Bioreversible Derivatives of Phenol. 2. Reactivity of Carbonate Esters with Fatty Acid-like Structures Towards Hydrolysis in Aqueous Solutions

Abstract
:Introduction
Results and Discussion
Synthesis

{kind=link}
{kind=link}
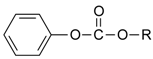
| Compound | R | kH (M-1s-1) | ko (s-1) | kOH (M-1s-1) | pKa | |
|---|---|---|---|---|---|---|
| 1 | -C2H5 | 1.4 × 10-6 | 6.8 × 10-8 | 1.3 | ||
| 2 | -C(CH3)3 | 1.8 × 10-3 | 4.1 × 10-4 | 3.3 × 10-2 | ||
| 3 | -C6H5 | 1.2 × 10-5 | 12.6 | |||
| 4 | -CH2COOH | 4.7 × 10-6 | 1.0 | 2.6; 2.42b) | ||
| 3.7 × 10-4a) | ||||||
| 5 | -(CH2)5COOH | 1.7 × 10-6 | 6.5 × 10-8 | 1.0 | 4.70b) | |
| 6 | -(CH2)7COOH | 1.0 | 4.79b) | |||
| 7 | -(CH2)11COOH | 1.0 | ||||
| 8 | -(CH2)15COOH | |||||
| 9 | Phenyl acetate | 3.2 × 10-4 | 3.9 × 10-8 | 4.1 | ||
- a)
- k’o (cf. Equation 2)
- b)
- pKa was determined by capillary electrophoresis at 25oC and μ = 0.050 M
Kinetics of chemical hydrolysis
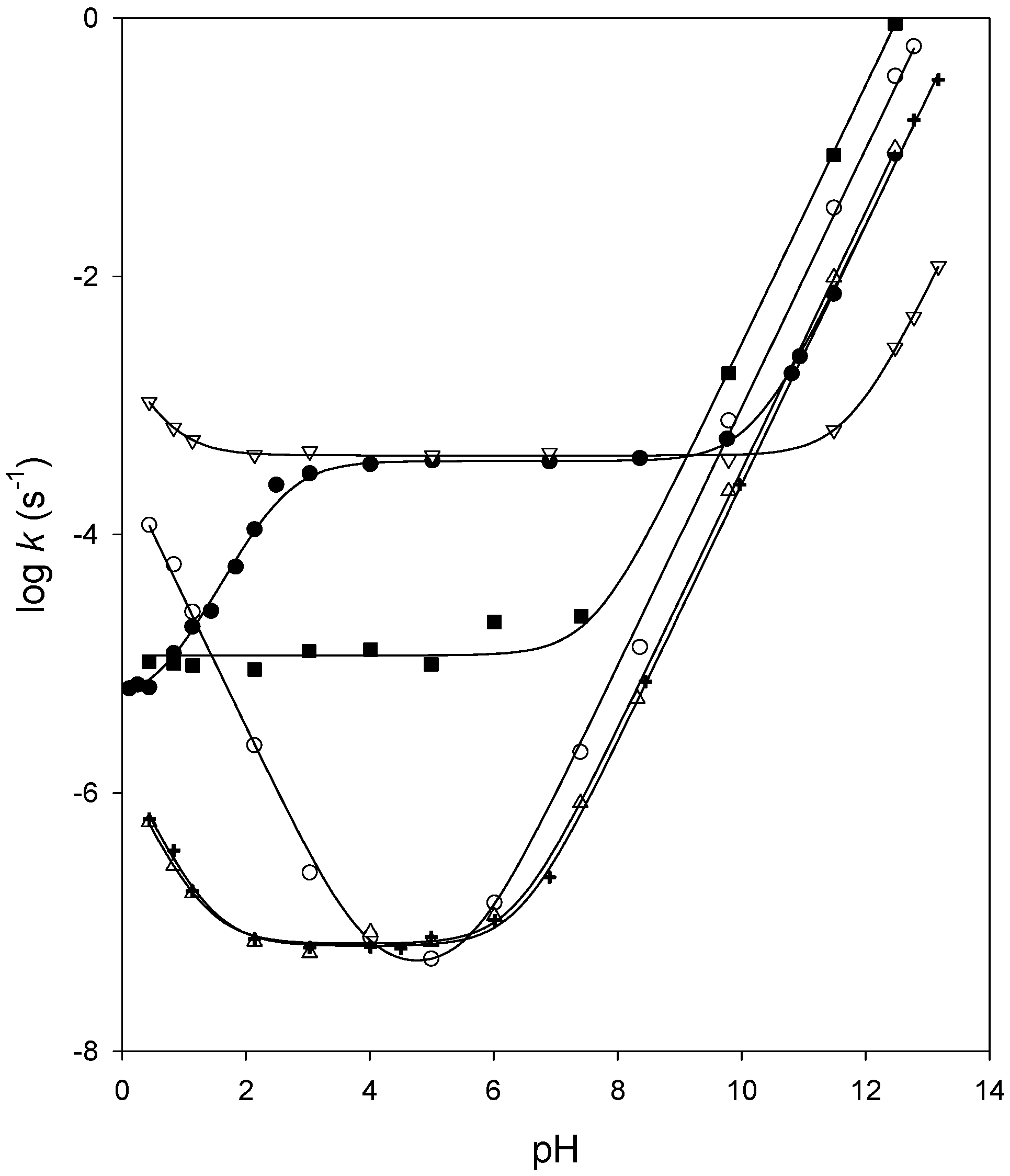
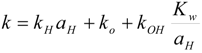
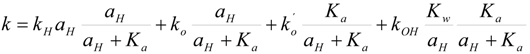
Reactivity of carbonate esters

Conclusions
Experimental
Chemicals
Apparatus
Preparation of carbonate esters
Kinetic measurements
Determination of pKa values by capillary electrophoresis
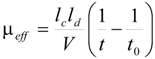
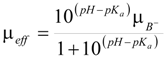
Acknowledgments
References
- Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 1997, 23, 3–25. [Google Scholar]
- Avdeef, A. Physicochemical profiling (Solubility, permeability and charge state). Curr. Topics Med. Chem. 2001, 1, 277–351. [Google Scholar]
- Muegge, I. Selection criteria for drug-like compounds. Med. Res. Rev. 2003, 23, 302–321. [Google Scholar]
- van de Waterbeemd, H.; Smith, D. A.; Beumont, K.; Walker, D. K. Property-based design: optimization of drug absorption and pharmacokinetics. J. Med. Chem. 2001, 44, 1313–1333. [Google Scholar]
- Walters, W. P.; Murcko, A.; Murcko, M. A. Recognizing molecules with drug-like properties. Curr. Opin. Chem. Biol. 1999, 3, 384–387. [Google Scholar]
- Bundgaard, H. Design of prodrugs: Bioreversible derivatives for various functional groups and chemical entities. In Design of prodrugs; Bundgaard, H., Ed.; Elsevier: Amsterdam, 1985; pp. 1–92. [Google Scholar]
- Larsen, C. S.; Østergaard, J. Design and application of prodrugs. In Textbook of drug design and discovery, 3rd ed.; Krogsgaard-Larsen, P., Liljefors, T., Madsen, U., Eds.; Taylor & Francis: London, 2002; pp. 410–458. [Google Scholar]
- Testa, B.; Mayer, J. M. Concepts in prodrug design to overcome pharmacokinetic problems. In Pharmacokinetic optimization in drug research; Testa, B., van de Waterbeemd, H., Folkers, G., Guy, R., Eds.; Verlag Helvetica Chimica Acta/Wiley-VCH: Zürich, 2001; pp. 85–95. [Google Scholar]
- Testa, B.; Mayer, J. M. Hydrolysis in drug and prodrug metabolism. Chemistry, biochemistry, and enzymology; Verlag Helvetica Chimica Acta: Zürich, 2003. [Google Scholar]
- Stella, V. J.; Borchardt, R. T.; Hageman, M. J.; Oliyai, R.; Maag, H.; Tilley, J. W. (Eds.) Prodrugs: challenges and rewards. Part 1; Springer-AAPS Press: New York, 2007.
- Testa, B.; Caldwell, J. Prodrugs revisited: "Ad hoc" approach as a complement to ligand design. Med. Res. Rev. 1996, 16, 233–241. [Google Scholar]
- Carter, D. C.; Ho, J. X. Structure of serum albumin. Adv. Protein Chem. 1994, 45, 153–203. [Google Scholar]
- Kragh-Hansen, U. Molecular aspects of ligand binding to serum albumin. Pharmacol. Rev. 1981, 33, 17–53. [Google Scholar]
- Kragh-Hansen, U. Structure and ligand binding properties of human serum albumin. Dan. Med. Bull. 1990, 37, 57–84. [Google Scholar]
- Kragh-Hansen, U.; Chuang, V. T. G.; Otagiri, M. Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol. Pharm. Bull. 2002, 25, 695–704. [Google Scholar]
- Peters, T., Jr. All about albumin. Biochemistry, genetics, and medical applications; Academic Press: San Diego, 1996. [Google Scholar]
- Vallner, J. J. Binding of drugs by albumin and plasma protein. J. Pharm. Sci. 1977, 66, 447–465. [Google Scholar]
- Jusko, W. J.; Gretch, M. Plasma and tissue protein binding of drugs in pharmacokinetics. Drug Metab. Rev. 1976, 5, 43–140. [Google Scholar]
- Kwong, T. C. Free drug measurements: methodology and clinical significance. Clin. Chim. Acta 1985, 151, 193–216. [Google Scholar]
- Meyer, M. C.; Guttmann, D. E. The binding of drugs by plasma proteins. J. Pharm. Sci. 1968, 57, 895–918. [Google Scholar]
- Kurtzhals, P.; Havelund, S.; Jonassen, I.; Kiehr, B.; Larsen, U. D.; Ribel, U.; Markussen, J. Albumin binding of insulins acylated with fatty acids: characterization of the ligand-protein interaction and correlation between binding affinity and timing of the insulin effect in vivo. Biochem. J. 1995, 312, 725–731. [Google Scholar]
- Kurtzhals, P.; Havelund, S.; Jonassen, I.; Markussen, J. Effect of fatty acids and selected drugs on the albumin binding of a long-acting, acylated insulin analogue. J. Pharm. Sci. 1997, 86, 1365–1368. [Google Scholar]
- Markussen, J.; Havelund, S.; Kurtzhals, P.; Andersen, A. S.; Halstrøm, J.; Hasselager, E.; Larsen, U. D.; Ribel, U.; Schäffer, L.; Vad, K.; Jonassen, I. Soluble, fatty acid acylated insulins bind to albumin and show protracted action in pigs. Diabetologia 1996, 39, 281–288. [Google Scholar]
- Knudsen, L. B.; Nielsen, P. F.; Huusfeldt, P. O.; Johansen, N. L.; Madsen, K.; Pedersen, F. Z.; Thøgersen, H.; Wilken, M.; Agersø, H. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J. Med. Chem. 2000, 43, 1664–1669. [Google Scholar]
- Oku, N.; Yamashita, S.; Sakuragi, n.; Doi, K.; Okada, S.; Shimidzu, K.; sumi, M.; Nadai, T.; Kusumoto, S.; Suda, Y. Therapeutic efficacy of 5-fluorouracil prodrugs using endogenous serum proteins as drug carriers: A new strategy in drug delivery system. Biol. Pharm. Bull. 1995, 18, 181–184. [Google Scholar]
- Østergaard, J.; Larsen, C. Bioreversible derivatives of phenol. I. The role of human serum albumin as related to the stability and binding properties of carbonate esters with fatty acid-like structures in aqueous solution and biological media. Molecules 2007, 12, 2380–2395. [Google Scholar] [CrossRef]
- Pond, S. M.; Tozer, T. N. First-pass elimination. Basic concepts and clinical consequenses. Clin. Pharmacokinet. 1984, 9, 1–25. [Google Scholar] [CrossRef]
- Ballinger, L. N.; Cross, S. E.; Roberts, M. S. Availability and mean transit times of phenol and its metabolites in the isolated perfused rat liver: normal and retrograde studies using tracer concentrations of phenol. J. Pharm. Pharmacol. 1995, 47, 949–956. [Google Scholar]
- Scott, D. O.; Lunte, C. E. In vivo microdialysis sampling in the bile, blood, and liver of rats to study the disposition of phenol. Pharm. Res. 1993, 10, 335–342. [Google Scholar]
- Cassidy, M. K.; Houston, J. B. In vivo assessment of extrahepatic conjugative metabolism in first pass effects using the model compound phenol. J. Pharm. Pharmacol. 1980, 32, 57–59. [Google Scholar]
- Cassidy, M. K.; Houston, J. B. In vivo capacity of hepatic and extrahepatic enzymes to conjugate phenol. Drug Metab. Disp. 1984, 12, 619–624. [Google Scholar]
- Hansen, J.; Mørk, N.; Bundgaard, H. Phenyl carbamates of amino acids as prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1992, 81, 253–261. [Google Scholar]
- Fredholt, K.; Mørk, N.; Begtrup, M. Hemiesters of aliphatic dicarboxylic acids as cyclization-activated prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1995, 123, 209–216. [Google Scholar]
- Thomsen, K. F.; Bundgaard, H. Cyclization-activated phenyl carbamate prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1993, 91, 39–49. [Google Scholar]
- Thomsen, K. F.; Strøm, F.; Sforzini, B. V.; Begtrup, M.; Mørk, N. Evaluation of phenyl carbamates of ethyl diamines as cyclization-activated prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1994, 112, 143–152. [Google Scholar]
- King, S. W.; Lum, V. R.; Fife, T. H. Interaction of carboxypeptidase A with carbamate and carbonate esters. Biochemistry 1987, 26, 2294–2230. [Google Scholar]
- Harned, H. S.; Hamer, W. J. The ionization constant of water and the dissociation of water in potassium chloride solutions from electromotive forces of cells without liquid junction. J. Am. Chem. Soc. 1933, 55, 2194–2206. [Google Scholar]
- van der Houwen, O. A. G. J.; de Loos, M. R.; Beinen, J. H.; Bult, A.; Underberg, W. J. M. Systematic interpretation of pH-degradation profiles. A critical review. Int. J. Pharm. 1997, 155, 137–152. [Google Scholar]
- Nøring, I.; Jensen, A.; Faurholt, C. Studies on monoalkyl carbonates. XII. The monoalkyl carbonates of ethylene glycol and ethylene chlorohydrin. Acta Chem. Scand. 1952, 6, 404–410. [Google Scholar] [CrossRef]
- Miller, N. F.; Case, L. O. The kinetics of the alkaline hydrolysis of ethyl carbonate and potassium ethyl carbonate. J. Am. Chem. Soc. 1935, 57, 810–814. [Google Scholar]
- Faurholt, C. Studien über monoalkylkarbonate. III. Über die Kinetik bei der Zersetzung der Monoalkylcarbonate und bei der bildung der Monoalkylkarbonate aus Bikarbonat in wässerigen alkalishen Lösungen. Z. Physik. Chem. 1927, 126, 211–226. [Google Scholar]
- Cooper, G. D.; Williams, B. Hydrolysis of simple aromatic esters and carbonates. J. Org. Chem. 1962, 27, 3717–3719. [Google Scholar]
- Cooper, G. D.; Johnson, H. T.; Williams, B. Substituent effects in hydrolysis of diaryl carbonates. J. Org. Chem. 1965, 30, 3989–3991. [Google Scholar]
- Jaffé, H. H. A reexamination of the Hammett equation. Chem. Rev. 1953, 53, 191–261. [Google Scholar]
- Johnson, S. L. General base and nucleophilic catalysis of ester hydrolysis and related reactions. Adv. Phys. Org. Chem. 1967, 5, 237–330. [Google Scholar]
- Marlier, J. F.; O'Leary, M. H. Carbon kinetic isotope effects on the hydrolysis of aryl carbonates. J. Am. Chem. Soc. 1990, 112, 5996–5998. [Google Scholar]
- Fife, T. H.; Hutchins, J. E. C. Effect of nucleophile basicity on intramolecular nucleophilic aminolysis reactions of carbonate diesters. J. Am. Chem. Soc. 1981, 103, 4194–4199. [Google Scholar]
- Hansch, C.; Leo, A.; Hoekman, D. Exploring QSAR. Hydrophobic, electronic, and steric constants, 1 ed.; ACS: Washington, 1995; Vol. 2. [Google Scholar]
- Dittert, L. W.; Higuchi, T. Rates of hydrolysis of carbamate and carbonate esters in alkaline solution. J. Pharm. Sci. 1963, 52, 852–857. [Google Scholar]
- Dittert, L. W.; Caldwell, H. C.; Ellison, T.; Irwin, G. M.; Rivard, D. E.; Swintosky, J. V. Carbonate ester prodrugs of salicylic acid. Synthesis, solubility characteristics, in vitro enzymatic hydrolysis rates, and blood levels of total salicylate following oral administration to dogs. J. Pharm. Sci. 1968, 57, 828–831. [Google Scholar] [CrossRef]
- Rattie, E. S.; Shami, E. G.; Dittert, L. W.; Swintosky, J. V. Acetaminophen prodrugs III. Hydrolysis of carbonate and carboxylic acid esters in aqueous buffers. J. Pharm. Sci. 1970, 59, 1738–1741. [Google Scholar] [CrossRef]
- Dittert, L. W.; Irwin, G. M.; Chong, C. W.; Swintosky, J. V. Acetaminophen prodrugs II. Effect of structure and enzyme source on enzymatic and nonenzymatic hydrolysis of carbonate esters. J. Pharm. Sci. 1968, 57, 780–783. [Google Scholar] [CrossRef]
- March, J. Advanced organic chemistry. Reactions, mechanisms, and structure, 4 ed.; John Wiley & Sons: New York, 1992. [Google Scholar]
- Nicholls, P. H.; Tillett, J. G. The acid-catalysed hydrolysis of diaryl carbonates. J. Chem. Soc., Perkin Trans. 2 1972, 1970–1971.
- Levin, I.; Pohoryles, L. A.; Sarel, S.; Usieli, V. Organic carbonates. Part VIII. The acid catalysed hydrolysis of ethylene, trimethylene, and tetramethylene carbonate. J. Chem. Soc. 1963, 3949–3954.
- Sarel, S.; Levin, I.; Pohoryles, L. A. Organic carbonates. Part V. The mechanism of hydrolysis of cyclic carbonates: tracer studies. J. Chem. Soc. 1960, 3079–3082.
- Faurholt, C.; Gjaldbæk, J. C. Studier over monoalkylkarbonater. VIII. Propylalkoholernes monoalkylkarbonater. Dansk Tidskr. Farm. 1943, 17, 213–227. [Google Scholar]
- Taft, R. W. Polar and steric substituent constants for aliphatic and o-benzoate groups from rates of esterification and hydrolysis of esters. J. Am. Chem. Soc. 1952, 74, 3120–3128. [Google Scholar]
- Cohen, S. G.; Schneider, A. Cleavage of the alkyl-oxygen bond in the hydrolysis of esters. t-butyl 2,4,6-trimethylbenzoate. J. Am. Chem. Soc. 1941, 63, 3382–3388. [Google Scholar] [CrossRef]
- Bunton, C. A.; Wood, J. L. Tracer studies on ester hydrolysis. Part II. The acid hydrolysis of tert.-butyl acetate. J. Chem. Soc. 1955, 1522–1525. [Google Scholar] [CrossRef]
- Salomaa, P. Kinetics of the uncatalyzed hydrolysis of tertiary butyl acetate. Suom. Kemi. 1969, B42, 134–135. [Google Scholar]
- Tillett, J. G.; Wiggins, D. E. Neighbouring hydroxyl group catalysis of carbonate esters. Tetrahedron Lett. 1971, 14, 911–914. [Google Scholar]
- Bruice, T. C.; Pandit, U. K. The effect of geminal substitution ring size and rotamer distribution on the intramolecular nucleophilic catalysis of the hydrolysis of monophenyl esters of dibasic acids and the solvolysis of intermediate anhydrides. J. Am. Chem. Soc. 1960, 82, 5858–5865. [Google Scholar]
- St Pierre, T.; Jencks, W. P. Intramolecular catalysis in the reactions of nucleophilic reagents with aspirin. J. Am. Chem. Soc. 1968, 90, 3817–3827. [Google Scholar]
- Connors, K. A. Reaction mechanisms in organic analytical chemistry; John Wiley & Sons: New York, 1973. [Google Scholar]
- Fersht, A. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding; W. H. Freeman and Company: New York, 1998. [Google Scholar]
- Hansen, L. B.; Christrup, L. L.; Bundgaard, H. Ketobemidone prodrugs for buccal delivery. Acta Pharm. Nord. 1991, 3, 77–82. [Google Scholar]
- Honkanen, E. Synthesis of some phenyl carboxymethyl esters of carbonic acid and its sulphur analogues. Acta Chem. Scand. 1970, 24, 1120–1122. [Google Scholar]
- Leonard, J.; Lygo, B.; Procter, G. Advanced practical organic chemistry, 2 ed.; Blackie Academic & Professional: London, 1995. [Google Scholar]
- Connors, K. A. Chemical kinetics. The study of reaction rates in solution; VCH Publishers: New York, 1990. [Google Scholar]
- Guggenheim, E. A. XLVI. On the determination of the velocity constant of a unimolecular reaction. Phil. Mag. 1926, 2, 538–543. [Google Scholar]
- Østergaard, J.; Hansen, S. H.; Larsen, C.; Schou, C.; Heegaard, N. H. H. Determination of octanol-water partition coefficients for carbonate esters and other small organic molecules by microemulsion electrokinetic chromatography. Electrophoresis 2003, 24, 1038–1046. [Google Scholar]
- Kok, W. Th. Thermal management. joule heating in CE. Chromatographia Suppl. 2000, 51, S-24–S-27. [Google Scholar]
- Ishihama, Y.; Oda, Y.; Asakawa, N. Microscale determination of dissociation constants of multivariant pharmaceuticals by capillary electrophoresis. J. Pharm. Sci. 1994, 83, 1500–1507. [Google Scholar]
- Sample Availability: Samples of the carbonate esters are available from the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Ostergaard, J.; Larsen, C. Bioreversible Derivatives of Phenol. 2. Reactivity of Carbonate Esters with Fatty Acid-like Structures Towards Hydrolysis in Aqueous Solutions. Molecules 2007, 12, 2396-2412. https://doi.org/10.3390/12102396
Ostergaard J, Larsen C. Bioreversible Derivatives of Phenol. 2. Reactivity of Carbonate Esters with Fatty Acid-like Structures Towards Hydrolysis in Aqueous Solutions. Molecules. 2007; 12(10):2396-2412. https://doi.org/10.3390/12102396
Chicago/Turabian StyleOstergaard, Jesper, and Claus Larsen. 2007. "Bioreversible Derivatives of Phenol. 2. Reactivity of Carbonate Esters with Fatty Acid-like Structures Towards Hydrolysis in Aqueous Solutions" Molecules 12, no. 10: 2396-2412. https://doi.org/10.3390/12102396
APA StyleOstergaard, J., & Larsen, C. (2007). Bioreversible Derivatives of Phenol. 2. Reactivity of Carbonate Esters with Fatty Acid-like Structures Towards Hydrolysis in Aqueous Solutions. Molecules, 12(10), 2396-2412. https://doi.org/10.3390/12102396