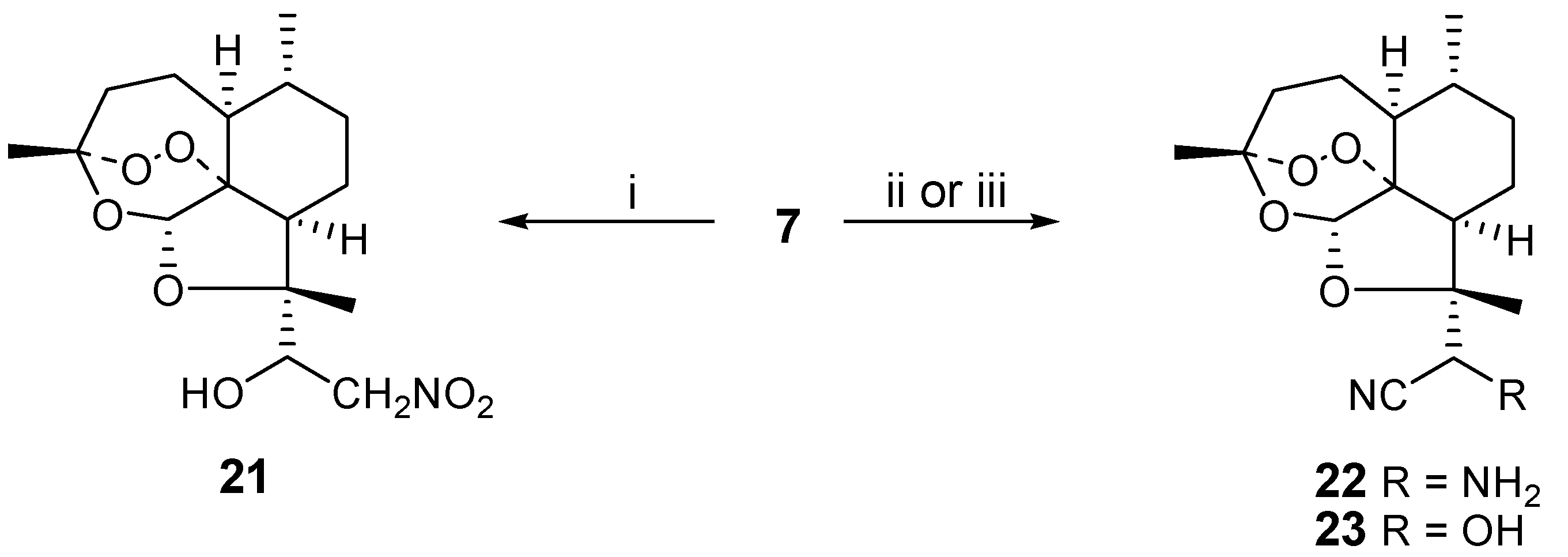Results and Discussion
The β-allyl derivative
6 was prepared in good yield starting from the benzoate of
2 using the literature procedure [
2]. The dehydrodeoxy analog
8 was formed at the same time as a side product (10 %). The acetate analog of
2, in comparison, gave much less of the desired
6, vinyl ether
8 being the major product (50 %). Compound
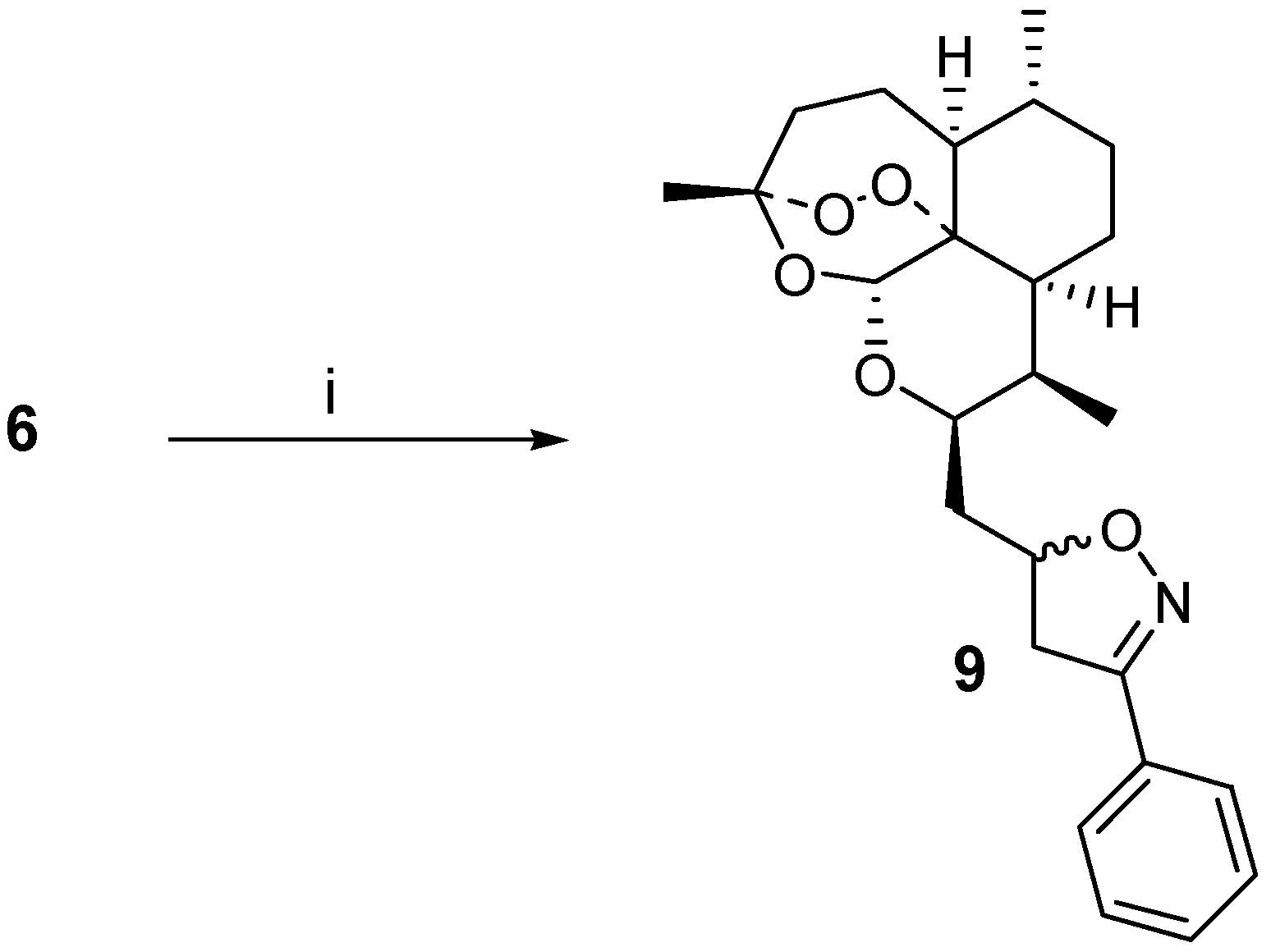
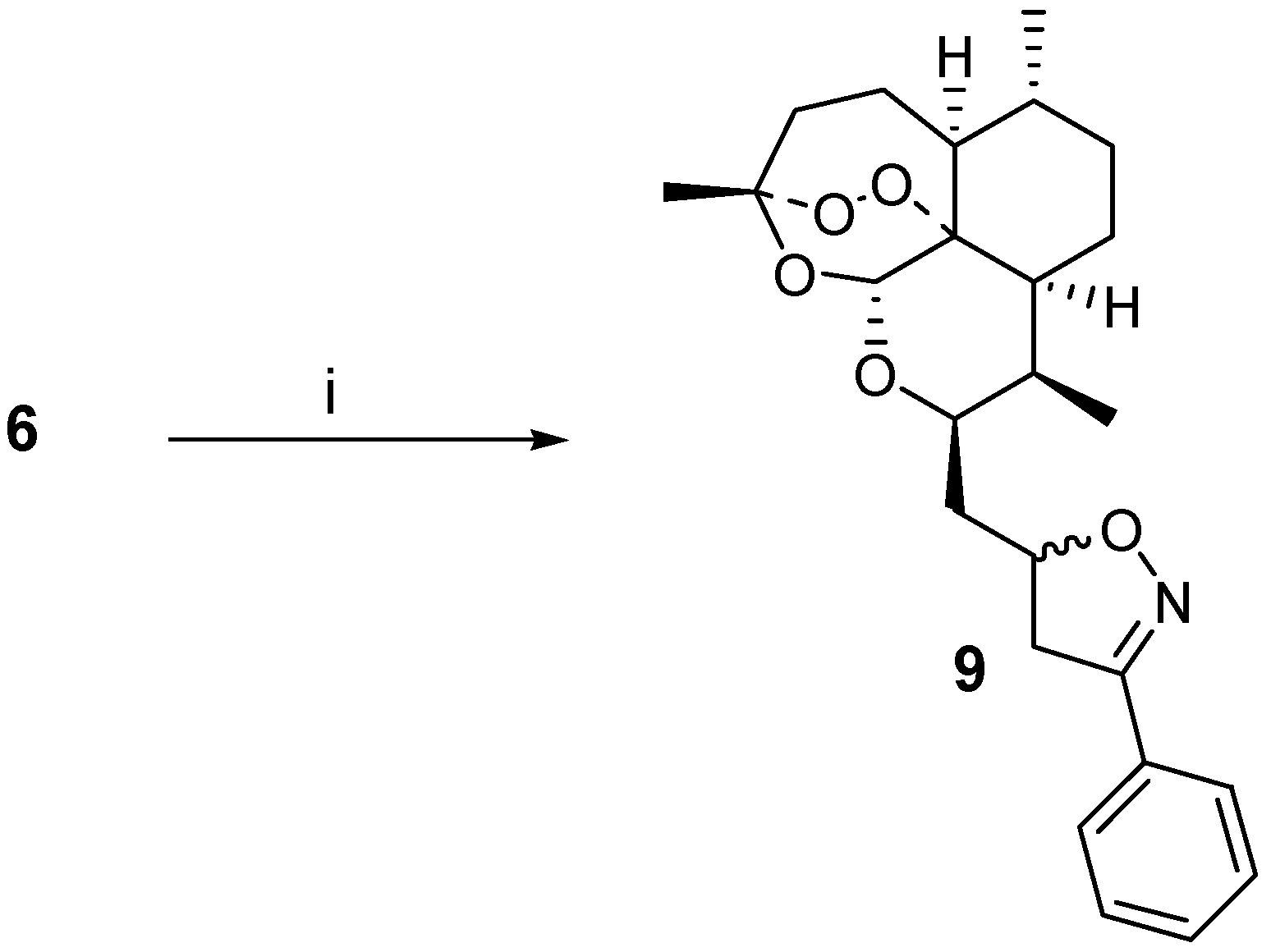
6 was then used as a dipolarophile in combination with
in situ generated [
4] benzonitrile oxide. A cycloadduct
9 was isolated in 29 % yield after careful chromatography, and from NMR-analysis it was found to be a 60:40 mixture of diastereoisomers (stereochemistry not assigned) (
Scheme 1). The vinyl ether
8, when combined with benzonitrile oxide under the same conditions, did not afford an isoxazoline adduct, but rather the starting material was recovered.
Scheme 1.
i) PhCH=NOH, NaOCl, Et3N, CH2Cl2, RT
In view of the low yield of isoxazoline
9, the lack of diastereoselectivity and the fact that
6 has been previously and extensively used by others [
5] to prepare carbon-connected artemisinin derivatives, we decided to shift our attention to the ring-contracted artemisinin derivative
7. This compound is readily available starting from the dehydro analog
8 by treatment with iodine and rearrangement in polar medium [
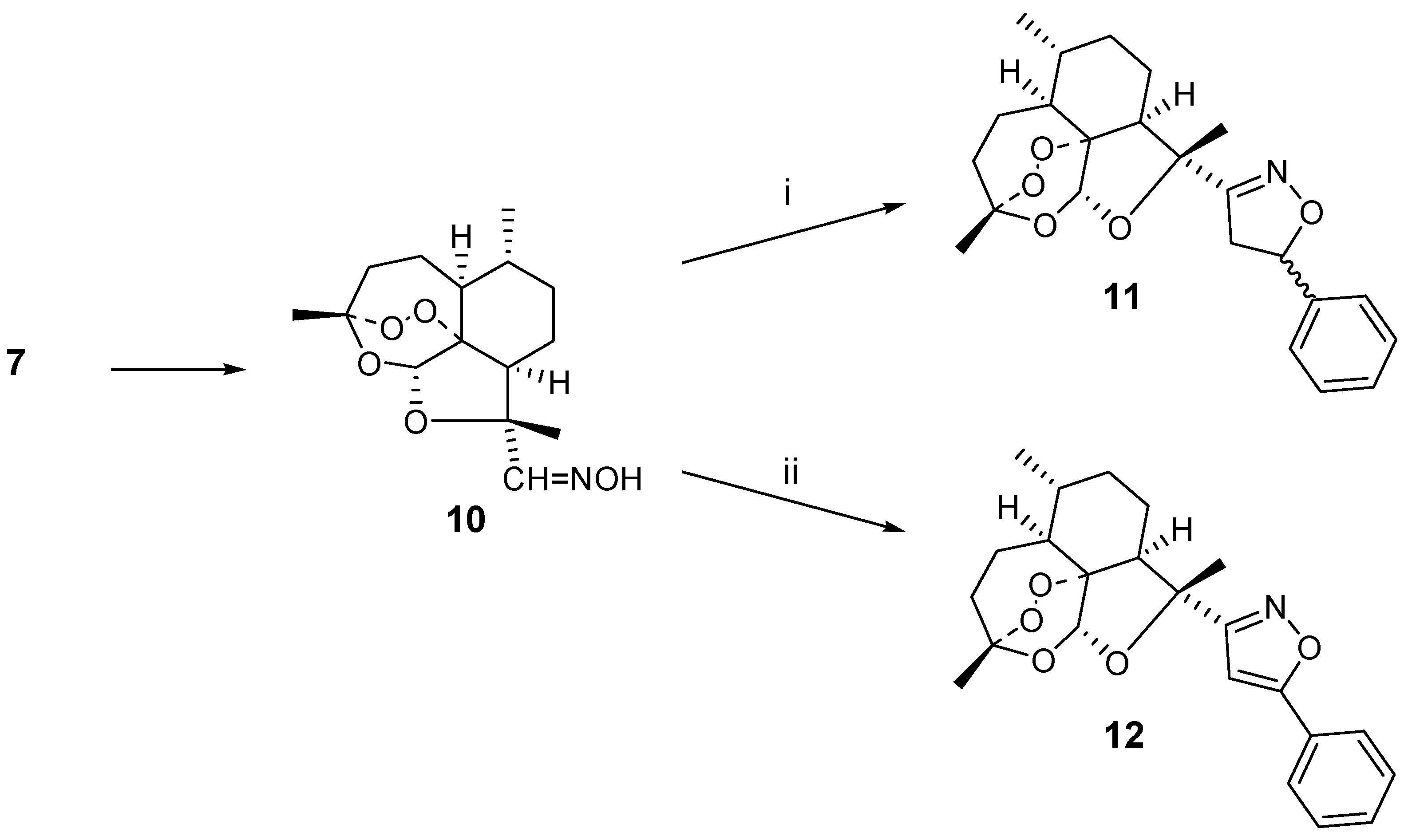
3]. In the first step we converted this aldehyde
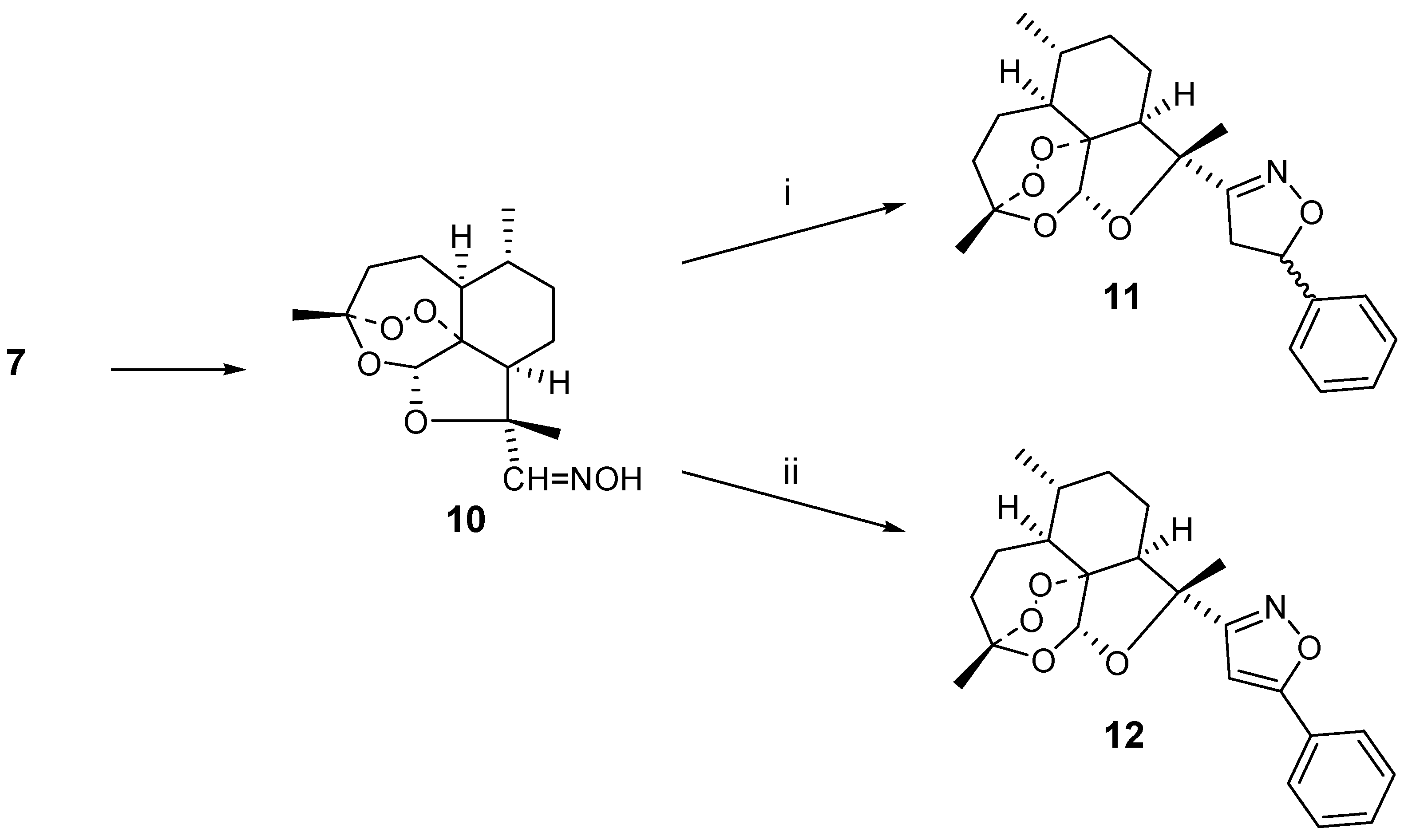
7 into the oxime derivative
10. This oxime
10 was then used to generate the nitrile oxide intermediate in the presence of excess styrene, affording 1,3-dipolar cycloadduct
11 in 40 % yield as a 1:1 mixture of diastereo-isomers. The diastereomer issue could be avoided when phenylacetylene was used as the dipolarophile. In this case, 21 % of the isoxazole
12 was obtained. The reaction is sensitive to steric hindrance. For instance, use of cyclohexene as the dipolarophile did not lead to the isolation of a cycloadduct and rather a mixture of unidentified compounds was formed (
Scheme 2)
Scheme 2.
i) styrene (10 eq.), NaOCl, Et3N, CH2Cl2;
ii) phenylacetylene (10 eq.), NaOCl, Et3N, CH2Cl2
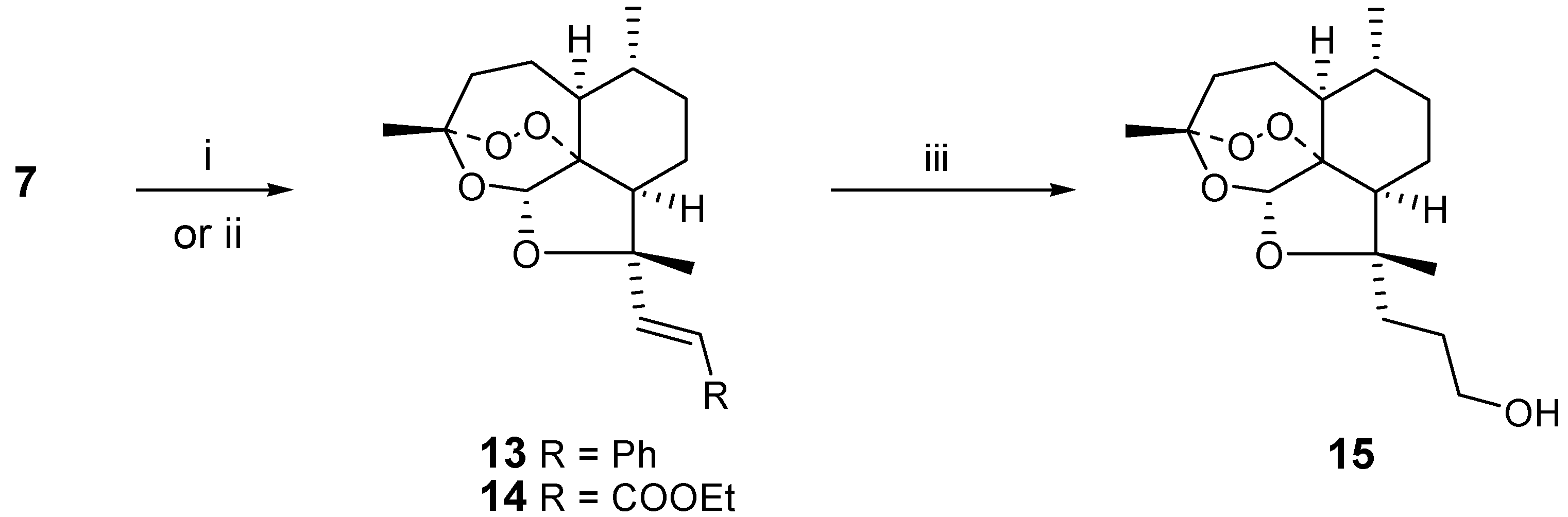
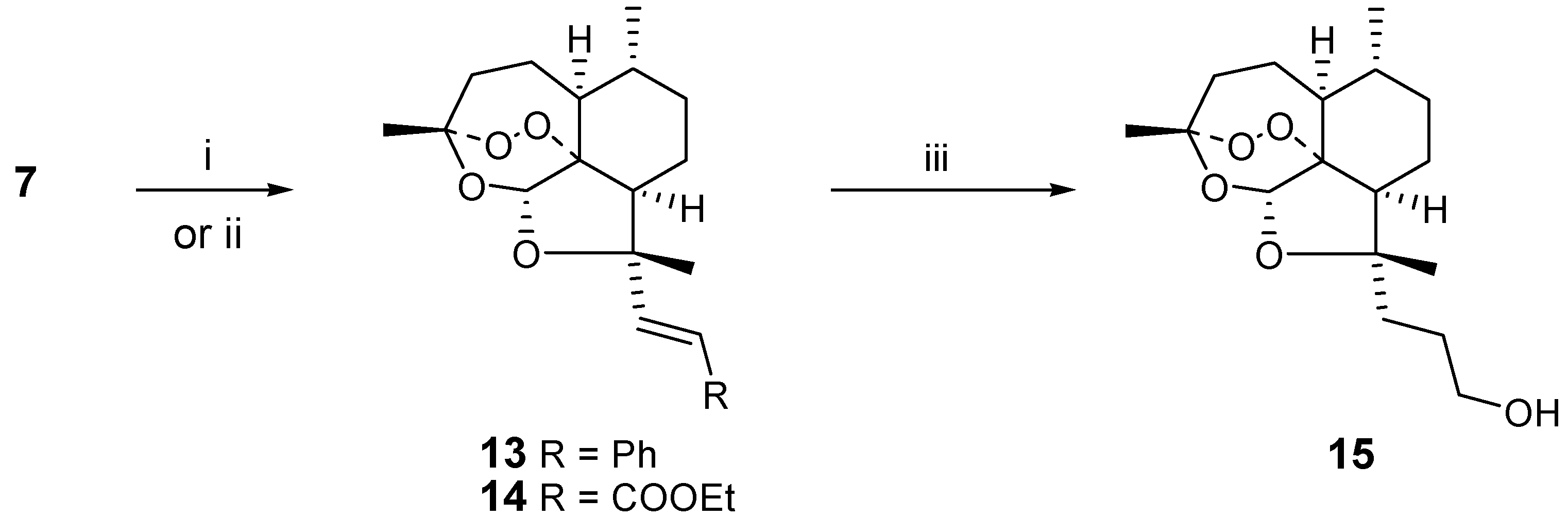
We decided to use the aldehyde
7 to introduce synthetic diversity, extending the functional group away from the artemisinin moiety, as a possible way to increase the reactivity. The Wittig reaction of
7 with benzylidene triphenylphosphorane gave a good yield of the corresponding (
E-)-styrene derivative
13. Analogously, Wittig reaction of
7 gave the (
E)-acrylate ester
14. Remarkably,
14 could be catalytically reduced to the propanol derivative
15 in 74 % yield without cleavage of the peroxide bridge. A reaction time of 12h was optimal and longer reaction times resulted in partial decomposition. (
Scheme 3)
Scheme 3.
i) PhCH2PPh3Br, BuLi, THF, reflux ; ii) EtOOCCH=PPh3, RT, THF, overnight;
iii) Pd/C (5%), H2, THF, RT, 12h
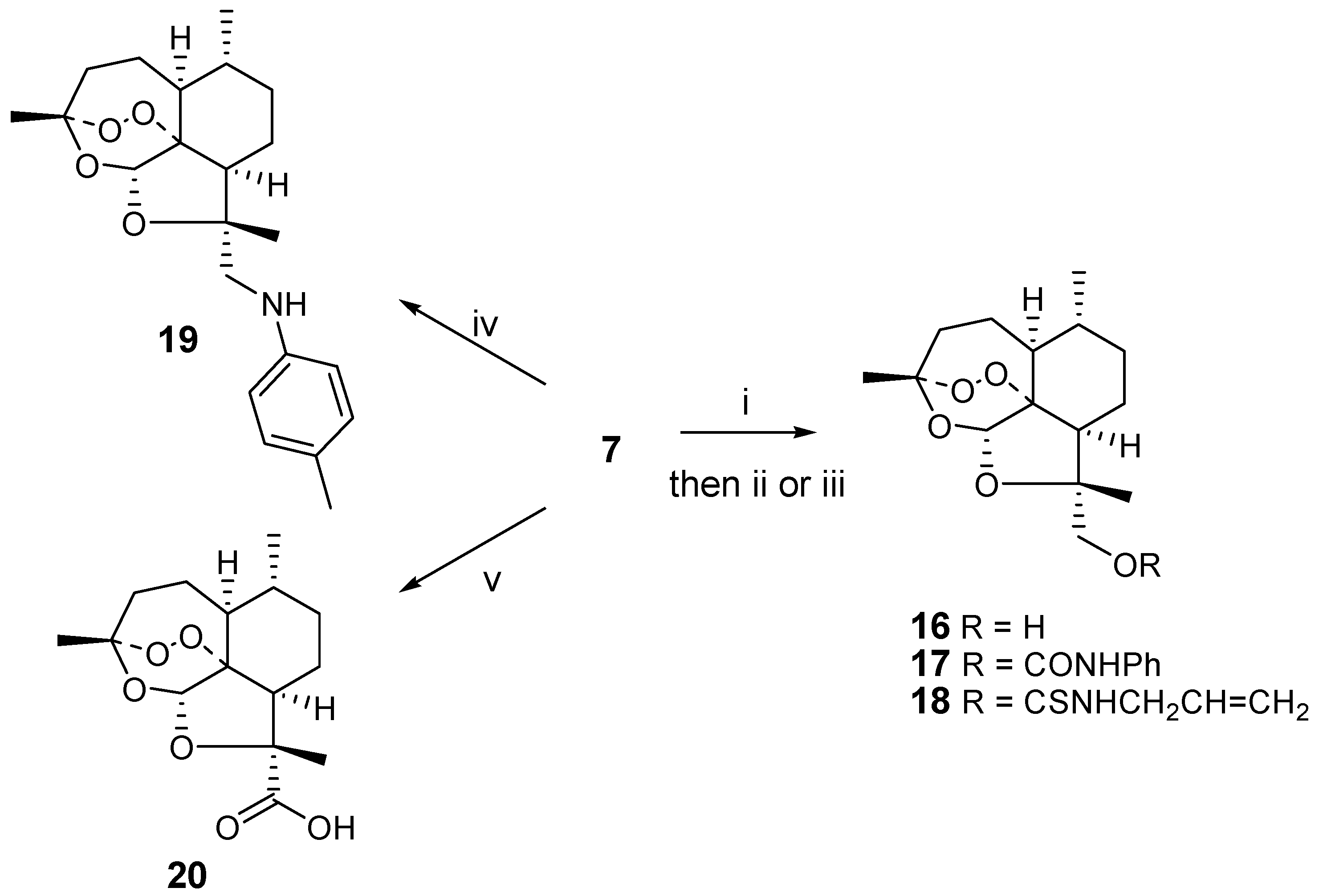
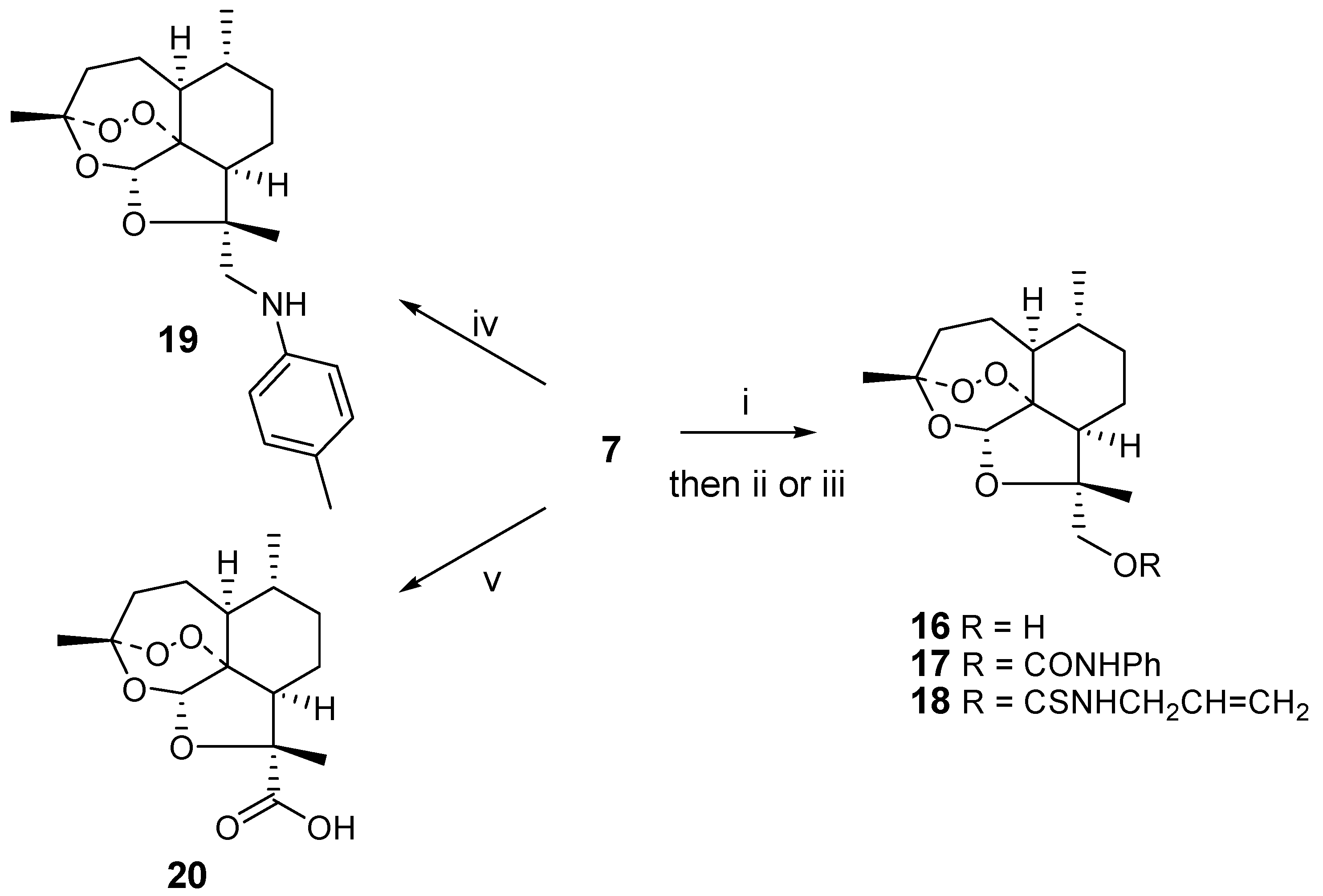
The methanol analogue
16 was prepared from
7 by reduction with excess sodium borohydride. Similar reactions were carried out by Venugopalan on the epimer [
6]. Reaction of
16 with phenyl isocyanate and allyl isothiocyanate resulted in the formation of the urea derivative
17 or thiourea derivative
18, respectively. Reductive amination of
7 with 4-methylaniline afforded a good yield of the secondary aniline
19. Oxidation of
7, on the other hand, gave the carboxylic acid
20. All reactions leading to
16-20 occurred in good yields (53-84 %) (
Scheme 4).
Scheme 4.
i) NaBH4 (4eq.), methanol, 0°C, 1h; ii) phenyl isocyanate, pyridine, RT, 14 h; iii) allyl isothiocyanate, pyridine, 60°C, 14h; iv) 4-methylaniline (4eq.), NaBH4 (4eq.), methanol, RT, 30 min.; v) aminosulfonic acid (4 eq.), sodium chlorite (4 eq.), water/dioxane, RT, 30 min.
Finally, we studied nucleophilic additions to aldehyde
7 as a way to introduce synthetic diversity. Earlier trifluoromethyl and enolate anions had been used [
3] to prepare ketone derivatives of
7. We reacted nitromethane and base with
7 to obtain the nitroaldol
21 in 67 % yield. Analogously, cyanide could be used as the nucleophile in the presence or absence of ammonia, affording aminoacetonitrile
22 or cyanohydrin
23 in 78 and 83 % yields, respectively. Only one of two possible diastereoisomers is obtained for
21-23, but the absolute stereochemistry of the new stereocenters has not yet been determined (
Scheme 5).
Scheme 5.
i) nitromethane (4 eq.), NaOMe (excess), ethanol, 0°C, 18h; ii) NaCN (5 eq.), NH4Cl, methanol/water, 45°C, 2h; iii) KCN (4 eq.), methanol, acetic acid, 18h, RT
Experimental
General
1H- and
13C-NMR spectra were recorded on Varian Mercury 300 MHz, Bruker Avance 300 MHz, or Bruker AMX3 400 spectrometers using tetramethylsilane as an internal standard. ES MS were measured on Micromass Mattro II instrument. Melting points were determined with a Boetius Block apparatus and are uncorrected. Compounds
6 [
2] and
7 [
3] were prepared according to the literature procedures.
10-((3-Phenyl-4,5-dihydroisoxazolyl)methyldeoxoartemisinin (9)
To allyldeoxoartemisinin 6 (0.100 g, 0.32 mmol) in CH2Cl2 (5 mL) was added benzaldehyde oxime (0.2 mL, 1.6 mmol), a solution of sodium hypochlorite (0.5 mL, 13 % active chlorine, 6.8 mmol) and a few drops of triethylamine. The mixture was stirred overnight at room temperature and afterwards washed with water (2 x 10 mL). The organic layer was dried (MgSO4) and evaporated to give a residue that was chromatographed over silica gel with CH2Cl2 as the eluent. The oily product 9 was obtained as a mixture of diastereoisomers (0.040 g, 29 %); MS (ES): 428 (M+); Major isomer: 1H-NMR (400 MHz, C6D6, δ): 0.88 (3H, d, J = 7 Hz, CH3-9), 0.97 (3H, d, J = 3 Hz, CH3-6), 1.26-2.34 (12H, m), 2.69 (1H, m, H-9), 3.13 (1H, dd, J = 9, 16 Hz, isoxazolo-H-4), 3.48 (1H, dd, J = 10, 16 Hz, oxazolo-H-4), 4.53 (1H, ddd, J = 2, 6, 10 Hz, H-10), 4.90 (1H, m, oxazolo-H-5), 5.33 (1H, s, H-12), 7.38-7.42 (5H, m, Ph); 13C-NMR (100 MHz, C6D6, δ): 12.7 (CH3), 20.1 (CH3), 26.0 (CH3), 30.3 (CH), 37.6 (CH), 40.6 (CH2), 44.1 (CH), 52.2 (CH), 72.0 (CH), 80.3 (CH), 80.9 (C), 89.3 (CH), 103.1 (C), 126.7 (2xCH), 128.6 (2xCH), 129.9 (CH), 156.9 (C). Minor isomer: 1H-NMR (400 MHz, C6D6, δ): 0.88 (3H, d, J = 7 Hz, CH3-9), 0.96 (3H, d, J = 3 Hz, CH3-6), 1.26-2.34 (12H, m), 2.69 (1H, m, H-9), 3.13 (1H, dd, J = 9, 16 Hz, isoxazolo-H-4), 3.42 (1H, dd, J = 10, 16 Hz, oxazolo-H-4), 4.35 (1H, ddd, J = 2, 6, 10 Hz, H-10), 5.04 (1H, m, oxazolo-H-5), 5.37 (1H, s, H-12), 7.38-7.42 (5H, m, Ph); 13C-NMR (100 MHz, C6D6, δ): 12.9 (CH3), 20.1 (CH3), 26.0 (CH3), 30.3 (CH), 37.5 (CH), 39.1 (CH2), 44.2 (CH), 52.3 (CH), 71.5 (CH), 79.9 (CH), 80.9 (C), 89.3 (CH), 103.1 (C), 126.7 (2xCH), 128.6 (2xCH), 129.9 (CH), 156.4 (C)
(1S,4R,5S,8R,9S,11S,12S)-1,5,9-Trimethyl-10,13,14,15-tetraoxatetracyclo[9.1.3.1.04,12.08,12]penta- decane-9α-carbaldehyde oxime (10)
To the aldehyde 7 (1.00 g, 3.5 mmol) in methanol (10 mL) was added hydroxylamine hydro-chloride (1.17 g, 16.5 mmol) and NaHCO3 (1.39 g, 16.5 mmol). The mixture was stirred overnight at room temperature and afterwards diluted with water and extracted with dichloromethane (3 x 30 mL). The organic layer was dried (MgSO4) and evaporated to give a yellow oil (0.84 g, 86 %); MS (ES): 298 (M+); 1H-NMR (300 MHz, CDCl3, δ): 0.99 (3H, d, J = 6 Hz, CH3-9), 1.26-2.34 (15H, m), 5.69 (1H, s, C-12), 7.48 (1H, s, OH), 8.05 (1H, s, H-10); 13C-NMR (75 MHz, CDCl3, δ): 14.7 (CH3), 19.8 (CH3), 24.6 (CH2), 25.3 (CH2), 25.5 (CH3), 32.5 (CH2), 36.9 (CH), 46.2 (CH), 48.2 (CH), 84.11 (C), 86.8 (C), 96.7 (CH), 103.7 (C), 159.6 (CH).
9-α-(5-Phenyl-4,5-dihydrooxazol-3-yl)-(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetra-oxatetracyclo[9.1.3.1.04,12.08,12]pentadecane (11)
To the oxime 10 (43 mg, 0.15 mmol) in CH2Cl2 (5 mL) was added styrene (0.17 mL, 1.45 mmol), a solution of sodium hypochlorite (0.5 mL, 13 % active chlorine, 6.8 mmol) and a few drops of triethylamine. The mixture was stirred overnight at room temperature and afterwards washed with water (2 times 10 mL). The organic layer was dried (MgSO4) and evaporated to give a residue that was chromatographed over silica gel with dichloromethane as the eluent. The oily product 11 was obtained as a mixture of diastereoisomers (0.023 g, 40 %); MS (ES): 400 (M+); Major isomer: 1H-NMR (400 MHz, CDCl3, δ): 1.01 (6H, d, J = 6 Hz, CH3-9), 1.26-2.34 (12H, m), 3.08 (1H, dd, J = 9, 17 Hz, isoxazole-H-4), 3.52 (1H, dd, J = 9, 17 Hz, isoxazole-H-4’), 3.65 (1H, dd, J = 9, 17 Hz, isoxazole-H-4’), 4.00 (1H, dd, J = 10, 17 Hz, isoxazole-H-4), 5.51 (1H, m, isoxazole-H-5’), 5.60 (1H, dd, J = 8, 10 Hz, isoxazole-H-5), 5.64 (2H, s, H-12), 7.45 (5H, m, Ph); 13C-NMR (100 MHz, CDCl3, δ): 19.8 (CH3), 23.9 (CH3), 24.6 (CH2), 25.5 (CH2), 25.7 (CH3), 32.4 (CH2), 37.0 (CH), 37.1 (CH2), 43.3 (CH2), 48.4 (CH), 52.1 (CH), 82.9 (CH), 83.2 (C), 96.4 (CH), 103.3 (C), 126.9 (2xCH), 127.9 (CH), 128.4 (2xCH), 166.5 (C). Minor isomer: 1H-NMR (400 MHz, CDCl3, δ): 0.99 (6H, d, J = 6 Hz, CH3-9), 1.26-2.34 (12H, m), 3.08 (1H, dd, J = 9, 17 Hz, isoxazole-H-4), 3.52 (1H, dd, J = 9, 17 Hz, isoxazole-H-4’), 3.65 (1H, dd, J = 9, 17 Hz, isoxazole-H-4’), 4.00 (1H, dd, J = 10, 17 Hz, isoxazole-H-4), 5.51 (1H, m, isoxazole-H-5’), 5.60 (1H, dd, J = 8, 10 Hz, isoxazole-H-5), 5.66 (2H, s, H-12), 7.34 (5H, m, Ph); 13C-NMR (100 MHz, CDCl3, δ): 19.8 (CH3), 23.5 (CH3), 24.6 (CH2), 25.5 (CH2), 25.7 (CH3), 32.4 (CH2), 37.0 (CH), 37.1 (CH2), 43.3 (CH2), 48.3 (CH), 52.5 (CH), 82.1 (CH), 83.2 (C), 96.4 (CH), 103.3 (C), 125.9 (2xCH), 127.9 (CH), 128.6 (2xCH), 166.5 (C).
9-α-(5-Phenyl-3-isoxazol-3-yl)-(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetraoxatetra-cyclo[9.1.3.1.04,12.08,12]pentadecane (12)
To the oxime 10 (200 mg, 0.67 mmol) in CH2Cl2 (20 mL) was added phenylacetylene (0.75 mL, 6.7 mmol), a solution of sodium hypochlorite (2 mL, 13 % active chlorine, 28 mmol) and a few drops of triethylamine. The mixture was stirred overnight at room temperature and afterwards washed with water (2 times 10 mL). The organic layer was dried (MgSO4) and evaporated to give a residue that was chromatographed over silica gel with 9/1 dichloromethane/ethyl acetate as the eluent. The product was obtained as a yellow oil (0.055 g, 21 %); MS (ES): 398 (M+); 1H-NMR (300 MHz, CDCl3, δ): 0.99 (3H, d, J = 6 Hz, CH3-9), 1.26-2.34 (18H, m), 5.77 (1H, s, H-12), 6.86 (1H, s, isoxazole-H), 7.41 – 7.70 (5H, m, Ph); 13C-NMR (75 MHz, CDCl3, δ): 14.5 (CH3), 19.8 (CH3), 25.4 (CH2), 25.6 (CH2), 25.7 (CH3), 32.3 (CH2), 36.9 (CH), 37.0 (CH2), 48.3 (CH), 52.7 (CH), 82.5 (C), 86.7 (C), 96.5 (CH), 98.6 (CH), 103.1 (C), 125.7 (2xCH), 128.0 (CH), 128.7 (2xCH), 129.6 (C), 168.1 (C), 172.5 (C).
2’-[(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetraoxatetracyclo[9.1.3.1.04,12.08,12]penta- decyl-9α]styrene (13)
To benzylidene triphenylphosphonium bromide (3.10 g, 7.10 mmol) in THF (20 mL) at -78 °C was added butyllithium (2M in hexane, 7.10 mmol). The mixture was stirred for one hour, brought to room temperature and the aldehyde 7 (0.5 g, 1.77 mmol) in THF (5 mL) was added dropwise. The mixture was stirred one hour under reflux conditions. Afterwards, the mixture was diluted with pentane (20 mL) and the resulting precipitate was removed by filtration. The organic layer was evaporated to give a residue that was chromatographed over silica gel with 85:15 CH2Cl2/petroleum ether as the eluent. The product was obtained as a yellow oil (0.40 g, 64 %); MS (ES): 357 (M+); 1H-NMR (300 MHz, CDCl3, δ): 0.99 (3H, d, J = 6 Hz, CH3-9), 1.26-2.34 (18H, m), 5.72 (1H, s, H-12), 6.51 (1H, d, J = 16 Hz, CH=CH-Ph), 6.78 (1H, d, J = 16 Hz, CH=CH-Ph), 7.27 (5H, m, Ph); 13C-NMR (75 MHz, CDCl3, δ): 20.0 (CH3), 23.5 (CH3), 24.8 (CH2), 25.6 (CH2), 26.2 (CH3), 31.1 (CH2), 37.1 (CH), 37.4 (CH2), 48.0 (CH), 53.6 (CH), 85.8 (C), 87.0 (C), 96.8 (CH), 103.5 (C), 125.9 (CH), 126.7 (CH), 127.0 (2xCH), 128.5 (2xCH), 137.9 (CH), 138.3 (C)
Ethyl-2-[(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetraoxatetracyclo [9.1.3.1.04,12.08,12]-pentadecyl-9α]acrylate (14)
To the aldehyde 7 (0.5 g, 1.77 mmol) in THF (15 mL) was added carbethoxymethylene triphenylphosphorane (2.35 g, 6.80 mmol). The mixture was stirred overnight at room temperature. The organic layer was evaporated to give a residue that was chromatographed over silica gel with 9:1 CH2Cl2/ethylacetate as the eluent. The product was obtained as white crystals (0.46 g, 77 %); MS (ES): 353 (M+); mp: 135-136 °C; 1H-NMR (300 MHz, CDCl3, δ): 0.99 (3H, d, J = 6 Hz, CH39), 1.26-2.34 (21H, m), 4.16 (2H, m, COOCH2CH3), 5.69 (1H, s, H-12), 6.06 (1H, d, J = 16 Hz, CH=CHCO), 7.22 (1H, d, J = 16 Hz, CH=CHCO); 13C-NMR (75 MHz, CDCl3, δ): 14.4 (CH3), 19.9 (CH3), 23.4 (CH3), 24.8 (CH2), 25.5 (CH2), 25.8 (CH3), 32.7 (CH2), 37.0 (CH), 37.2 (CH2), 48.7 (CH), 52.7 (CH), 60.3 (CH2), 85.1 (C), 86.4 (C), 96.8 (CH), 103.5 (C), 117.7 (CH), 154.3 (CH), 167.3 (CO)
3-[(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetraoxatetracyclo[9.1.3.1.04,12.08,12]-pentadecyl-9α]-propanol (15)
To the aldehyde 7 (0.025 g, 0.0087 mmol) in THF (5 mL) was added 5% Pd/C-catalyst (0.010 g). Air was removed and hydrogen was added. The mixture was stirred overnight, then filtered over Celite®. The organic layer was dilute with diethyl ether (5 mL), washed with brine, dried (MgSO4) and evaporated to give a residue that was chromatographed over silica gel with 9:1 CH2Cl2/ethyl acetate as the eluent. The product was obtained as yellow oil (0.020 g, 74 %); MS (ES): 313 (M+); 1H-NMR (300 MHz, CDCl3, δ): 0.90 (2H, m, CH2-10), 0.91 (3H, d, J = 6 Hz, CH3-9), 1.26-2.34 (20H, m), 4.21 (2H, dd, J=2, 6 Hz, CH2-OH), 5.62 (1H, s, H-12); 13C-NMR (75 MHz, CDCl3, δ): 18.7 (CH3), 22.0 (CH3), 24.0 (CH2), 25.6 (CH2), 26.5 (CH3), 29.4 (CH2), 31.0 (CH2), 33.0 (CH2), 35.2 (CH), 36.2 (CH2), 46.8 (CH), 51.1 (CH), 60.6 (CH2), 86.5 (C), 89.3 (C), 94.2 (CH), 103.7 (C)
(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetraoxatetracyclo[9.1.3.1.04,12.08,12]penta- decyl-9α-methanol (16)
To the aldehyde 7 (0.050 g, 0.18 mmol) in methanol (5 mL) was added sodium borohydride (0.027 g, 0.72 mmol) in small portions. The mixture was stirred for one hour at 0 °C. Afterwards, HCl (0.2 mL, 7.5 N) was added in a dropwise manner and the mixture was stirred for 30 minutes and then evaporated. The residue was diluted with water (10 mL) and extracted with diethyl ether (3 x 15 mL). The organic layer was washed with NaHCO3, dried (MgSO4) and evaporated. The product was obtained as white crystals (0.042 g, 84 %); MS (ES): 285 (M+); mp: 123 °C; 1H-NMR (300 MHz, CDCl3, δ): 0.99 (3H, d, J = 6 Hz, CH3-9), 1.26-2.34 (18H, m), 3.55 (1H, J = 8, 11 Hz, CH2OH ), 4.04 (1H, dd, J = 8, 11 Hz, CH2OH), 5.61 (1H, s, H-12); 13C-NMR (75 MHz, CDCl3, δ): 20.1 (CH3), 20.3 (CH3), 24.4 (CH2), 25.5 (CH2), 26.2 (CH3), 32.8 (CH2), 36.9 (CH), 37.4 (CH2), 48.1 (CH), 49.3 (CH), 69.2 (CH2), 85.4 (C), 89.8 (C), 96.2 (CH), 103.7 (C)
O-[(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetraoxatetracyclo[9.1.3.1.04,12.08,12]penta- decyl-9α-methyl]-N-phenylcarbamate (17)
To the alcohol 16 (0.082 g, 0.29 mmol) in pyridine (5 mL) was added phenyl isocyanate (0.078 mL, 0.72 mmol). The mixture was stirred for 14 hours by 60 °C. Afterwards, the mixture was extracted with petroleum ether (3 x 10 mL). The organic layer was washed with HCl (1N) and brine, dried (MgSO4) and evaporated to give a residue that was chromatographed over silica gel with 95:5 CH2Cl2/ethyl acetate as the eluent. The product was obtained as an oil (0.070 g, 67 %); MS (ES): 404 (M+); 1H-NMR (300 MHz, CDCl3, δ): 0.99 (3H, d, J=6, CH3-9), 1.26-2.34 (18H, m), 4.50 (2H, dd, J = 10, 11 Hz, CH2COONHPhH), 5.67 (1H, s, H-12), 6.80-7.40 (5H, m, Ph); 13C-NMR (75 MHz, CDCl3, δ): 17.7 (CH3), 19.7 (CH3), 24.5 (CH2), 25.4 (CH2), 26.2 (CH3), 32.7 (CH2), 36.9 (CH), 37.3 (CH2), 48.7 (CH), 49.0 (CH), 73.6 (CH2), 84.3 (C), 86.6 (C), 96.6 (CH), 103.8 (C), 121.3 (CH), 122.3 (2XCH), 123.4 (2xCH), 129.2 (C), 138.1 (CO)
O-[(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetraoxatetracyclo[9.1.3.1.04,12.08,12]-pentadecyl-9α-methyl]-N-allylthiocarbamate (18)
To the alcohol 16 (0.58 g, 2.04 mmol) in pyridine (25 mL) was added allyl isothiocyanate (0.50 g, 5.09 mmol). The mixture was stirred for 14 hours by 60 °C and then extracted with petroleum ether (3 x 30 mL). The organic layer was washed with HCl (1N) and brine, dried (MgSO4) and evaporated to give a residue that was chromatographed over silica gel with 95:5 CH2Cl2/ethyl acetate as the eluent. The product was obtained as an oil (0.42 g, 53 %); MS (ES): 384 (M+); 1H-NMR (300 MHz, CDCl3, δ): 0.99 (3H, d, J = 6 Hz, CH3-9), 1.26-2.34 (18H, m), 4.20 (2H, t, NHCH2CH=CH2), 4.78 (2H, m, H-10), 5.22 (2H, m, NHCH2CH=CH2), 5.65 (1H, s, H-12), 5.87 (1H, m, NHCH2CH=CH2), 6.45 (1H, s, NH); 13C-NMR (75 MHz, CDCl3, δ): 17.7 (CH3), 19.7 (CH3), 24.6 (CH2), 24.8 (CH2), 25.4 (CH3), 32.4 (CH2), 37.0 (CH), 37.2 (CH2), 47.9 (CH), 49.0 (CH), 49.1 (CH2), 75.5 (CH2), 85.5 (C), 86.5 (C), 96.6 (CH), 103.8 (C), 117.5 (CH2), 132.8 (CH), 157.9 (CS)
N-(4-methylphenyl)-(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetraoxatetracyclo[9.1.3.1.04,12.08,12]-pentadecyl-9α-methylamine (19)
To the aldehyde 7 (0.5 g, 1.77 mmol) in methanol (15 mL) was added p-toluidine (0.79 g, 7.08 mmol) and sodium cyanoborohydride (0.44g, 7.08 mmol). The mixture was stirred for 30 minutes at room temperature, afterwards KOH (0.4 g, 7.08 mmol) was added in small portions and stirring continued for 30 minutes. The mixture was diluted with NH4Cl-solution (10 mL) and extracted with ethyl acetate (3 x 15 mL). The organic layer was washed with brine, dried (MgSO4) and evaporated. The residue was chromatographed over silica gel with 95:5 CH2Cl2/ethyl acetate as the eluent. The product was obtained as a yellow oil (0.49 g, 74 %); MS (ES): 374 (M+); 1H-NMR (300 MHz, CDCl3, δ): 0.98 (3H, d, J = 6 Hz, CH3-9), 1.26-2.34 (21H, m), 3.43 (1H, dd, J = 4, 12 Hz, CH2-10), 3.58 (1H, dd, J = 3, 13 Hz, CH2-10), 5.63 (1H, s, H-12), 6.66 (2H, d, J = 8 Hz, o-Ph), 6.97 (2H, d, J = 8 Hz, m-Ph); 13C-NMR (75 MHz, CDCl3, δ): 14.3 (CH3), 21.2 (CH3), 24.4 (CH2), 25.5 (CH2), 26.4 (CH3), 31.0 (CH2), 36.9 (CH), 37.4 (CH2), 47.3 (CH), 53.6 (CH), 60.5 (CH2), 85.2 (C), 87.0 (C), 96.3 (CH), 103.8 (C), 113.7 (2xCH), 117.6 (CH3), 120.7 (C), 129.8 (2xCH), 171.3 (C)
(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetraoxatetracyclo[9.1.3.1.04,12.08,12]penta-decyl-9α-acid (20)
To the aldehyde 7 (0.250 g, 0.87 mmol) in 1,4-dioxane (10 mL) was added aminosulfonic acid (0.34 g, 3.55 mmol) and water (3.5 mL). The mixture was stirred for 20 hours at 0 °C, then NaClO2 (0.32 g, 3.55 mmol) and water (2.5 mL) were added. After stirring for 30 minutes Na2SO4 (0.250 g) was added and the mixture was diluted with water (10 mL). The mixture was extracted with diethyl ether (3 x 20 mL) and the organic layer was washed with brine, dried (MgSO4) and evaporated. The product was obtained as a brown solid (0.225 mg, 80 %); mp: 137 °C; 1H-NMR (300 MHz, CDCl3, δ): 1.01 (3H, d, J = 6 Hz, CH3-9), 1.26-2.34 (18H, m), 5.72 (1H, s, H-12), 9.47 (1H, s, COOH); 13C-NMR (75 MHz, CDCl3, δ): 19.9 (CH3), 21.1 (CH3), 24.6 (CH2), 25.0 (CH2), 25.7 (CH3), 32.4 (CH2), 36.7 (CH), 37.2 (CH2), 47.6 (CH), 50.5 (CH), 85.4 (C), 86.7 (C), 97.6 (CH), 103.7 (C), 175.6 (CO)
1’-[(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetraoxatetracyclo[9.1.3.1.04,12.08,12]penta-decyl-9α]-2-nitro-ethanol (21)
To the aldehyde 7 (0.050 g, 0.18 mmol) in ethanol (5 mL) was added nitromethane (0.038 mL, 0.72 mmol), the mixture was cooled to 0 °C and sodium methoxide (1 g) in methanol (5 mL) was added dropwise. The mixture was stirred for 18 minutes at 0 °C, then diluted with ethyl acetate and washed with brine. The organic layer was dried and evaporated and the product was obtained as an oil (0.040 g, 67 %); 1H-NMR (300 MHz, CDCl3, δ): 0.99 (3H, d, J = 6 Hz, CH3-9), 1.15 (3H, s, CH3-6), 1.26-2.34 (15H, m), 4.44 (1H, dd, J = 4, 10 Hz, H-10), 4.93 (1H, dd, J = 4, 10 Hz, CH2NO2), 5.12 (1H, dd, J = 4, 10 Hz, CH2NO2), 5.59 (1H, s, H-12); 13C-NMR (75 MHz, CDCl3, δ): 16.6 (CH3), 20.0 (CH3), 24.4 (CH2), 25.3 (CH2), 26.3 (CH3), 32.7 (CH2), 36.9 (CH), 47.6 (CH), 49.1 (CH), 60.6 (CH), 72.4 (CH2), 85.1 (C), 86.9 (C), 96.6 (CH), 103.9 (C)
(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetraoxatetracyclo[9.1.3.1.04,12.08,12]penta-decyl-9α-2’-aminoacetonitrile (22)
To the aldehyde 7 (1.4 g, 4.90 mmol) in methanol (25 mL) was added a solution of sodium cyanide (0.98 g, 20 mmol) in water (25 mL) and NH4Cl. The mixture was stirred for 2 hours at 45 °C. Afterwards the mixture was diluted with water (10 mL) and extracted with toluene (3 x 50 mL). The organic layer was washed with brine, dried (MgSO4) and evaporated. The product was obtained as an oil (1.2 g, 78 %); MS (ES): 309 (M+); 1H-NMR (300 MHz, CDCl3, δ): 1.01 (3H, d, J = 6 Hz, CH3-9), 1.26-2.34 (18H, m), 5.17 (1H, s, H-10), 5.63 (1H, s, H-12); 13C-NMR (75 MHz, CDCl3, δ): 18.2 (CH3), 19.8 (CH3), 24.2 (CH2), 25.2 (CH2), 25.8 (CH3), 32.5 (CH2), 37.1 (CH2), 47.2 (CH), 48.6 (CH), 48.9 (CH), 84.6 (C), 86.9 (C), 96.7 (CH), 103.9 (C)
(1S,4R,5S,8R,9S,11S,12S)-1,5,9-trimethyl-10,13,14,15-tetraoxatetracyclo[9.1.3.1.04,12.08,12]penta-decyl-9α-2’-hydroxyacetonitrile (23)
To the aldehyde 7 (0.050 g, 0.18 mmol) in methanol (5 mL) was added acetic acid (0.10 mL, 70%) and potassium cyanide (0.046 g, 0.71 mmol). The mixture was stirred for 18 hours at room temperature and then evaporated to give a residue that was diluted with ethyl acetate (10 mL) and the resulting precipitate was removed by filtration. The organic layer was evaporated to give a residue that was chromatographed over silica gel with 9:1 ethyl acetate/methanol as the eluent. The product was obtained as an oil (0.045 g, 83 %); MS (ES): 310 (M+); 1H-NMR (300 MHz, CDCl3, δ): 1.00 (3H, d, J = 6 Hz, CH3-9), 1.26-2.34 (18H, m), 5.16 (1H, s, H-10), 5.62 (1H, s, H-12); 13C-NMR (75 MHz, CDCl3, δ): 17.8 (CH3), 19.6 (CH3), 23.5 (CH2), 24.9 (CH2), 25.0 (CH3), 32.2 (CH2), 36.30 (CH), 36.7 (CH2), 46.6 (CH), 48.5 (CH), 86.6 (C), 89.2 (C), 95.9 (CH), 103.0 (C)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
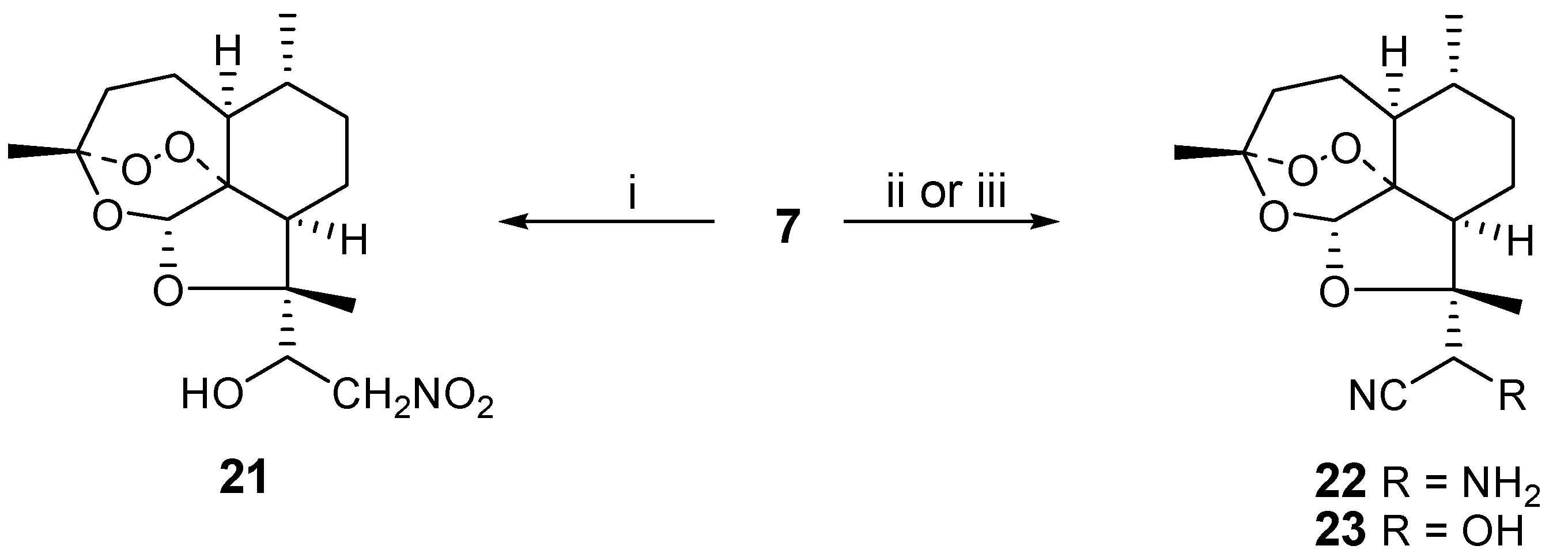{kind=link}