Hantzsch Synthesis of 2,6-Dimethyl-3,5-dimethoxycarbonyl-4-(o-methoxyphenyl)-1,4-dihydropyridine; a Novel Cyclisation Leading to an Unusual Formation of 1-Amino-2-methoxycarbonyl-3,5-bis(o-methoxyphenyl)-4-oxa-cyclohexan-1-ene

Abstract
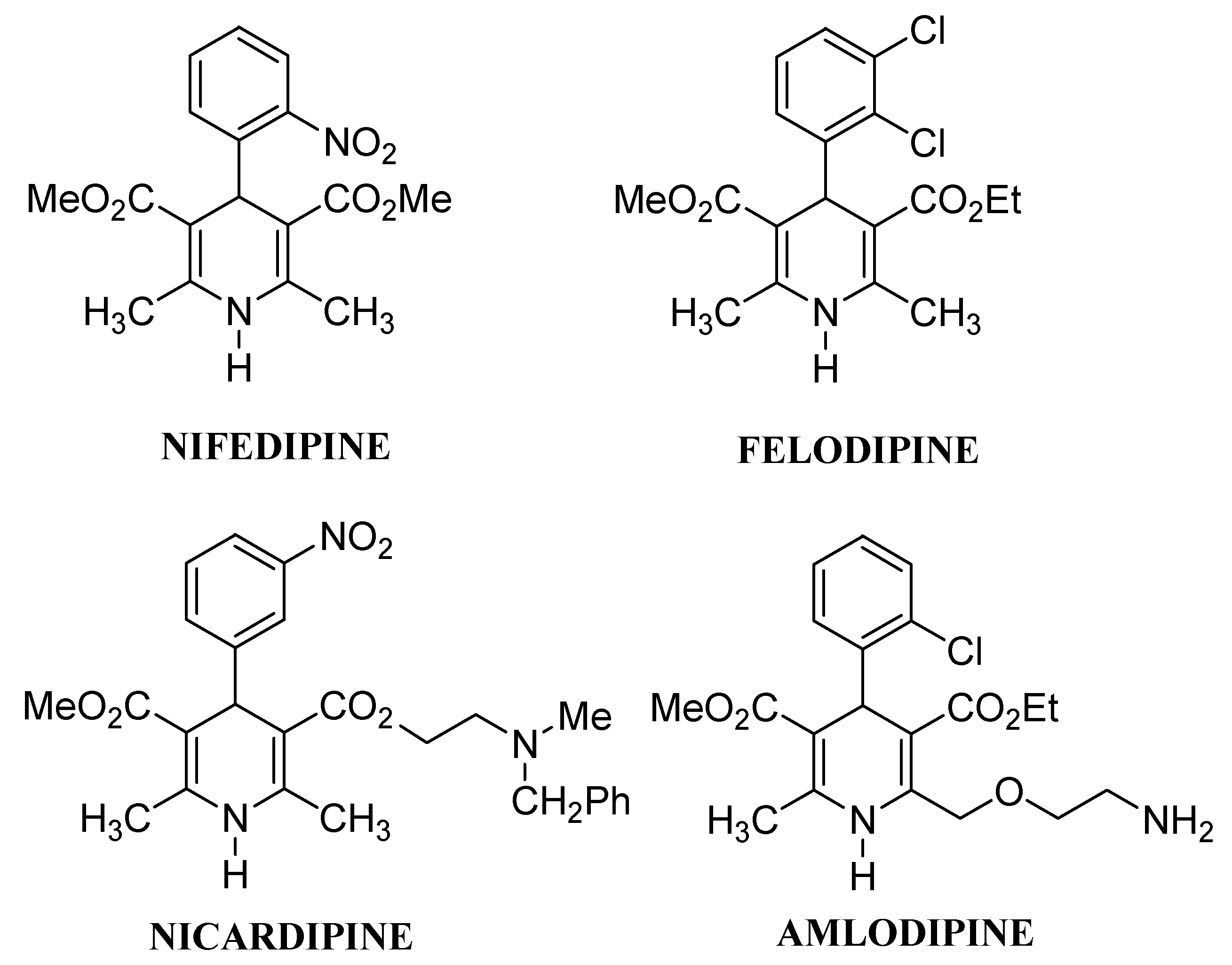
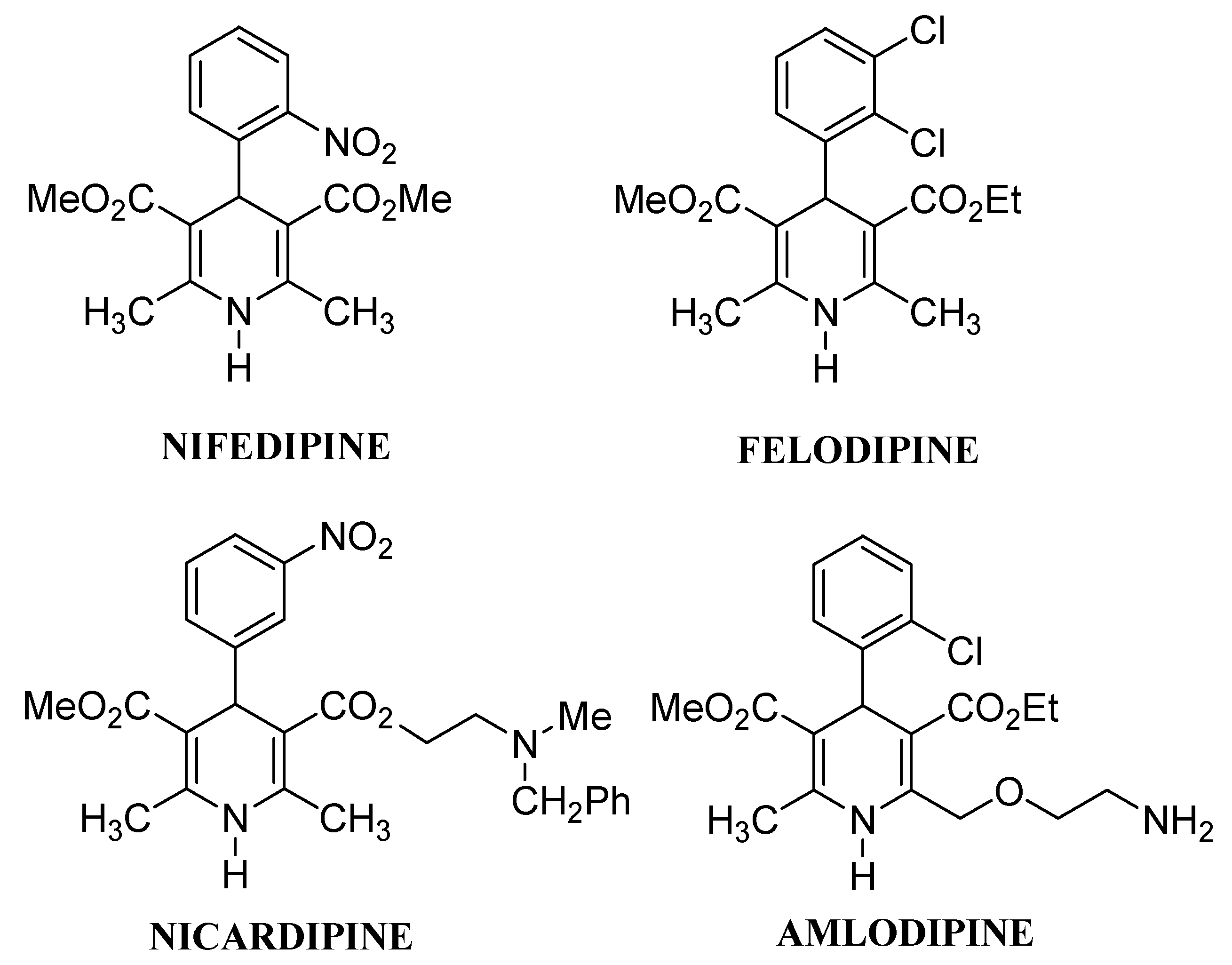
:Introduction
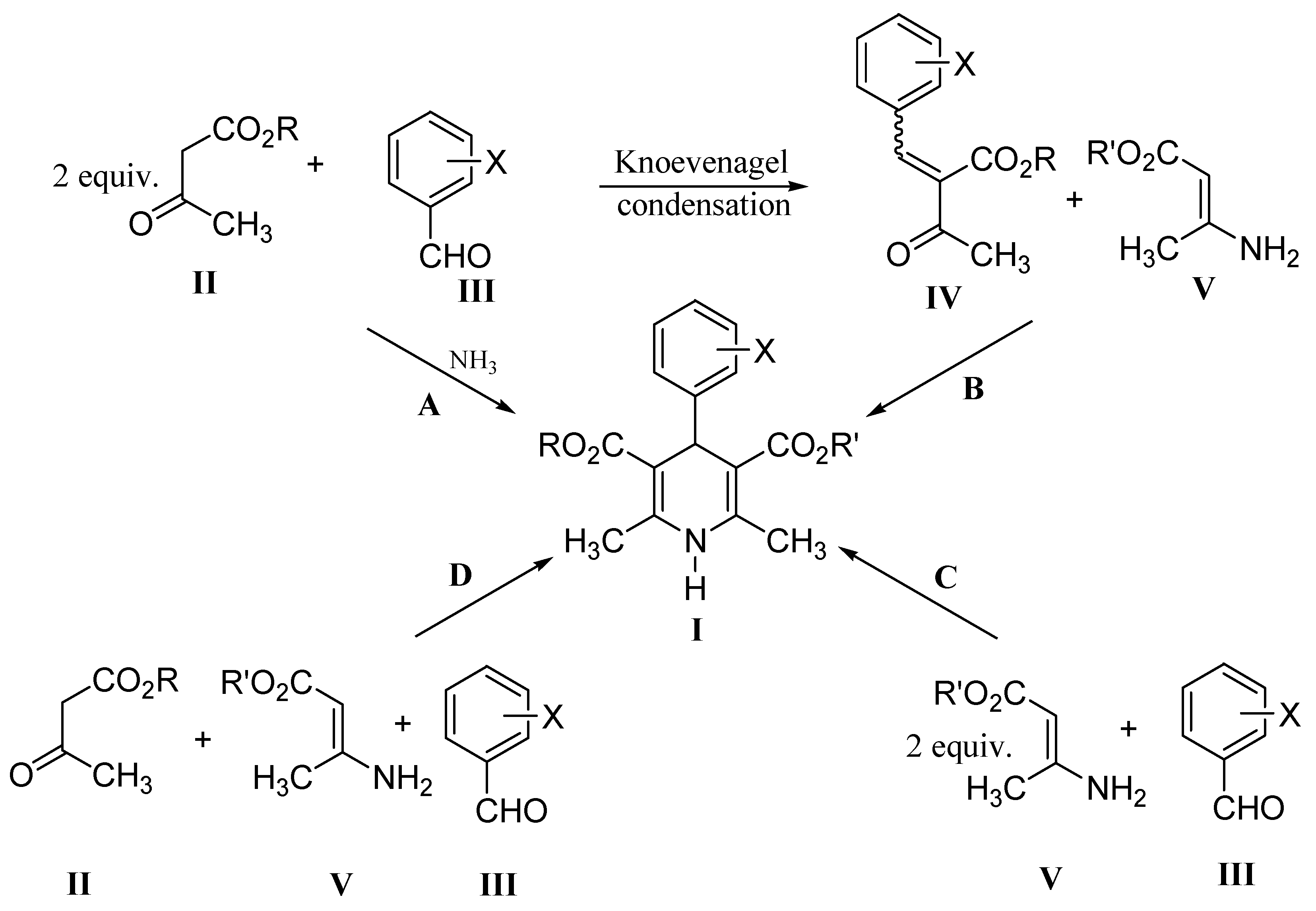
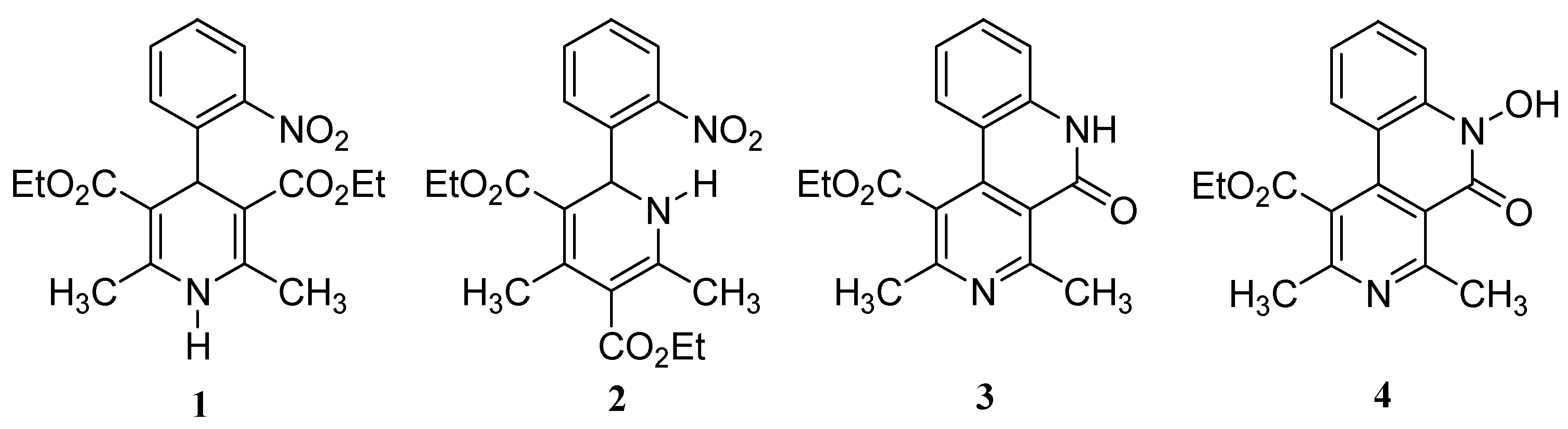
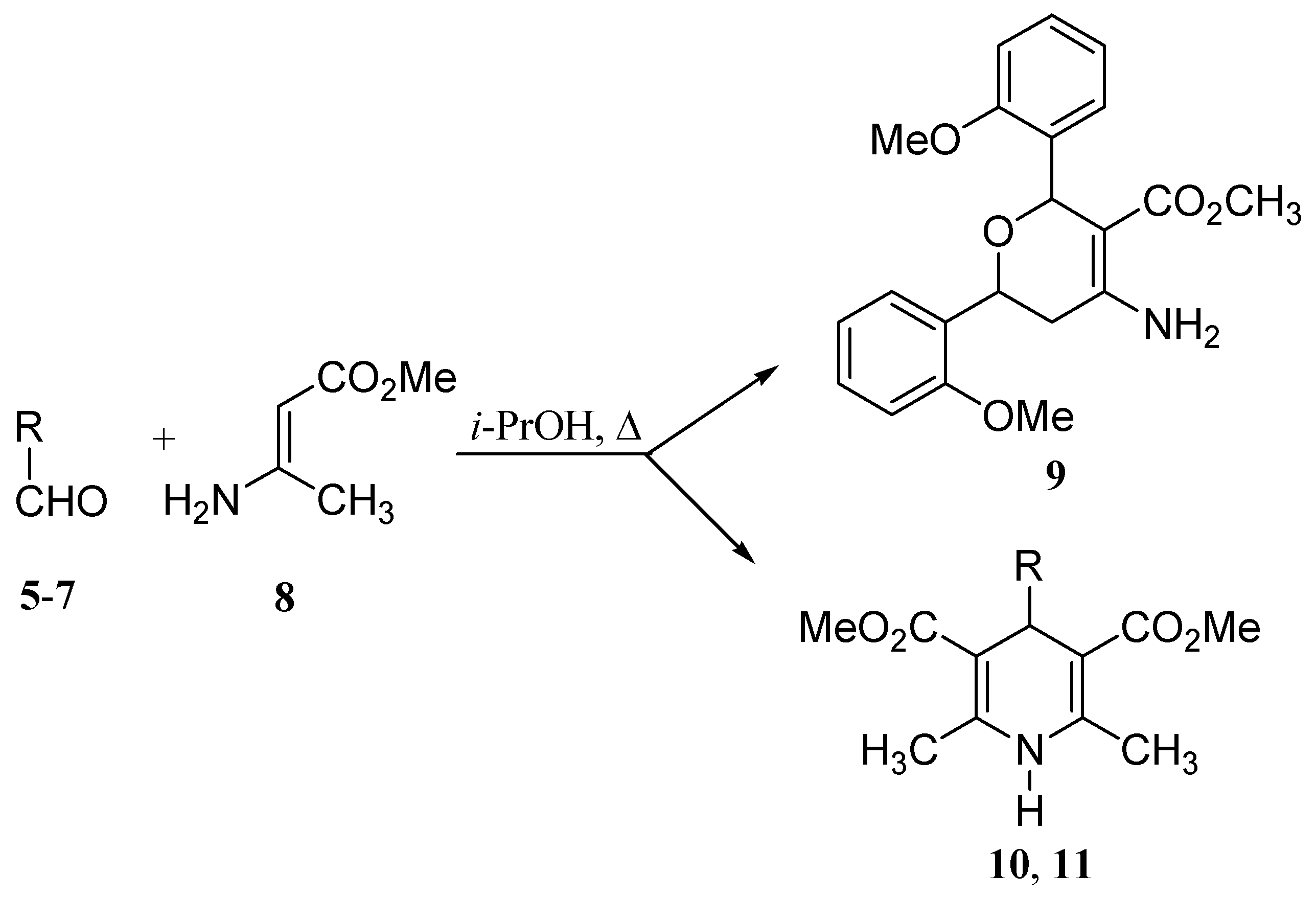
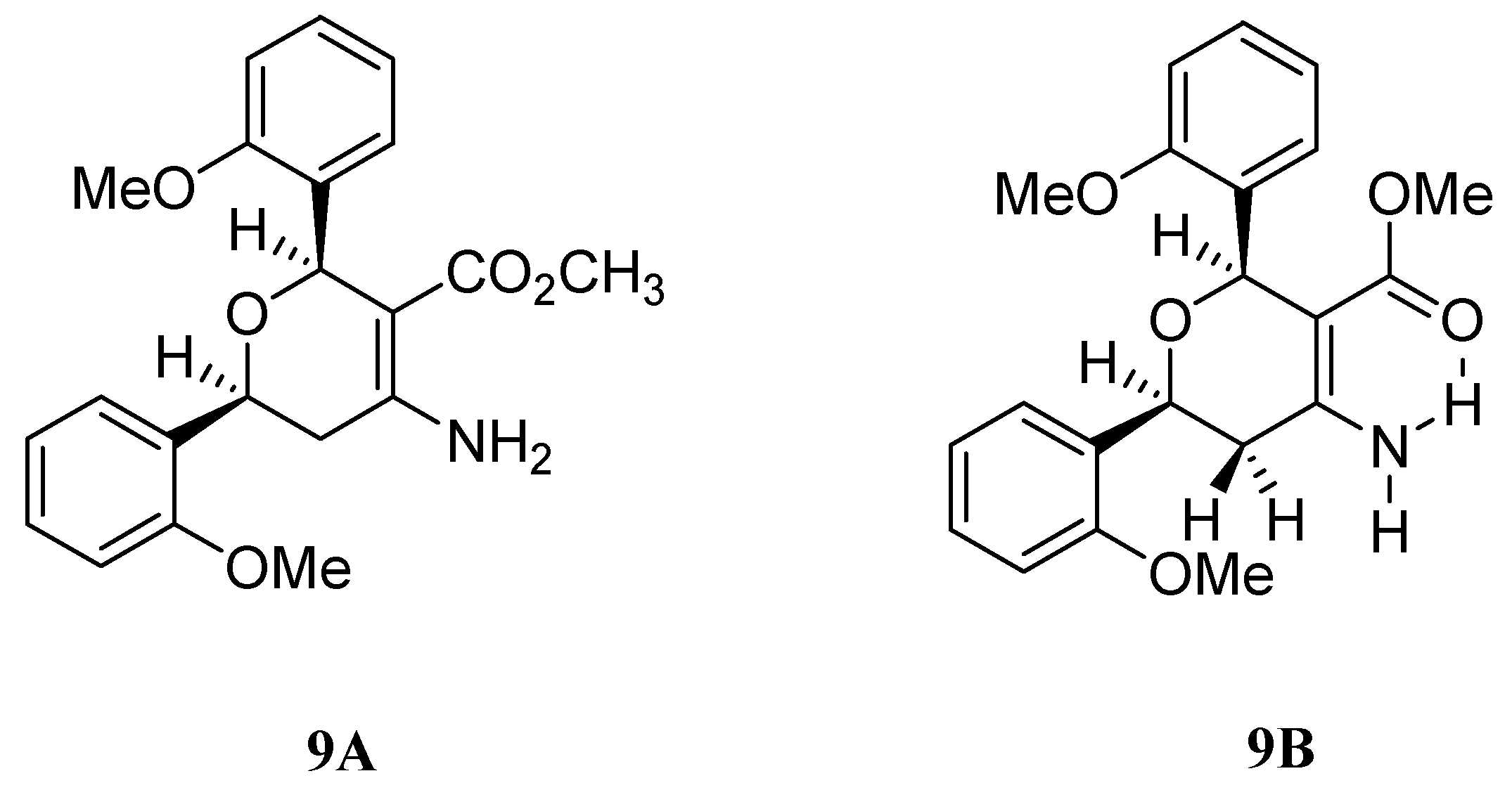
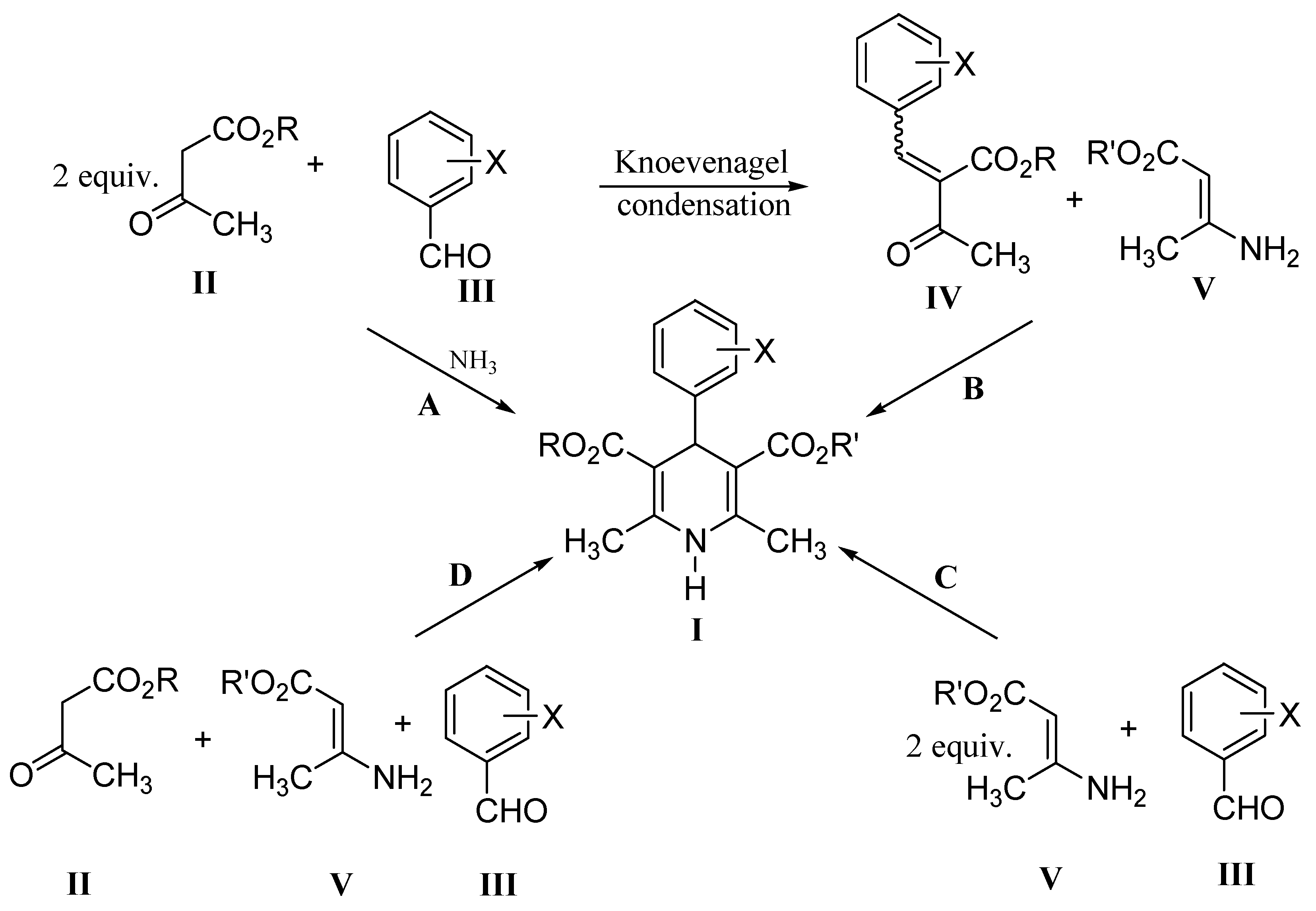
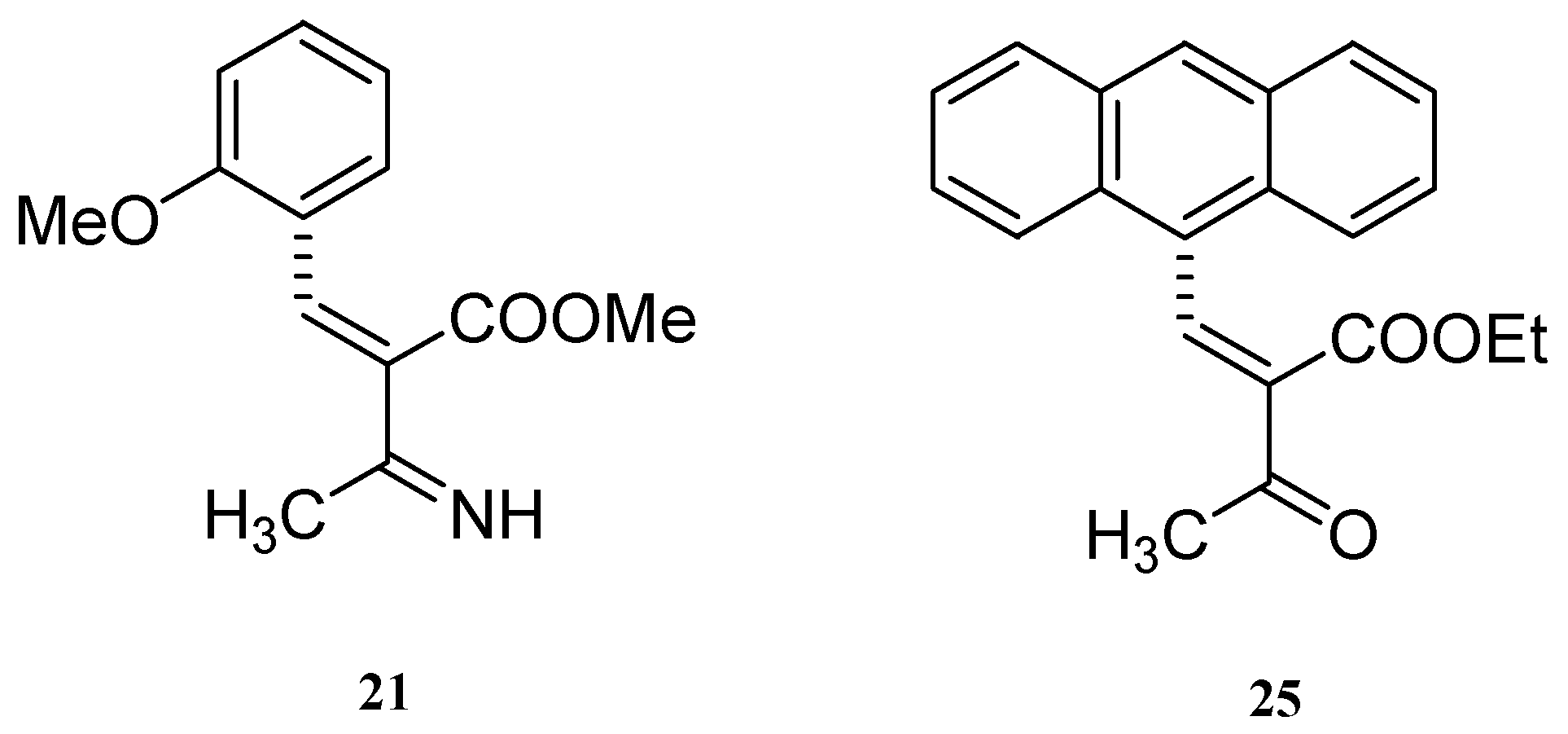
Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
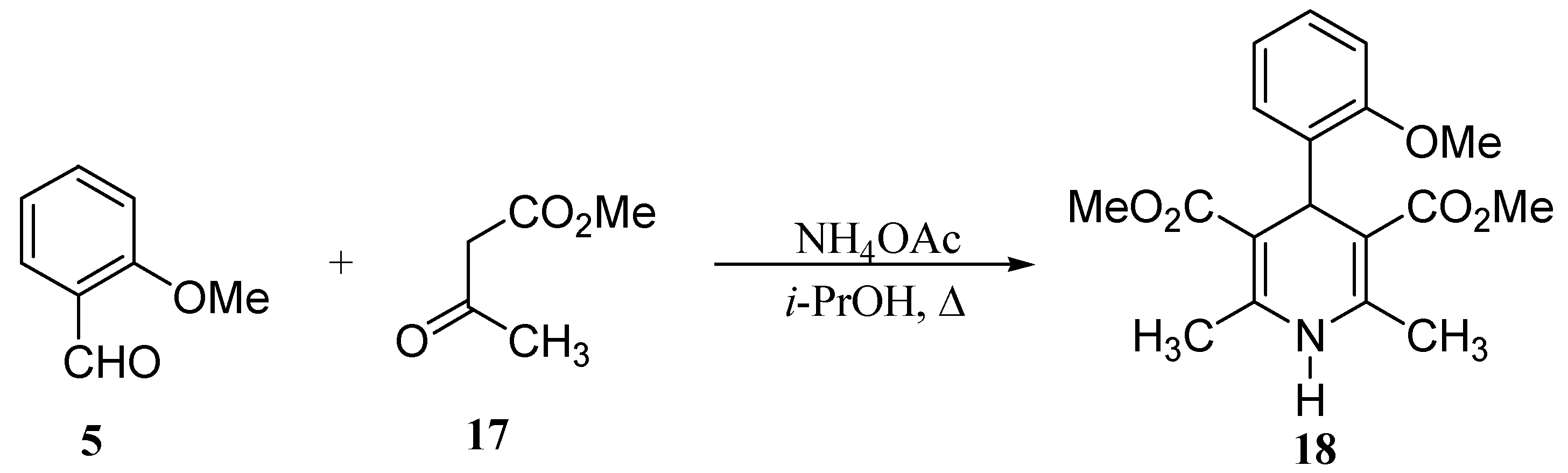{kind=link}
{kind=link}
| Entry | R | Product | Reaction time[h]a | Yields after crystallisation [%] | mp | Rf (TLC) |
|---|---|---|---|---|---|---|
| 1 | o-MeOC6H4 | 9 | 84 | 25.6 | 170.0 - 171.5 | 0.11b |
| 2 | m-MeOC6H4 | 10 | 23 | 28.8 | 168.0 - 170.5 | 0.39c |
| 3 | p-MeOC6H4 | 11 | 17 | 15.3 | 172.5 - 175.0 | 0.47c |
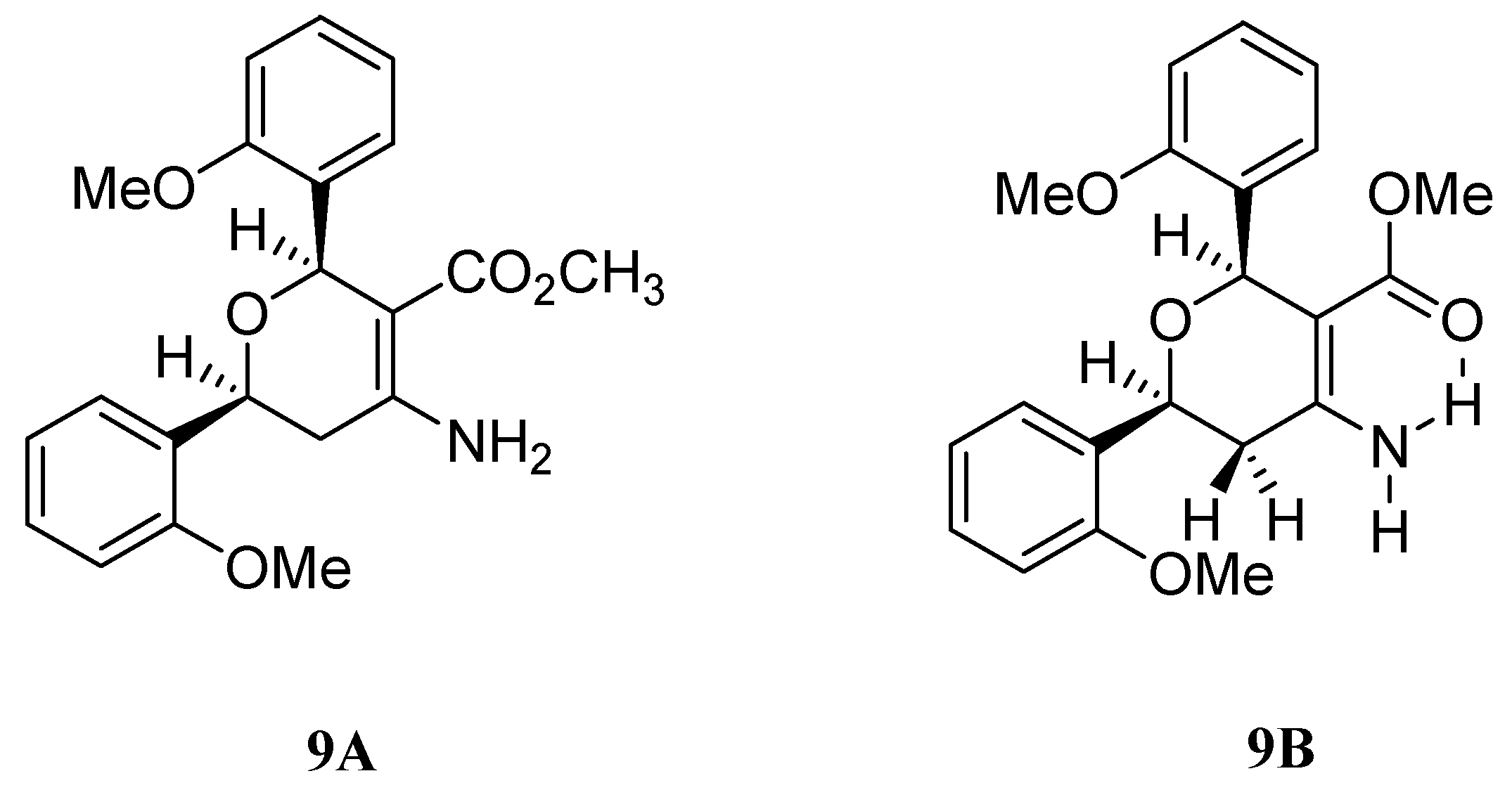

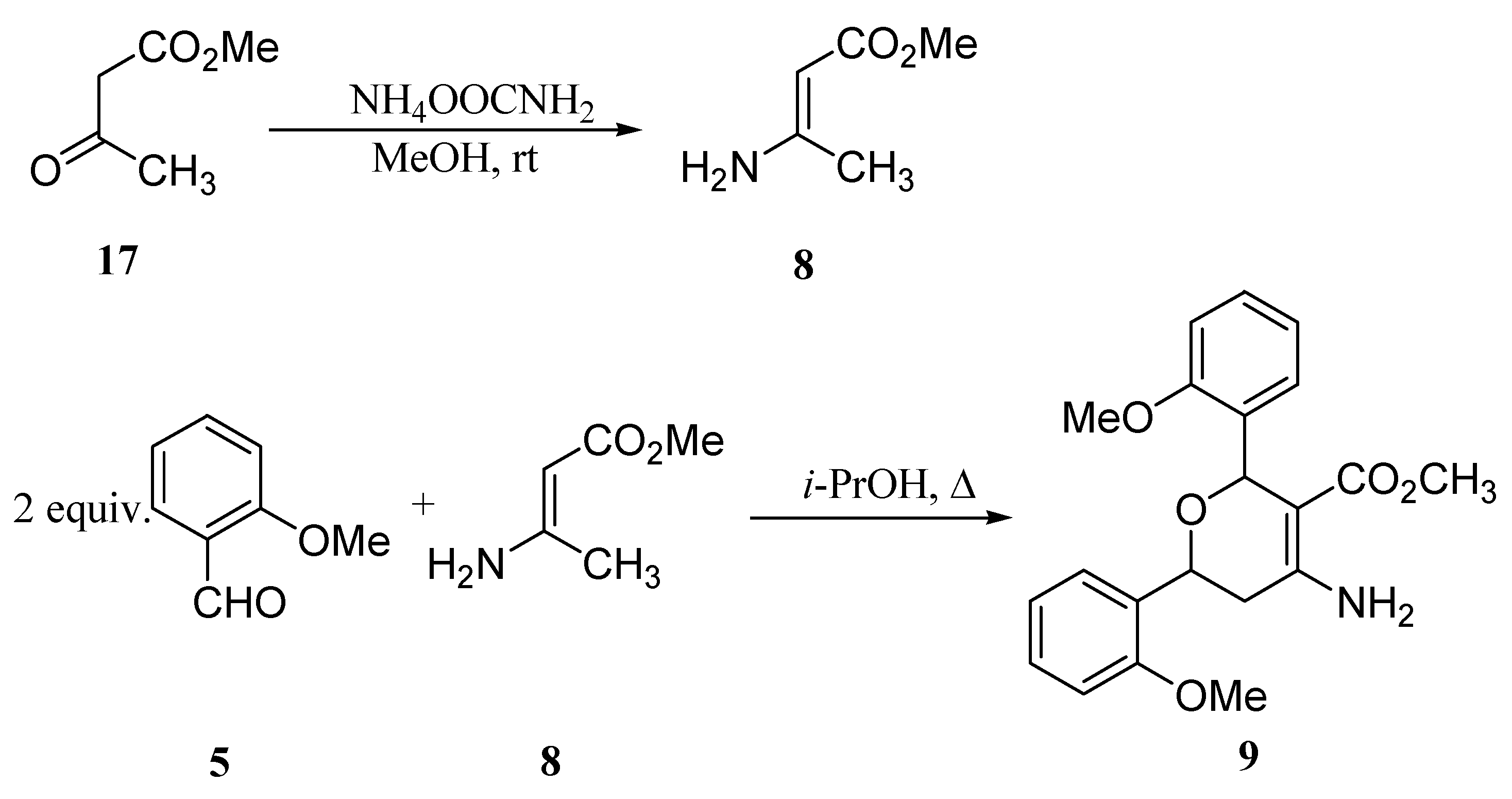
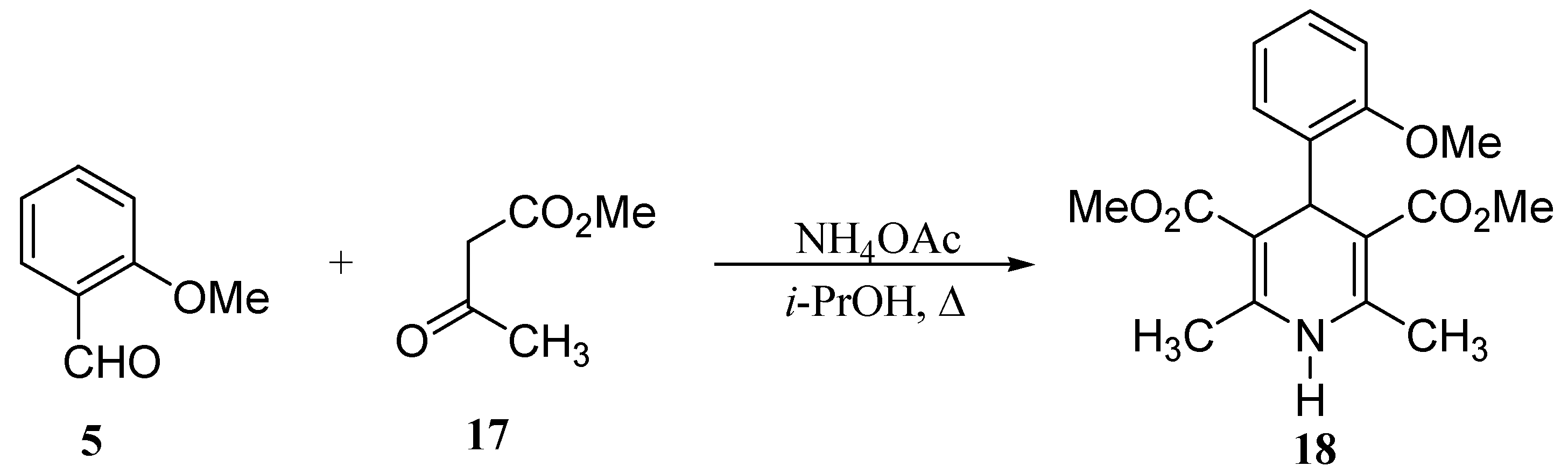
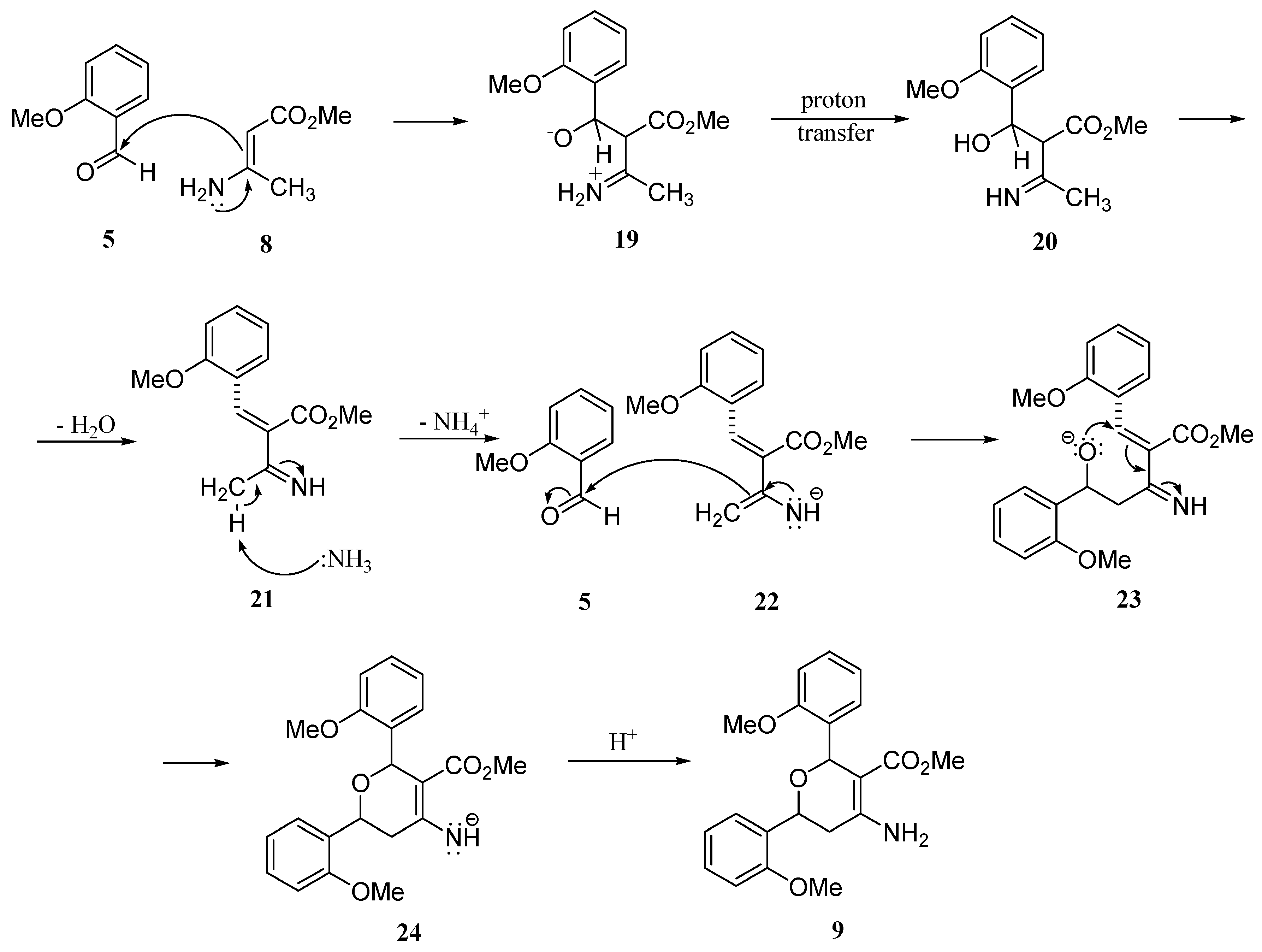
Conclusions
Experimental
General
General procedure for the Hantzsch condensation
Acknowledgments
References
- Hantzsch, A. Condensationprodukte aus Aldehydammoniak und ketoniartigen Verbindungen. Chem. Ber. 1881, 14, 1637–1638. [Google Scholar]
- Eisner, U.; Kuthan, J. The Chemistry of Dihydropyridines. Chem. Rev. 1972, 72, 1–42. [Google Scholar] Stout, D. M.; Meyers, A. I. Recent Advances in the Chemisty of Dihydropyridines. Chem. Rev. 1982, 82, 223–243. [Google Scholar] Sausinš, A.; Duburs, G. Synthesis of 1,4-Dihydropyridines by Cyclocondensation Reactions. Heterocycles 1988, 27, 269–289. [Google Scholar] Kuthan, J.; Kurfürst, A. Development in Dihydropyridine Chemistry. Ind. Eng. Chem. Prod. Res. Dev. 1982, 21, 191–261. [Google Scholar] Zolfigol, M. A.; Salehi, P.; Safaiee, M. An Efficient and Eco-Friendly Procedure for the Synthesis of Hantzsch Ethyl 1,4-Dihydro-2,6-Dimethylpyridine-3,5-Dicarboxylates Under Mild and Green Conditions. Lett. Org. Chem. 2006, 3, 153–156. [Google Scholar] Zolfigol, M. A.; Salehi, P.; Khorramabadi-Zad, A.; Shayegh, M. Iodine-catalysed synthesis of novel Hantzsch N-hydroxyethyl 1,4-dihydropyridines under mild conditions. J. Mol. Catal. A: Chem. 2007, 261, 88–92. [Google Scholar] Zolfigol, M. A.; Safaiee, M. Synthesis of 1,4-Dihydropyridines under Solvent-free Conditions. Synthesis 2004, 827–828. [Google Scholar]
- The Merck Index, 12th edition; Merck Research Laboratories: Rahway, NJ, 1996; p. 1121.
- Boström, S. L.; Ljung, B.; Mårdh, S.; Forsen, S.; Thulin, E. Interaction of the antihypertensive drug felodipine with calmodulin. Nature 1981, 292, 777–778. [Google Scholar] [CrossRef]
- Iwanami, M.; Shibanuma, T.; Fujimoto, M.; Kawai, R.; Tamazawa, K.; Takenaka, T.; Takahashi, K.; Murakami, M. Synthesis of New Water-soluble Dihydropyridine Vasodilatators. Chem. Pharm. Bull. 1979, 27, 1426–1440. [Google Scholar] [CrossRef]
- Arrowsmith, J. E.; Campbell, S. F.; Cross, P. E.; Stubbs, J. K.; Burges, R. A.; Gardiner, D. G.; Blackburn, K. J. Long-Acting Dihydropyridine Calcium Antagonists. 1. 2-Alkoxymethyl Derivatives Incorporating Basic Substituents. J. Med. Chem. 1986, 29, 1696–1702. [Google Scholar] [CrossRef]
- Berkels, B.; Roesen, R.; Dhein, S.; Fricke, U.; Klaus, W. Dihydropyridine Calcium Antagonist-Induced Modulation of Endothelial Function: A Reviw. Cardiovasc. Drug Rev. 1999, 17, 179–186. [Google Scholar] [CrossRef]
- Tsuruo, T.; Iida, H.; Nojiri, M.; Tsukagoshi, S.; Sakurai, Y. Circumvention of Vincristine and Adriamycin Resistance in Vitro and in Vivo by Calcium Influx Blockers. Cancer Res. 1983, 43, 2905–2910. [Google Scholar]
- Chapman, R. W.; Danko, G.; Siegels, M. I. Effect of Extra- and Intracellular Calcium Blockers on Histamine and Antigen-Induced Bronchospasm in Guinea Pigs and Rats. Pharmacology 1984, 29, 282–291. [Google Scholar] [CrossRef]
- Malaise, W. J.; Mathias, P. C. F. Stimulation of Insulin Release by Organic Calcium-Agonist. Diabetologia 1985, 28, 153–156. [Google Scholar]
- Krauze, A.; Germane, S.; Eberlins, O.; Sturms, I.; Klusa, V.; Duburs, G. Derivatives of 3-cyano-6-phenyl-4-(3’-pyridyl)pyridine-2(1H)-thione and their neurotropic activity. Eur. J. Med. Chem. 1999, 34, 301–310. [Google Scholar] [CrossRef]
- Peri, R.; Padmanabhan, S.; Singh, S.; Rutledge, A.; Triggle, D. J. Permanently Charged Chiral 1,4-Dihydropyridines: Molecular Probes of L-Type Calcium Channels. Synthesis and Pharmacological Characterization of Methyl (-Trimethylalkylammonium) 1,4-Dihydro-2,6-dimethyl-4-(3-nitrophenyl)-3,5- pyridinedicarboxylate Iodide, Calcium Channel Antagonists. J. Med. Chem. 2000, 43, 2906–2914. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, L.; Tseng, E.; Scott-Ramsay, E.; Schentag, J. J.; Coburn, R. A.; Morris, M. E. New 4-aryl-1,4-dihydropyridines and 4-arylpyridines as P-glycoprotein inhibitors. Drug. Metab. Dispos. 2005, 33, 321–328. [Google Scholar]
- Loev, B.; Goodman, M. M.; Snader, K. M.; Tedeschi, R.; Macko, E. "Hantzsch-Type" Dihydropyridine Hypotensive Agents. J. Med. Chem. 1974, 17, 956–965. [Google Scholar] [CrossRef]
- Meyer, H.; Bossert, F.; Horstmann, H. Dihydropyridine, II. Synthese Von 1,4-Dihydropyridinen mit Brückenkopf-N-atom. Liebigs Ann. Chem. 1977, 1888–1894. [Google Scholar]
- Phillips, A. P. Hantzsch's Pyridine Synthesis. J. Am. Chem. Soc. 1949, 71, 4003–4007. [Google Scholar] [CrossRef]
- Meyer, H.; Bossert, F.; Wehinger, E.; Stoepel, K.; Vater, W. Synthese und vergleichende pharmakologische Untersuchungen von 1,4-Dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridin-3,5-dicarbonsäuereestern mit nicht-identischen Esterfunktionen. Arzneim. Forsch. (Drug Res.) 1981, 31, 407–409. [Google Scholar]
- Wong, W. C.; Chiu, G.; Wetzel, J. M.; Marzabadi, M. R.; Nagarathnam, D.; Wang, D.; Fang, J.; Miao, S. W.; Hong, X.; Forray, C.; Vaysse, P. J. J.; Branchek, T. A.; Gluchowski, C. Identification of a Dihydropyridine as a Potent α1a Adrenoreceptor-Selective Antagonist That Inhibits Phenylephrine-Induced Contraction of the Human Prostate. J. Med. Chem. 1998, 41, 2643–2650. [Google Scholar] [CrossRef]
- Nagarathnam, D.; Wetzel, J. M.; Miao, S. W.; Marzabadi, M. R.; Chiu, G.; Wong, W. C.; Hong, X.; Fang, J.; Forray, C.; Branchek, T. A.; Heydorn, W. E.; Chang, R. S. L.; Broten, T.; Schorn, T. W.; Gluchowski, C. Design and Synthesis of Novel α1a Adrenoreceptor-Selective Antagonist for the Treatment of Benign Prostatic Hyperplasia. J. Med. Chem. 1998, 41, 5320–5333. [Google Scholar]
- Khadilkar, B. M.; Jaisinghani, H. G.; Saraf, M. N.; Desai, S. K. Synthesis and pharmacological studies of new derivatives of dimethyl-1,4-dihydro-2,6-dimethyl-3,5-pyridine dicarboxylate. Ind. J. Chem. 2001, 40B, 82–86. [Google Scholar]
- Wang, G.-W.; Miao, C.-B. Environmentally benign one-pot multi-component approaches to the synthesis of novel unsymmetrical 4-arylacridiones. Green Chem. 2006, 8, 1080–1095. [Google Scholar] [CrossRef]
- Angeles, E.; Santillán, H.; Martínez, I.; Ramírez, A.; Velásquez, A.; López-Castañares, R.; Martínez, R. Rearrangement of o-Nitrobenzaldehyde in the Hantzsch Reaction. Molecules 2001, 6, 683–693. [Google Scholar] [CrossRef]
- Görlitzer, K.; Fabian, J.; Trittmacher, J. Dimere aus o-Nitrobenzylidenacetessigestern. Pharmazie 2005, 60, 571–573. [Google Scholar]
- Litvić, M.; Cepanec, I.; Filipan, M.; Kos, K.; Bartolinčić, A.; Drušković, V.; Tibi, M. M.; Vinković, V. Mild, Selective and High-Yield Oxidation of Hantzsch 1,4-Dihydropyridines with Lead(IV) Acetate. Heterocycles 2005, 65, 23–36. [Google Scholar] [CrossRef]
- Litvić, M.; Cepanec, I.; Vinković, V. A convenient Hantzsch synthesis of 1,4-dihydropyridines using tetraethyl orthosilicate. Heterocycl. Commun. 2003, 9, 385–390. [Google Scholar]
- Litvić, M.; Filipan, M.; Pogorelić, I.; Cepanec, I. Ammonium carbamate; mild, selective and efficient ammonia source for the preparation of β-amino-α,β-unsaturated esters at room temperature. Green Chem. 2005, 7, 771–774. [Google Scholar] [CrossRef]
- Kim, S. C.; Choi, K. M.; Cheong, C. S. Synthesis of Amlodipine Using Aza Diels-Alder Reaction. Bull. Kor. Chem. Soc. 2002, 23, 143–144. [Google Scholar] [CrossRef]
- Katritzky, A. R.; Ostercamp, D. L.; Yousaf, T. I. The Mechanism of the Hantzsch Pyridine Synthesis: A Study by 15N and 13C Spectroscopy. Tetrahedron 1986, 42, 5729–5738. [Google Scholar] [CrossRef]
- Treibs, W.; Beger, J. 4-Aryl-3-carboxy-pyridine als ausgangssubstanzen für N-methyl-1,2-benzisoazalene. Liebigs Ann. Chem. 1962, 652, 192–203. [Google Scholar] [CrossRef]
- Sample Availability: Available from the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Filipan-Litvić, M.; Litvić, M.; Cepanec, I.; Vinković, V. Hantzsch Synthesis of 2,6-Dimethyl-3,5-dimethoxycarbonyl-4-(o-methoxyphenyl)-1,4-dihydropyridine; a Novel Cyclisation Leading to an Unusual Formation of 1-Amino-2-methoxycarbonyl-3,5-bis(o-methoxyphenyl)-4-oxa-cyclohexan-1-ene. Molecules 2007, 12, 2546-2558. https://doi.org/10.3390/12112546
Filipan-Litvić M, Litvić M, Cepanec I, Vinković V. Hantzsch Synthesis of 2,6-Dimethyl-3,5-dimethoxycarbonyl-4-(o-methoxyphenyl)-1,4-dihydropyridine; a Novel Cyclisation Leading to an Unusual Formation of 1-Amino-2-methoxycarbonyl-3,5-bis(o-methoxyphenyl)-4-oxa-cyclohexan-1-ene. Molecules. 2007; 12(11):2546-2558. https://doi.org/10.3390/12112546
Chicago/Turabian StyleFilipan-Litvić, Mirela, Mladen Litvić, Ivica Cepanec, and Vladimir Vinković. 2007. "Hantzsch Synthesis of 2,6-Dimethyl-3,5-dimethoxycarbonyl-4-(o-methoxyphenyl)-1,4-dihydropyridine; a Novel Cyclisation Leading to an Unusual Formation of 1-Amino-2-methoxycarbonyl-3,5-bis(o-methoxyphenyl)-4-oxa-cyclohexan-1-ene" Molecules 12, no. 11: 2546-2558. https://doi.org/10.3390/12112546
APA StyleFilipan-Litvić, M., Litvić, M., Cepanec, I., & Vinković, V. (2007). Hantzsch Synthesis of 2,6-Dimethyl-3,5-dimethoxycarbonyl-4-(o-methoxyphenyl)-1,4-dihydropyridine; a Novel Cyclisation Leading to an Unusual Formation of 1-Amino-2-methoxycarbonyl-3,5-bis(o-methoxyphenyl)-4-oxa-cyclohexan-1-ene. Molecules, 12(11), 2546-2558. https://doi.org/10.3390/12112546