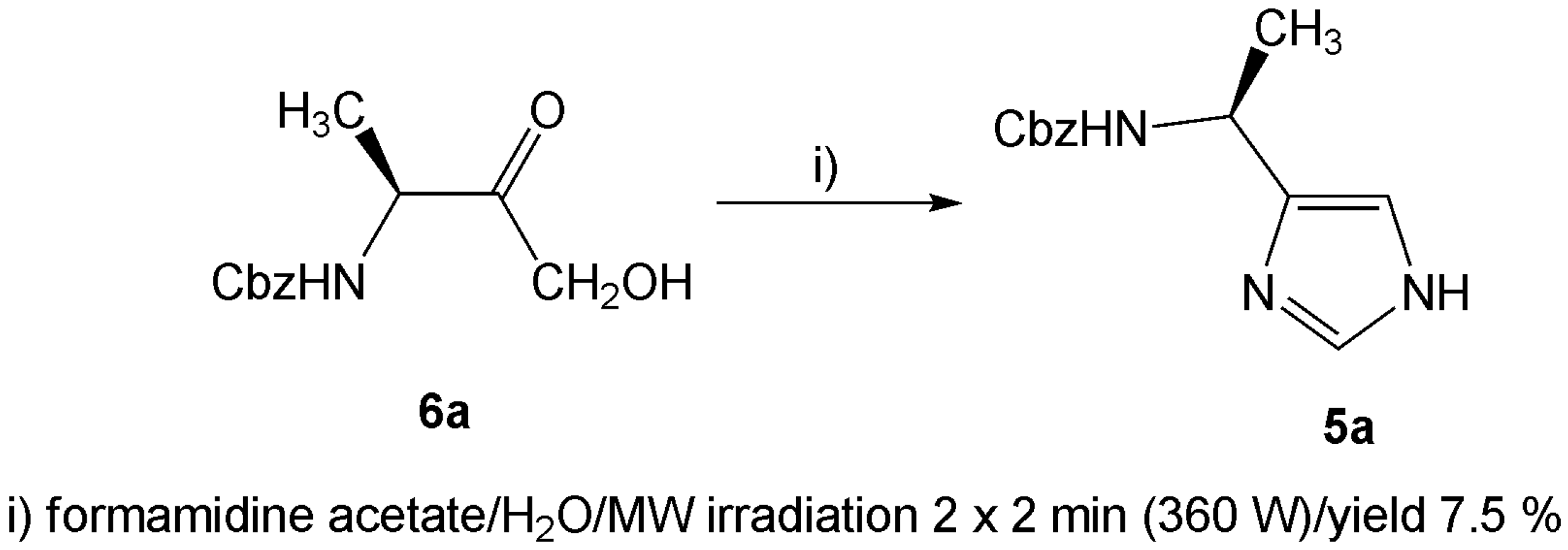
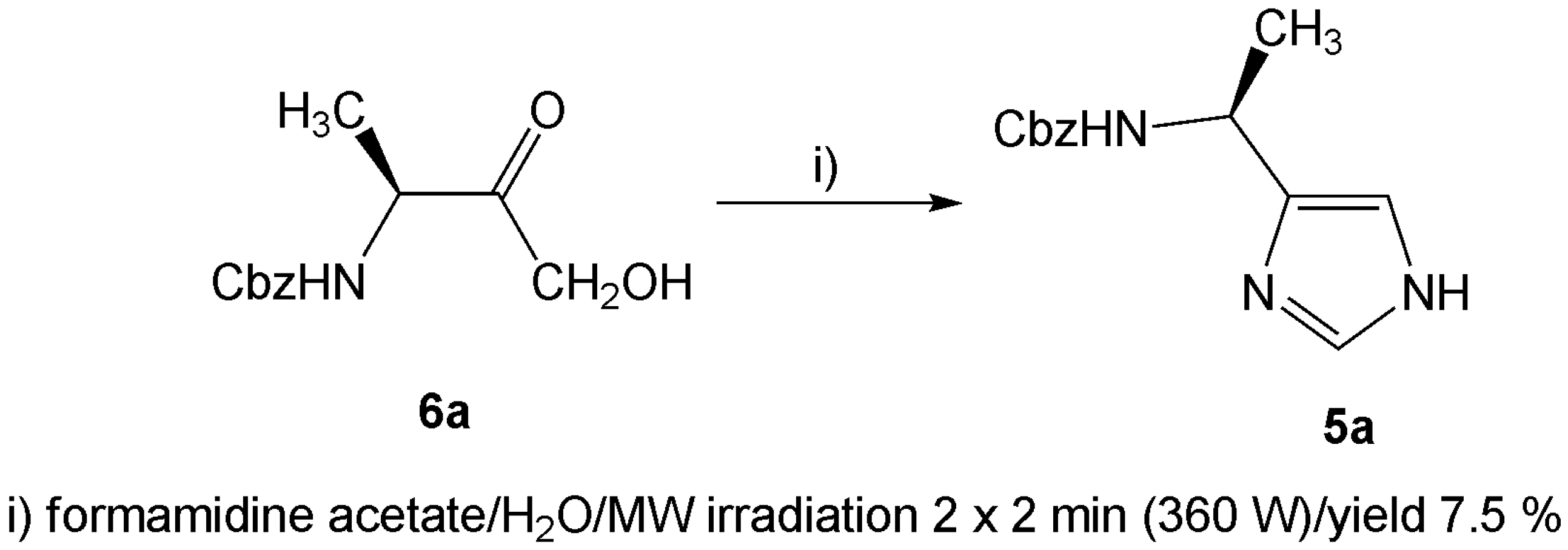{kind=link}
| Comp. | R / Starting amino acid | Yield[a] [%] | e.e. [%] | [α]D20 |
|---|---|---|---|---|
| 5a | CH3 / (S)-Alanine | 81/69 | 99 | -15.8 (c 1, MeOH) |
| 5b | (CH3)2CH / (S)-Valine | 49/42 | 99 | -31.7 (c 0.52, MeOH) |
| 5c | (CH3)2CHCH2 / (S)-Leucine | 69/57 | 99 | -28.5 (c 0.46, MeOH) |
| 5d | CH3CH2(CH3)CH / (S)-Isoleucine | 72/56 | 99 | -35.6 (c 1, MeOH) |
| 5e | PhCH2 / (S)-Phenylalanine | 40/37 | 99 | -29.0 (c 1, MeOH) |
Conclusions
Experimental
General
General method for the condensations in liquid ammonia
Microwave assisted condensation
Acknowledgments
References
- Katritzky, A. R.; Pozharskii, A. F. Handbook of Heterocyclic Chemistry, 2nd Edition ed; Pergamon: New York, 2000. [Google Scholar]
- Kamaraj, K.; Kim, E.; Galliker, B.; Zakharov, L. N.; Rheingold, A. R.; Zuberbuhler, A. D.; Karlin, K. D. Copper(I) and copper(II) complexes possessing cross-linked imidazole-phenol ligands: Structures and dioxygen reactivity. J. Am. Chem. Soc. 2003, 15, 6028–6029. [Google Scholar]
- Moore, L. R.; Cooks, S. M.; Anderson, M. S.; Schanz, H.-J.; Griffin, S. T.; Rogers, R. D.; Kirk, M. C.; Shaughnessy, K. H. Synthesis and characterization of water-soluble silver and palladium imidazol-2-ylidene complexes with noncoordinating anionic substituents. Organometallics 2006, 25, 5151–5158. [Google Scholar]
- Wiglenda, T.; Gust, R. Structure-Activity Relationship study to understand the estrogen receptor-dependent gene activation of aryl- and alkyl-substituted 1H-imidazoles. J. Med. Chem. 2007, 50, 1475–1484. [Google Scholar]
- Baran, P. S.; O’Malley, D. P.; Zografos, A. L. Sceptrin as a potential biosynthetic precursor to complex pyrrole–imidazole alkaloids: The total synthesis of ageliferin. Angew. Chem. Int. Ed. 2004, 43, 2674–2677. [Google Scholar]
- O’Malley, D. P.; Li, K.; Maue, M.; Zografos, A. L.; Baran, P. S. Total synthesis of dimeric pyrrole-imidazole alkaloids: Sceptrin, ageliferin, nagelamide E, oxysceptrin, nakamuric acid, and the axinellamine carbon skeleton. J. Am. Chem. Soc. 2007, 129, 4762–4775. [Google Scholar]
- Wang, R.; Xiao, J.-C.; Twamley, B.; Shreeve, J. M. Efficient Heck reactions catalyzed by a highly recyclable palladium(II) complex of a pyridyl-functionalized imidazolium-based ionic liquid. Org. Biomol. Chem. 2007, 5, 671–678. [Google Scholar]
- Kan, H.-C.; Tseng, M.-C.; Chu, Y.-H. Bicyclic imidazolium-based ionic liquids: Synthesis and characterization. Tetrahedron 2007, 63, 1644–1653. [Google Scholar]
- Yang, C.-G.; Wang, J.; Jiang, B. Enantioselective synthesis of the aminoimidazole segment of Dragmacidin D. Tetrahedron Lett. 2002, 43, 1063–1066. [Google Scholar]
- Santagostini, L.; Gullotti, M.; Pagliarin, R.; Bianchi, E.; Casella, L.; Monzani, E. Functional mimics of copper enzymes. Synthesis and stereochemical properties of the copper(II) complexes of a trinucleating ligand derived from L-Histidine. Tetrahedron: Asymmetry 1999, 10, 281–295. [Google Scholar]
- You, J.-S.; Yu, X.-Q.; Zhang, G.-L.; Xiang, Q.-X.; Lan, J.-B.; Xie, R.-G. Novel chiral imidazole cyclophane receptors: synthesis and enantioselective recognition for amino acid derivatives. Chem. Commun. 2001, 1816–1817. [Google Scholar]
- Suwinski, J.; Szczepankiewicz, W.; Swierczek, K.; Walczak, K. Synthesis of chiral imidazole derivatives as purine precursors. Eur. J. Org. Chem. 2003, 1080–1084. [Google Scholar]
- Jiang, H.-Y.; Zhou, C.-H.; Luo, K.; Chen, H.; Lan, J.-B.; Xie, R.-G. Chiral imidazole metalloenzyme models: Synthesis and enantioselective hydrolysis for α-amino acid esters. J. Mol. Catal. A – Chem. 2006, 260, 288–294. [Google Scholar]
- Bures, F.; Kulhanek, J. Chiral imidazole derivatives synthesis from enantiopure N-protected α-amino acids. Tetrahedron: Asymmetry 2005, 16, 1347–1354. [Google Scholar]
- Bures, F.; Szotkowski, T.; Kulhanek, J.; Pytela, O.; Ludwig, M.; Holcapek, M. Novel nitrogen ligands based on imidazole derivatives and their application in asymmetric catalysis. Tetrahedron: Asymmetry 2006, 17, 900–907. [Google Scholar]
- Sellier, C.; Buschauer, A.; Elz, S.; Schunack, W. Synthesis of (Z)- and (E)-3-(1H)-imidazol-4-yl)-2-propenamine and some 3-(1H-imidazol-4-yl)propanamines. Liebigs Ann. Chem. 1992, 317–323. [Google Scholar]
- Leschke, C.; Altman, J.; Schunack, W. Bis[1H-imidazol-4(5)-yl]ethane and bis(1-tritylimidazol-4-yl)alkanes. Synthesis 1993, 197–198. [Google Scholar]
- Elz, S.; Schunack, W. An alternative synthesis of Homohistamine and structurally related (imidazol-4-yl)alkylamines. Z. Naturforsch. 1987, 42b, 238–242. [Google Scholar]
- Griffith, R. K.; DiPietro, R. A. An improved preparation of imidazole-4(5)-methanol hydrochloride. Synthesis 1983, 576. [Google Scholar]
- Siendt, H.; Tschamber, T.; Streith, J. Improved double epimerisation of (D)-glucose into (D)-gulose and the synthesis of (D)-xylo-imidazolopiperidinose. Tetrahedron Lett. 1999, 40, 5191–5192. [Google Scholar]
- Streith, J.; Boiron, A.; Frankowski, A.; Le Nouen, D.; Rudyk, H.; Tschamber, T. On the way to glycoprocessing inhibotors: A general one-pot synthesis of imidazolosugars. Synthesis 1995, 944–946. [Google Scholar]
- Tschamber, T.; Rudyk, H.; Le Nouen, D. Expeditious syntheses of imidazole C-nucleosides (=C-glycosylimidazoles) from carbohydrates and formamidine acetate. Helv. Chim. Acta 1999, 82, 2015–2019. [Google Scholar]
- Ramtohul, Y. K.; James, M. N. G.; Vederas, J. C. Synthesis and evaluation of keto-glutamine analogues as inhibitors of Hepatitis A virus 3C proteinase. J. Org. Chem. 2002, 67, 3169–3178. [Google Scholar]
- Sample Availability: Samples of the compounds 5a-e are available from the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Marek, A.; Kulhanek, J.; Ludwig, M.; Bures, F. Facile Synthesis of Optically Active Imidazole Derivatives. Molecules 2007, 12, 1183-1190. https://doi.org/10.3390/12051183
Marek A, Kulhanek J, Ludwig M, Bures F. Facile Synthesis of Optically Active Imidazole Derivatives. Molecules. 2007; 12(5):1183-1190. https://doi.org/10.3390/12051183
Chicago/Turabian StyleMarek, Ales, Jiri Kulhanek, Miroslav Ludwig, and Filip Bures. 2007. "Facile Synthesis of Optically Active Imidazole Derivatives" Molecules 12, no. 5: 1183-1190. https://doi.org/10.3390/12051183
APA StyleMarek, A., Kulhanek, J., Ludwig, M., & Bures, F. (2007). Facile Synthesis of Optically Active Imidazole Derivatives. Molecules, 12(5), 1183-1190. https://doi.org/10.3390/12051183