Synthesis of 2,5-Disubstituted Octahydroquinolin-4-ones via anIntramolecular Hetero Diels-Alder Reaction

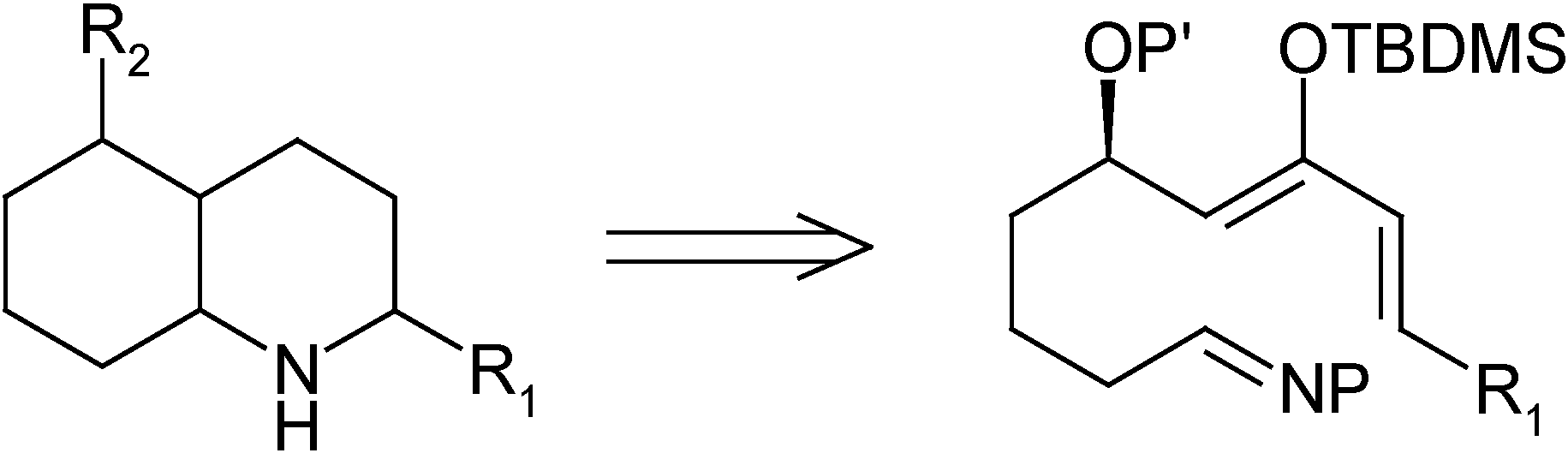{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
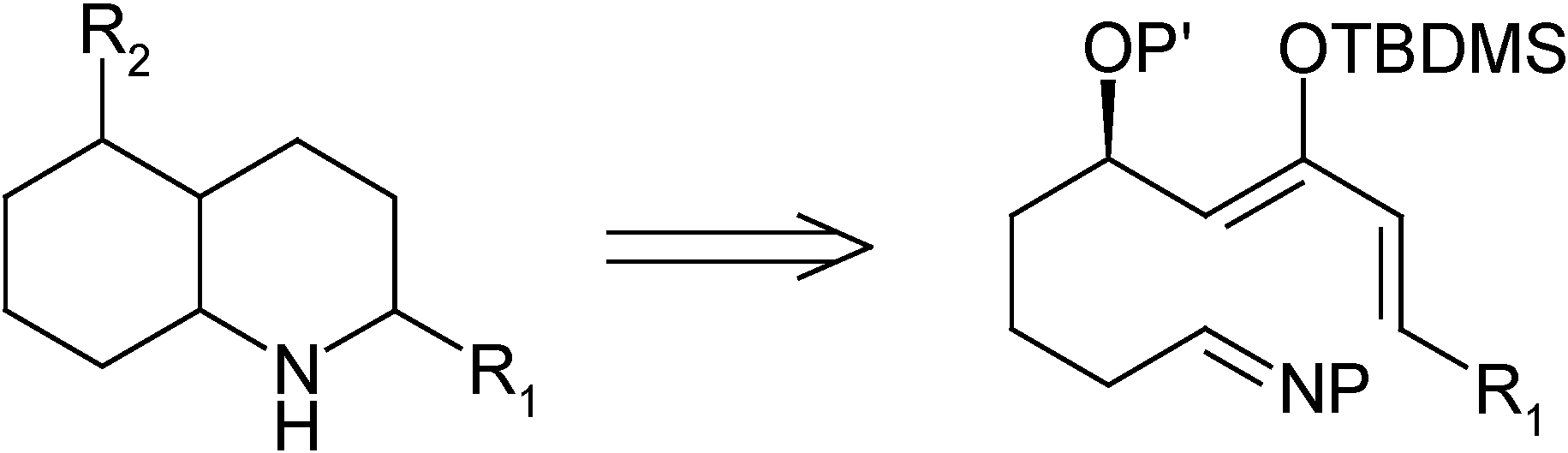
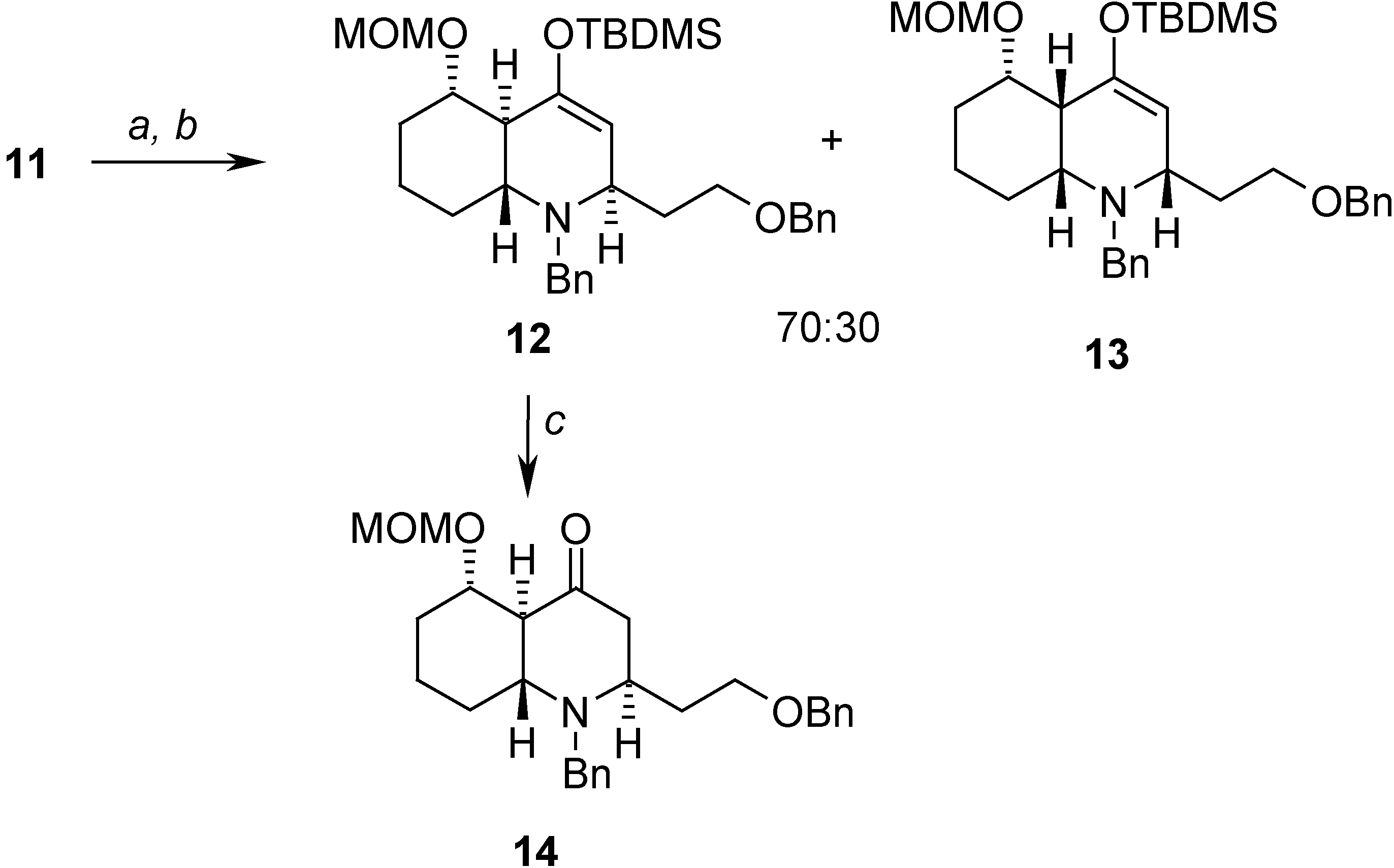
Results and Discussion
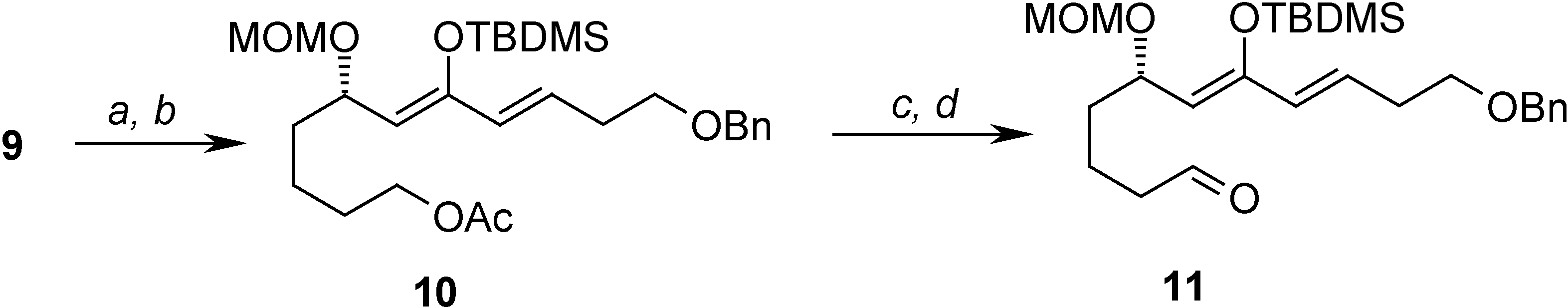
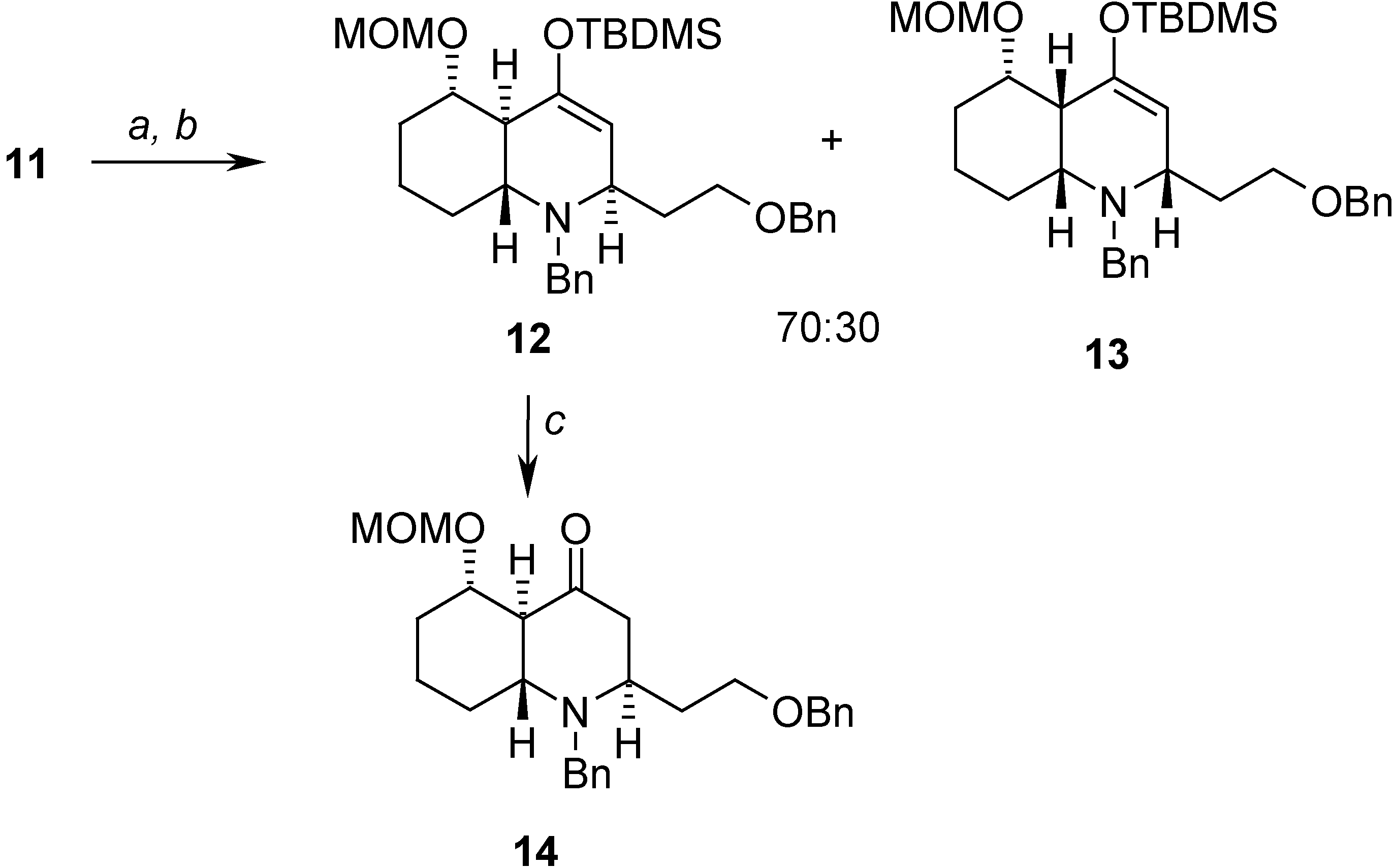
Conclusions
Experimental
General
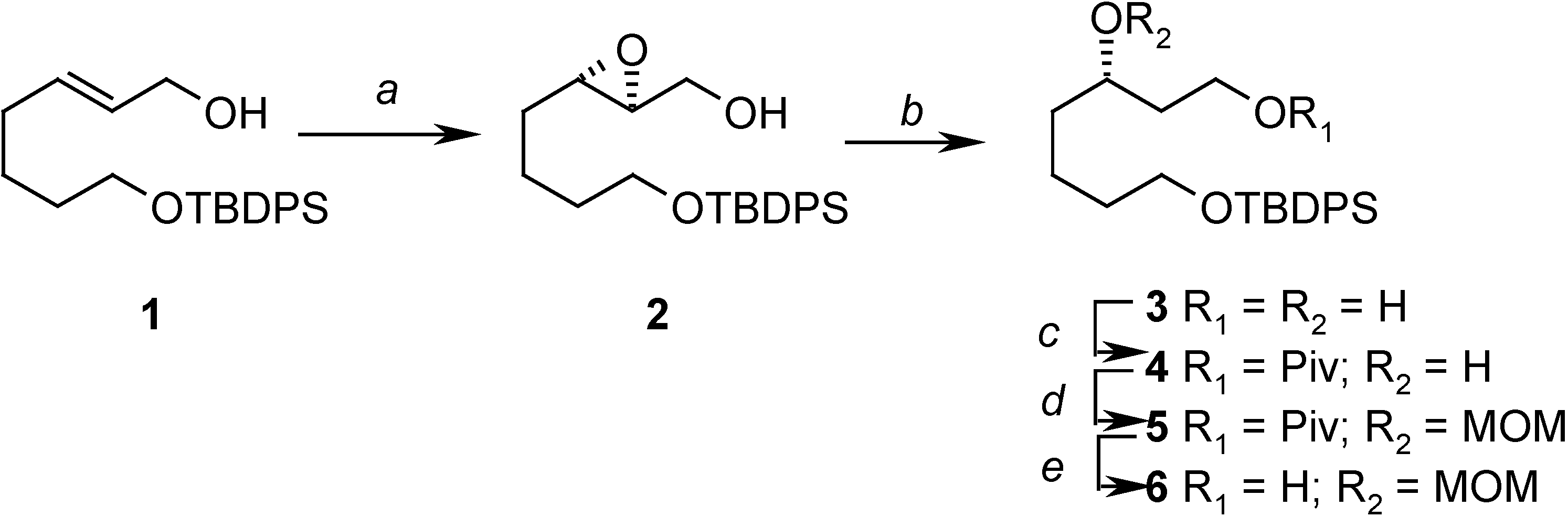
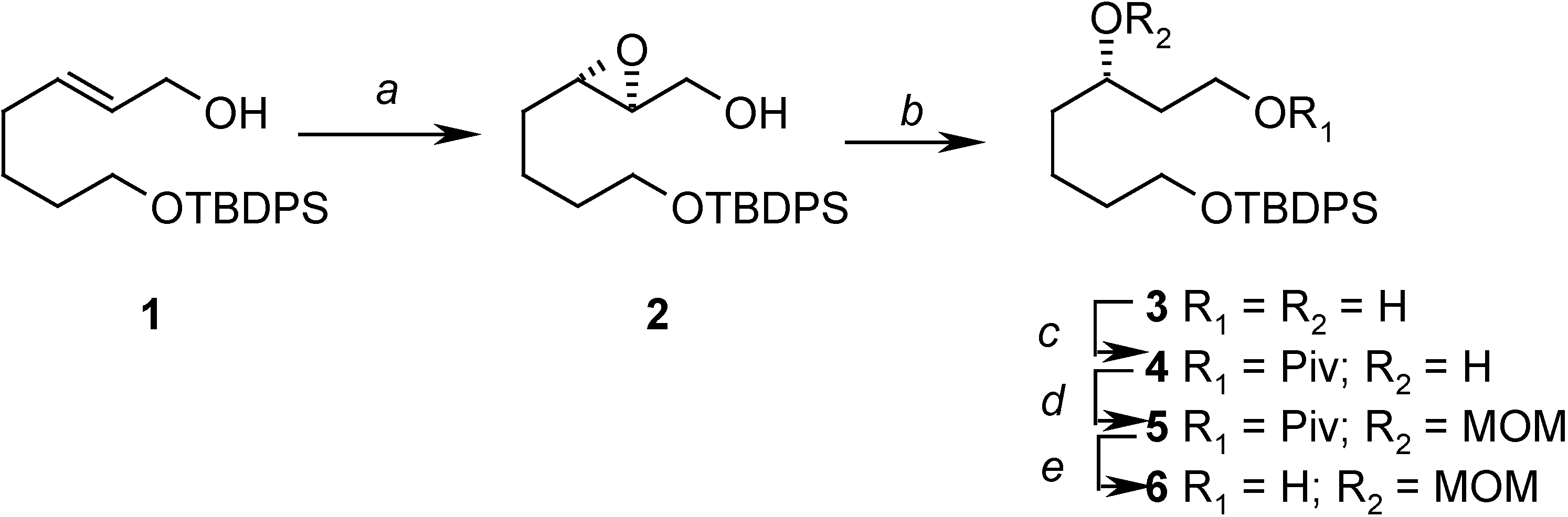
Preparation of ((2S, 3S)-3-(4-(tert-butyldiphenylsilyloxy)butyl)oxiran-2-yl)methanol (2)
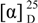 = -14.23 (c 2.08, CHCl3); 1H- NMR (CDCl3) δ 7.70-7.68 (m, 4H), 7.43-7.38 (m, 6H), 3.90 (dd, J = 1.7 Hz, 12.5 Hz, 1H), 3.69 (m, 2H), 3.60 (dd, J = 4.4 Hz, 12.5 Hz, 1H), 3.50 (bs, 1H), 2.95-2.91 (m, 2H), 1.64-1.57 (m, 6H), 1.08 (s, 9H); 13C-NMR (CDCl3) δ 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 63.6 (t), 61.8 (t), 58.6 (d), 56.0 (d), 32.2 (t), 31.3 (t), 26.9 (q), 22.3 (t), 19.2 (s).
= -14.23 (c 2.08, CHCl3); 1H- NMR (CDCl3) δ 7.70-7.68 (m, 4H), 7.43-7.38 (m, 6H), 3.90 (dd, J = 1.7 Hz, 12.5 Hz, 1H), 3.69 (m, 2H), 3.60 (dd, J = 4.4 Hz, 12.5 Hz, 1H), 3.50 (bs, 1H), 2.95-2.91 (m, 2H), 1.64-1.57 (m, 6H), 1.08 (s, 9H); 13C-NMR (CDCl3) δ 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 63.6 (t), 61.8 (t), 58.6 (d), 56.0 (d), 32.2 (t), 31.3 (t), 26.9 (q), 22.3 (t), 19.2 (s).Preparation of (S)-7-(tert-butyldiphenylsilyloxy)heptane-1,3-diol (3)
Preparation of (S)-7-(tert-butyldiphenylsilyloxy)-3-hydroxyheptyl pivalate (4)
= +1.08 (c 3.5, CHCl3); 1H-NMR (CDCl3) δ 7.71-7.76 (m, 4H), 7.45-7.38 (m, 6H), 4.35 (m, 1H), 4.16 (m, 1H), 3.71 (t, J = 6.0 Hz, 2H), 3.66 (m, 1H), 2.45 (s, 1H), 1.84-1.80 (m, 1H), 1.70-1.52 (m, 7H), 1.20 (s, 9H), 1.04 (s, 9H); 13C-NMR (CDCl3) δ 179.0 (s), 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 68.6 (d), 63.8 (t), 61.7 (t), 38.8 (s), 37.1 (t), 36.4 (t), 32.0 (t), 27.2 (q), 26.9 (q), 21.9 (t), 19.2 (s); EIMS m/z (%): 413 (M+-tBu, 5.0), 395 (M+-tBu-H2O, 2.0); HRMS (EI): C24H33O4Si (M+-tBu): 413.2148, found 413.2174.Preparation of (S)-7-(tert-butyldiphenylsilyloxy)-3-(methoxymethoxy)heptyl pivalate (5)
= +12.13 (c 2.06, CHCl3); 1H-NMR (CDCl3) δ 7.69-7.67 (m, 4H), 7.43-7.37 (m, 6H), 4.64 (d, J = 6.9 Hz, 1H), 4.62 (d, J = 6.9 Hz, 1H), 4.18 (m, 2H), 3.66 (m, 3H), 3.36 (s, 3H), 1.85-1.40 (m, 8H), 1.21 (s, 9H), 1.06 (s, 9H); 13C-NMR (CDCl3) δ 178.5 (s), 135.6 (d), 134.0 (s), 129.5 (d), 127.6 (d), 95.6 (t), 74.4 (d), 63.7 (t), 61.2 (t), 55.6 (q), 38.7 (s), 34.3 (t), 33.4 (t), 32.6 (t), 27.2 (q), 26.9 (q), 21.5 (t), 19.2 (q); EIMS m/z (%): 457 (M+-tBu, 3.1); HRMS (EI): C26H37O5Si (M+-tBu): 457.2410, found 457.2404.Preparation of (S)-7-(tert-butyldiphenylsilyloxy)-3-(methoxymethoxy)heptan-1-ol (6)
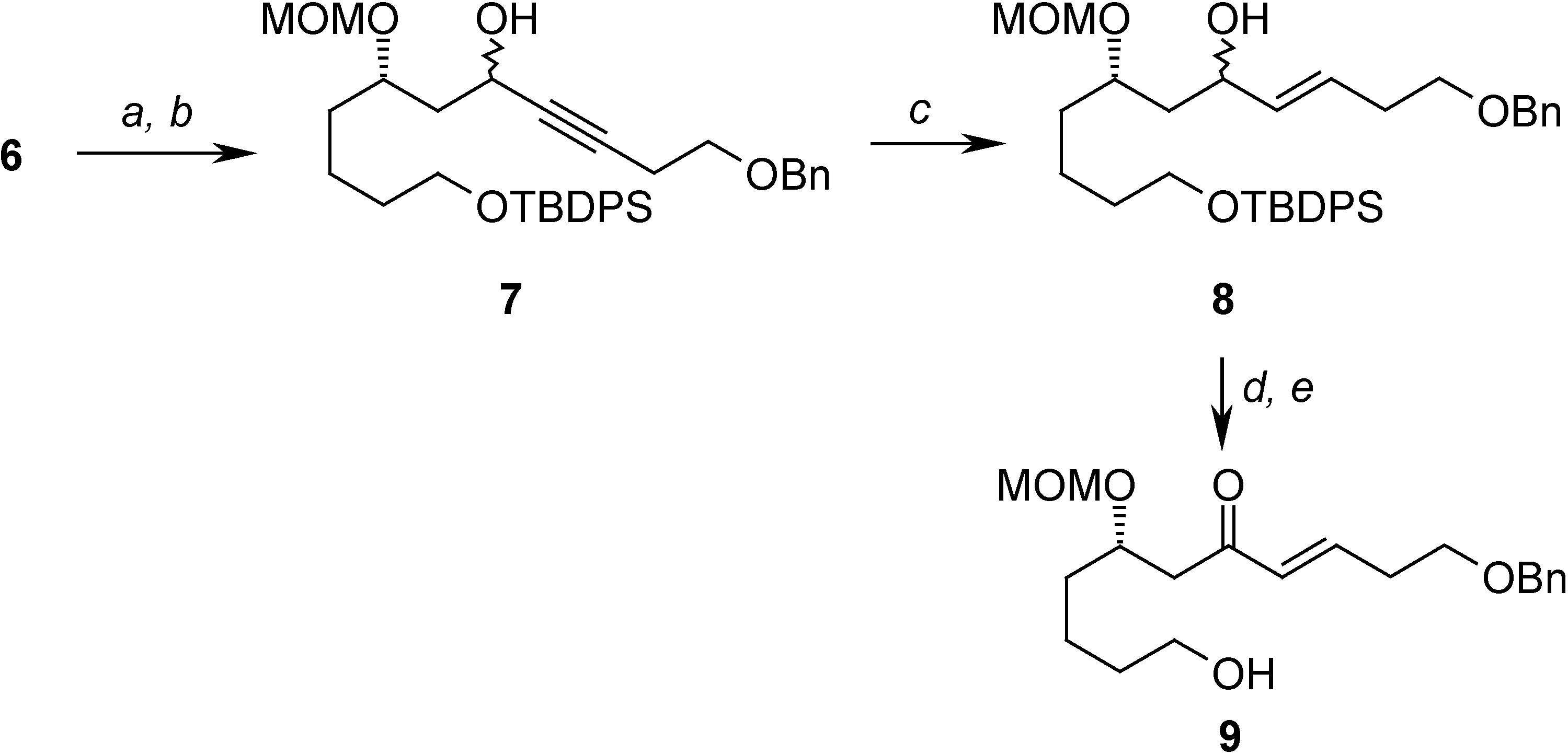
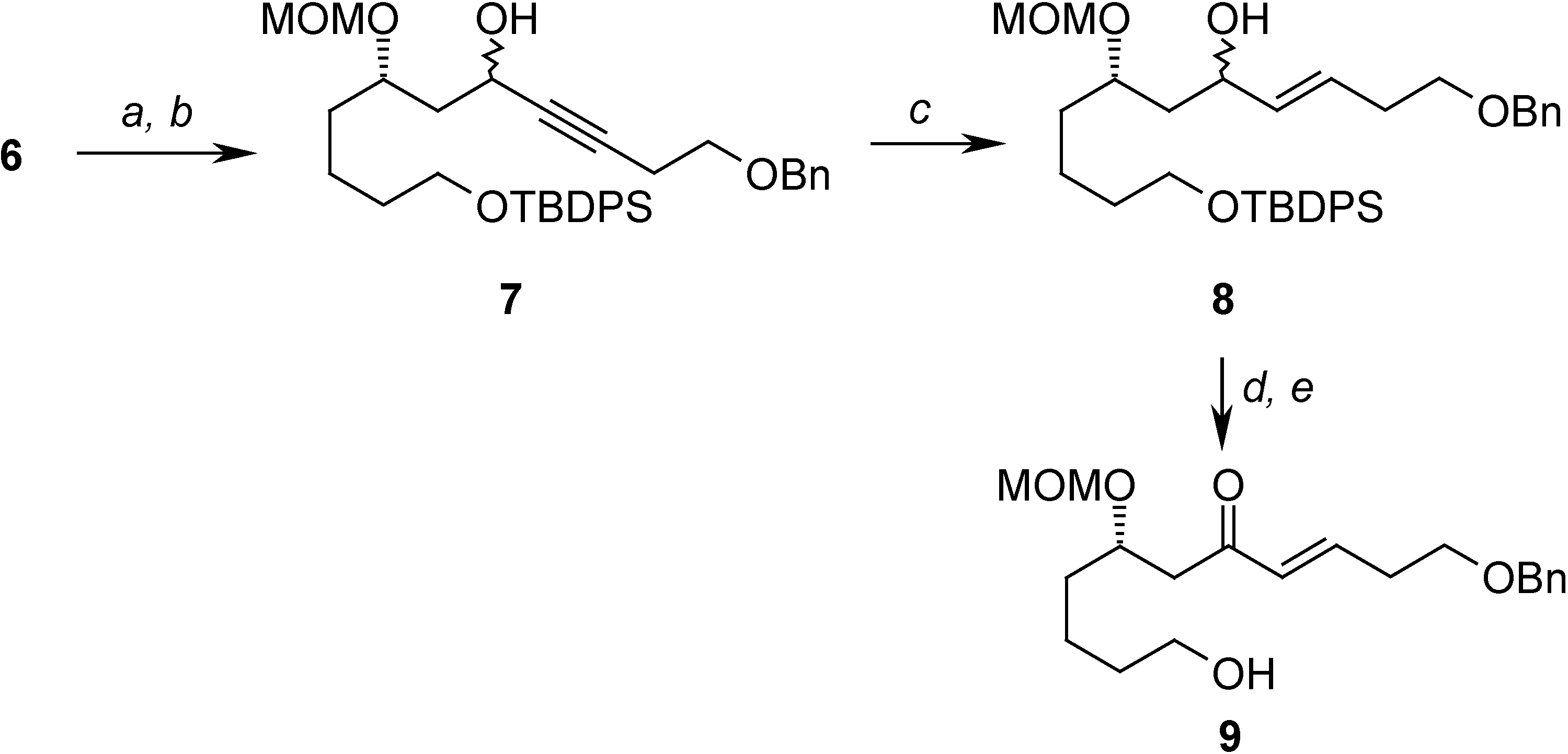
= +22.67 (c 2.67, CHCl3); 1H-NMR (CDCl3) δ 7.68-7.61 (m, 4H), 7.43-7.36 (m, 6H), 4.67 (d, J = 6.8 Hz, 1H), 4.65 (d, J = 6.8 Hz, 1H), 3.81-3.65 (m, 4H), 3.39 (s, 3H), 1.67-1.40 (m, 8H), 1.04 (s, 9H); 13C-NMR (CDCl3) δ 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 95.9 (t), 76.4 (d), 63.7 (t), 59.8 (t), 55.7 (q), 35.6 (t), 34.4 (t), 32.6 (t), 26.9 (q), 21.6 (t), 19.2 (s); EIMS m/z (%): 342 (M+-tBu-OCH3, 1.85).(7S)-1-(benzyloxy)-11-(tert-butyldiphenylsilyloxy)-7-(methoxymethoxy)undec-3-yn-5-ol (7)
(7S,E)-1-(benzyloxy)-11-(tert-butyldiphenylsilyloxy)-7-(methoxymethoxy)undec-3-en-5-ol (8)
(S,E)-1-(benzyloxy)-11-hydroxy-7-(methoxymethoxy)undec-3-en-5-one (9)
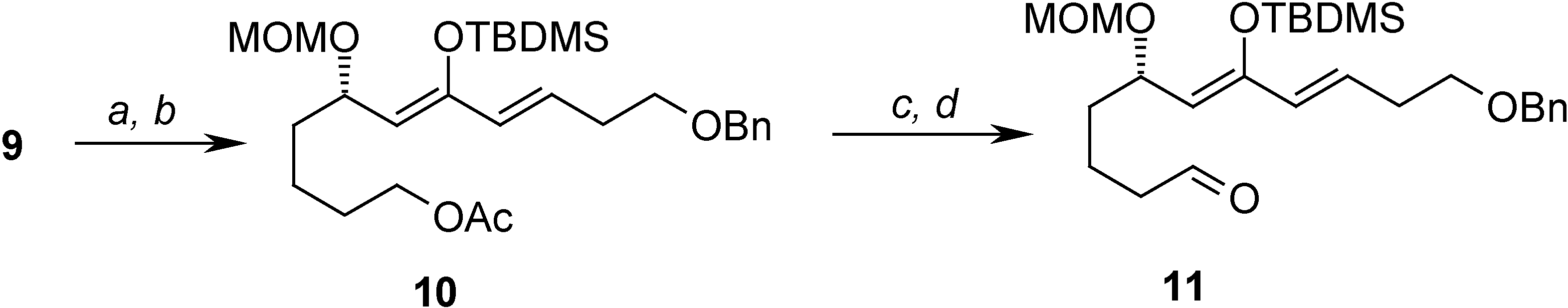
= +1.44 (c 3.60, CHCl3); 1H-NMR (CDCl3) δ 7.37-7.26 (m, 5H), 6.86 (dt, J = 6.9 Hz, 15.9 Hz, 1H), 6.17 (d, J = 15.9 Hz, 1H), 4.61 (d, J = 6.9 Hz, 1H), 4.63 (d, J = 6.9 Hz, 1H), 4.52 (s, 2H), 4.09 (m, 1H), 3.61 (m, 4H), 3.32 (s, 3H), 2.90 (dd, J = 6.9 Hz, 10.0 Hz, 1H), 2.62 (dd, J = 5.9, 10.0 Hz, 1H), 2.55-2.50 (m, 2H), 1.68-1.42 (m 6H); 13C-NMR (CDCl3) δ 198.3 (s), 144.2 (d), 137.7 (s), 131.9 (d), 128.2 (d), 127.5 (d), 127.4 (d), 95.8 (t), 74.1 (d), 72.8 (t), 68.3 (t), 62.2 (t), 55.3 (d), 45.0 (t), 34.5 (t), 32.6 (t), 32.3 (t), 21.1 (t).Preparation of (S,6Z,8E)-11-(benzyloxy)-7-(tert-butyldimethylsilyloxy)-5-(methoxymethoxy)undeca-6,8-dienyl acetate (10)
= -0.85 (c 2.60, CHCl3); 1H-NMR (CDCl3) δ 7.36 (m, 5H), 6.85 (dt, J = 6.8 Hz, 15.9 Hz, 1H), 6.17 (d, J = 15.9 Hz, 1H), 4.64 (d, J = 6.8 Hz, 1H), 4.58 (d, J = 6.8 Hz, 1H), 4.51 (s, 2H), 4.05 (m, 3H), 3.58 (t, J = 6.3 Hz, 2H), 3.31 (s, 3H), 2.88 (dd, J = 6.8 Hz, 15.9 Hz, 1H), 2.61-2.48 (m, 3H), 2.02 (s, 3H), 1.75-1.38 (m, 6H); 13C-NMR (CDCl3) δ 198.3 (s), 171.1 (s), 144.3 (d), 138.0 (s), 132.1 (d), 128.4 (d), 127.7 (d), 96.1 (t), 74.2 (d), 73.0 (t), 68.2 (t), 64.3 (t), 55.5 (q), 45.3 (t), 34.7 (t), 32.9 (t), 28.6 (t), 21.7 (t), 20.9 (q). tert-Butyldimethylsilyl triflate (0.51 mL, 2.23 mmol) was added to a stirred solution of the previously prepared compound (624 mg, 1.59 mmol) in THF (16 mL) at -78 °C under argon. After 15 min a solution of LDA (0.24 M in THF, 7.9 mL, 1.9 mmol) was added. The reaction mixture was stirred at -78 °C for 30 min, and then a saturated aqueous solution of NH4Cl (15 mL) was added. The reaction was allowed to reach room temperature and extracted with Et2O. The combined organic phases were dried (MgSO4), concentrated, and chromatographed on silica gel (80:20 Hex-EtOAc) yielding 500 mg (62%) of 10 as a colorless oil; = -49.39 (c 4.90, CHCl3); 1H-NMR (C6D6) δ 7.37-7.10 (m, 5H), 6.02 (dt, J = 6.7 Hz, 15.9 Hz, 1H), 5.88 (d, J = 15.9 Hz, 1H), 4.86 (d, J = 6.6, 1H), 4.75 (m, 2H), 4.46 (d, J = 6.6 Hz, 1H), 4.28 (s, 2H), 4.04-3.96 (m, 2H), 3.30 (t, J = 6.4 Hz, 2H), 3.24 (s, 3H), 2.30-2.26 (m, 2H), 1.67 (s, 3H), 1.66-1.50 (m, 6H), 0.99 (s, 9H), 0.20 (s, 3H), 0.14 (s, 3H); 13C-NMR (CDCl3) δ 169.5 (s), 150.1 (s), 138.5 (s), 129.7 (d), 128.2 (d), 127.9 (d), 127.4 (d), 113.4 (d), 93.2 (t), 72.4 (t), 69.5 (d), 69.2 (t), 63.7 (t), 54.5 (q), 35.3 (t), 32.6 (t), 28.5 (t), 25.6 (q), 21.9 (t), 19.9 (q), 18.0 (s), -4.1 (q).Preparation of (S,6Z,8E)-11-(benzyloxy)-7-(tert-butyldimethylsilyloxy)-5-(methoxymethoxy)undeca-6,8-dienal (11)
Hetero Diels-Alder Reaction
= -23.08 (c 1.30, CHCl3); 1H-NMR (CDCl3) δ 7.32-7.13 (m, 10H), 4.81 (m, 2H), 4.61 (d, J = 6.9 Hz, 1H), 4.13 (AB system, J = 11.6, 8.8 Hz, 2H), 3.84 (d, J = 13.3 Hz, 1H), 3.43 (m, 1H), 3.38 (s, 3H), 3.26 (d, J = 13.3 Hz, 1H), 3.22 (m, 1H), 3.08 (m, 1H), 2.67 (m, 1H), 2.15 (m, 2H), 1.80 (m, 1H), 1.75-1.69 (m, 3H), 1.60-1.55 (m, 2H), 1.44 (m, 2H), 0.98 (s, 9H), 0.22 (s, 3H), 0.20 (s, 3H); 13C-NMR (CDCl3) δ 151.5 (s), 140.8 (s), 138.6 (s), 129.1 (d), 128.2 (d), 128.0 (d), 127.8 (d), 127.3 (d), 126.7 (d), 108.0 (d), 97.5 (t), 80.5 (d), 72.7 (t), 68.0 (t), 56.2 (d), 55.3 (q), 53.7 (d), 51.1 (t), 44.2 (d), 35.4 (t), 34.4 (t), 30.4 (t), 26.2 (q), 23.0 (t), 18.7 (s), -4.9 (q).Preparation of (2R,4aS,5S,8aR)-1-benzyl-2-(2-benzyloxy)ethyl)-5-(methoxymethoxy)octahydro-quinolin-4(1H)-one (14)
Acknowledgments
References and Notes
- Daly, J.W.; Spande, T.F.; Garraffo, H.M. Alkaloids from amphibian skin. A tabulation of over eight-hundred compounds. J. Nat. Prod. 2005, 68, 1556–1575. [Google Scholar]
- Toyooka, N.; Nemoto, H. Synthetic studies on dart-poison frog alkaloids. Rec. Res. Dev. Org. Chem. 2002, 6, 611–624. [Google Scholar]
- Banner, E.J.; Stevens, E.D.; Trudell, M.L. Stereoselective synthesis of the cis-275B decahydroquinoline ring system. Tetrahedron Lett. 2004, 45, 4411–4414. [Google Scholar] Abe, H.; Aoyagi, S.; Kibayashi, C. First total synthesis of the marine alkaloids (±)-Fasicularine and (±)-Lepadiformine based on stereocontroled intramolecular acylnitroso-Diels-Alder. J. Am. Chem. Soc. 2000, 122, 4583–4592. [Google Scholar] Comins, D.L.; Al-Awar, R.S. An intramolecular Diels-Alder / retro-Mannich approach to the cis-perhydroquinoline ring system. Model studies toward the synthesis of Lycopodium alkaloids. J. Org. Chem. 1992, 57, 4098–4103. [Google Scholar] Oppolzer, W.; Flaskamp, E.; Bieber, L.W. Efficient asymmetric synthesis of pumiliotoxin C via intramolecular [4+2] cycloaddition 141-145. Helv. Chim. Acta 2001, 84, 141–145. [Google Scholar]
- Grieco, P.A.; Parker, D.T. Octahydroquinoline synthesis via immonium ion based Diels-Alder chemistry. Synthesis of (-)-8a-epipumiliotoxin C. J. Org. Chem. 1988, 53, 3658–3662. [Google Scholar]
- Adam, W.; Gläser, J.; Peters, K.; Prein, M. Highly like-selective [4+2] cycloadditions of chiral dienols: The importance of 1,3-allylic strain in the hydroxy-directed stereocontrol. J. Am. Chem. Soc. 1995, 117, 9190–9193. [Google Scholar] Roush, W.R.; Kageyama, M.; Riva, R.; Brown, B.B.; Warmus, J.S.; Moriarty, K.J. Enantioselective synthesis of the bottom half of chlorothricolide. 3. Studies of the steric directing group strategy for stereocontrol in intramolecular Diels-Alder reactions. J. Org. Chem. 1991, 56, 1192–1210. [Google Scholar]
- Katsuki, T.; Sharpless, K.B. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar] [CrossRef]
- Murphy, J.A.; Rasheed, F.; Roome, S.J.; Scott, K.A.; Lewis, N. Intramolecular termination of radical polar crossover reactions. J. Chem. Soc. Perkin Trans. 1. 1998, 15, 2331–2340. [Google Scholar] [CrossRef]
- Stonehouse, J.; Adell, P.; Keeler, J.; Shaka, A.J. Ultrahigh-quality NOE spectra. J. Am. Chem. Soc. 1994, 116, 6037–6038. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds described in this paper are available from authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Ruiz, J.M.; Afonso, M.M.; Palenzuela, J.A. Synthesis of 2,5-Disubstituted Octahydroquinolin-4-ones via anIntramolecular Hetero Diels-Alder Reaction. Molecules 2007, 12, 194-204. https://doi.org/10.3390/12020194
Ruiz JM, Afonso MM, Palenzuela JA. Synthesis of 2,5-Disubstituted Octahydroquinolin-4-ones via anIntramolecular Hetero Diels-Alder Reaction. Molecules. 2007; 12(2):194-204. https://doi.org/10.3390/12020194
Chicago/Turabian StyleRuiz, Juan M., Maria M. Afonso, and J. Antonio Palenzuela. 2007. "Synthesis of 2,5-Disubstituted Octahydroquinolin-4-ones via anIntramolecular Hetero Diels-Alder Reaction" Molecules 12, no. 2: 194-204. https://doi.org/10.3390/12020194
APA StyleRuiz, J. M., Afonso, M. M., & Palenzuela, J. A. (2007). Synthesis of 2,5-Disubstituted Octahydroquinolin-4-ones via anIntramolecular Hetero Diels-Alder Reaction. Molecules, 12(2), 194-204. https://doi.org/10.3390/12020194