Introduction
The pyrrolo[3,2-
c]quinoline skeleton can be found in Nature in the alkaloids martinelline and martinellic acid, isolated from South American plant root extracts [
1,
2,
3]. These alkaloids are also known for their biological activity as antagonists of the bradykinine B
1 and B
2 receptors and their antibiotic activity against both Gram-positive and Gram-negative bacteria [
4]. These properties have led to the investigation of possible synthetic pathways leading to these substances. Intramolecular cycloaddition of sarcosine (
N-methylglycine) to
N-(2-formylphenyl)-
N-allyl-4-toluenesulfonamide in refluxing DMF led to
cis–fused pyrrolino[3,2-
c]quinoline derivatives
via an azomethine ylide formed
in situ. The cycloaddition was accompanied with decarboxylation, which was very important for the subsequent synthetic steps. These attempts were further developed into a total synthesis of the martinelline skeleton [
5,
6].
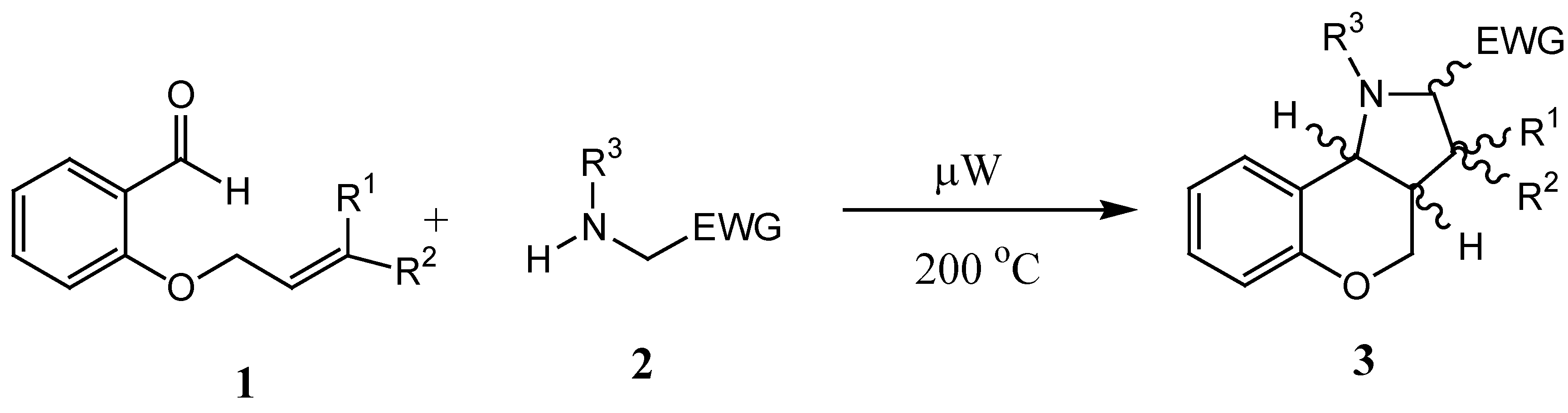
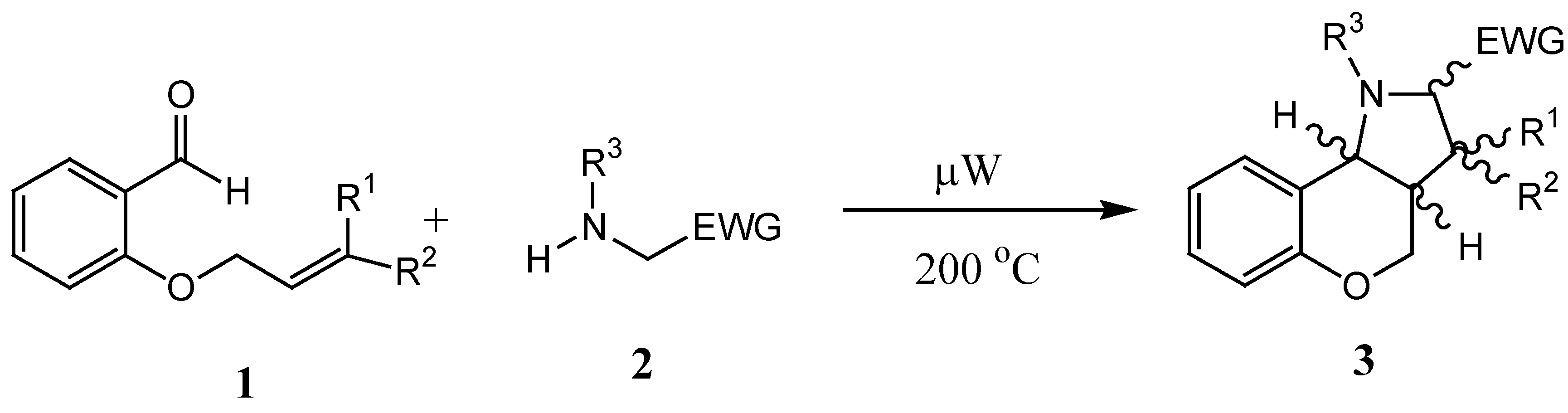
Generally, application of microwaves in synthesis leads to substantial reductions in reaction times, yield increases, solvent and waste minimization, and sometimes, to chemo-, regio- and stereoselectivity changes. During microwave irradiation the 2,450 MHz frequency is typically used and, since the individual oriented polar molecules present cannot match the resulting very fast changes in the electric field, this results in the transformation of part of the applied energy into heat, thus producing a temperature increase [
7,
8]. In the past we have investigated intramolecular microwave - assisted 1,3-dipolar cycloaddition reactions of
in situ generated azomethine ylides leading to hexahydrochromeno-[4,3-
b]pyrroles
3 [
9,
10]. A simple protocol was developed for these reactions, whereby the ylides were generated under microwave irradiation by the reaction of
ortho-(3-alkenyl)oxybenzaldehydes
1 with secondary amines
2, prepared in turn by
N-alkylation of alkyl aminoacetates. The subsequent cycloadditions gave the products
3 in very good yields and selectivity (
Scheme 1).
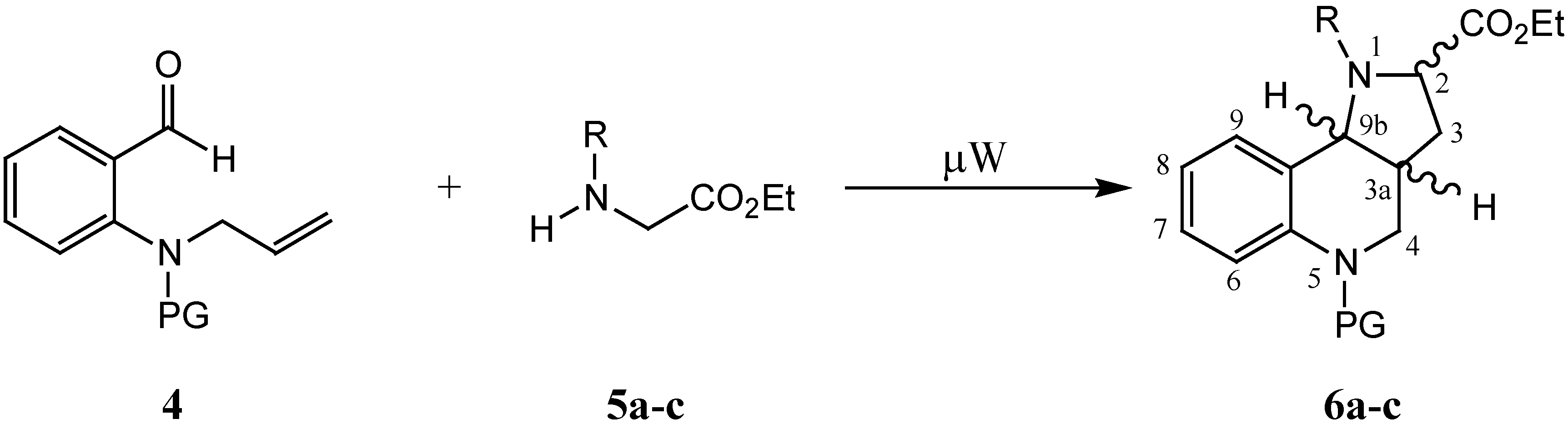
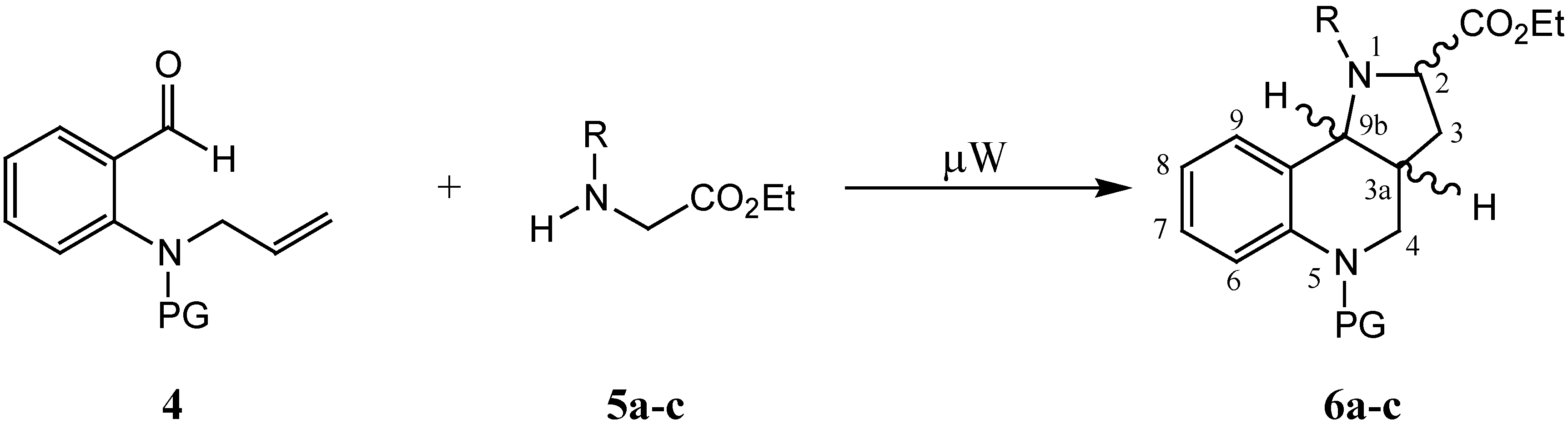
In addition, the effect of various electron-withdrawing groups (EWG) on the stability and reactivity of the dipoles was established. In this paper we would like to report the behaviour of protected
ortho-(3-alkenyl)amino benzaldehyde
4 in a similar microwave assisted reaction with secondary amines
5. The aim of our study of these reactions under microwave conditions was the creation of ylides capable of undergoing a subsequent intramolecular 1,3-dipolar cycloaddition (
Scheme 2).
Results and Discussion
The secondary amines
5 needed for the proposed reactions were synthesized from ethyl bromoacetate in acetonitrile at low temperature according to the literature [
11]. Their yields are presented in
Table 1.
Table 1.
Yields of the amino components 5 prepared for the azomethine ylide synthesis.
Table 1.
Yields of the amino components 5 prepared for the azomethine ylide synthesis.
| Amine | R | Yield [%] |
|---|
| 5a | n-Bu | 84 |
| 5b | CH3 | 85 |
| 5c | Bn | 78 |
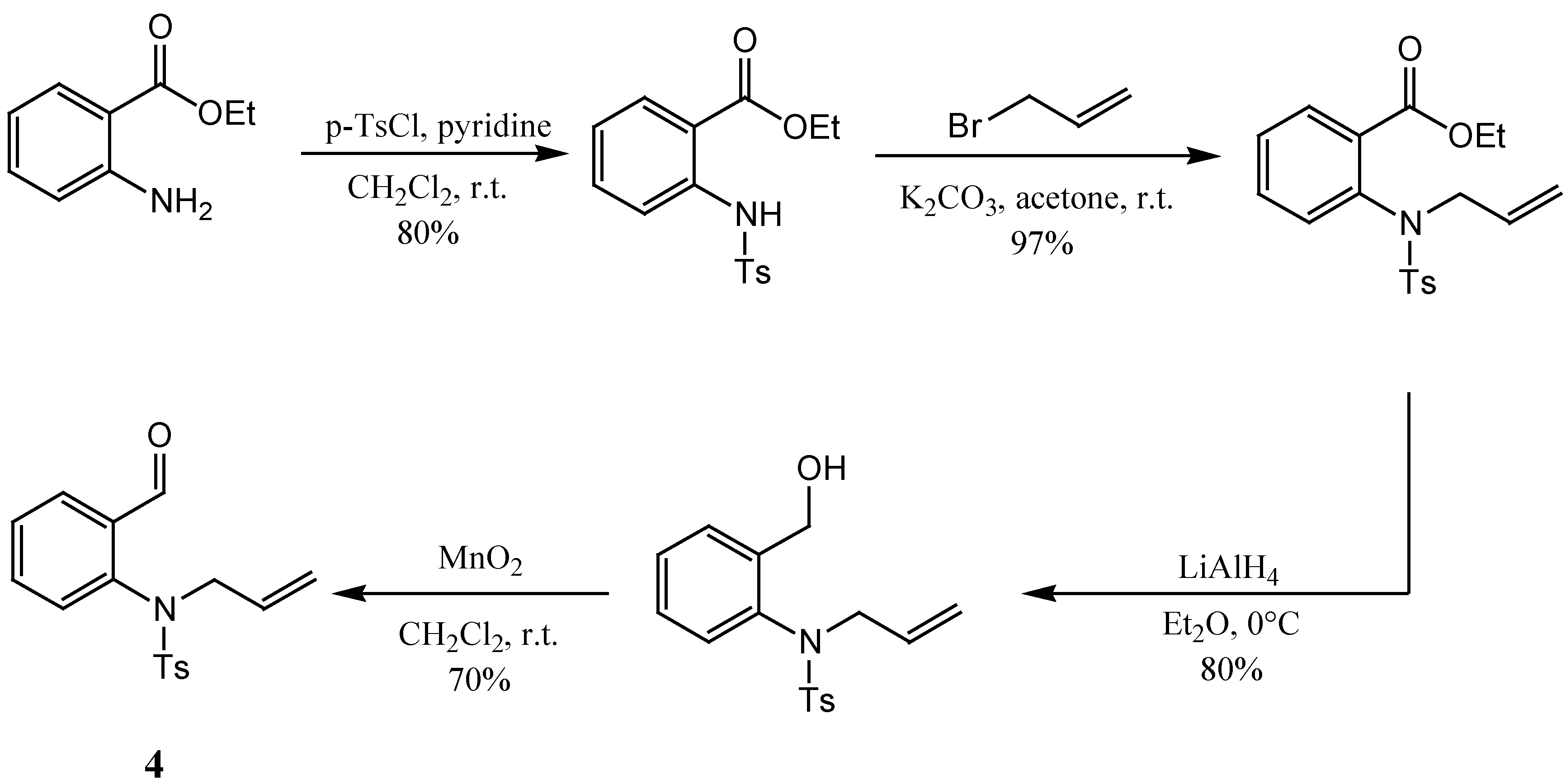
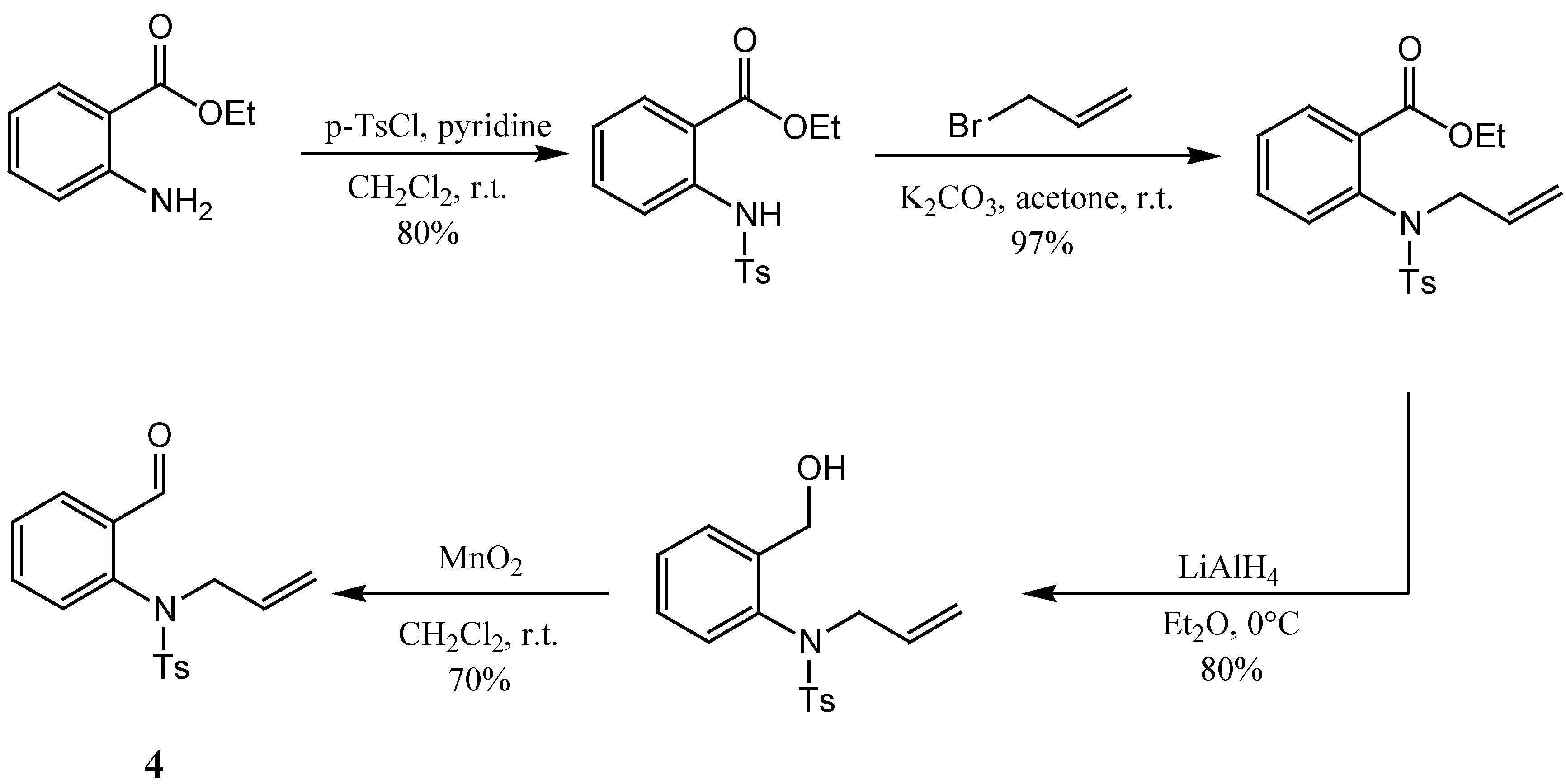
The preparation of the second component − aldehyde
4 − started with 2-nitrobenzaldehyde. Our initial intention was to reduce the nitro group after protection of the aldehyde moiety by transformation into an acetal, but although several reported nitro group reduction methods were applied [
12,
13,
14,
15], we did not succeed in preparing the target in good yields. As an alternative, the preparation of aldehyde
4 was then carried out
via a multi-step synthesis starting from ethyl 2-aminobenzoate. First, the amino group was protected as the corresponding (toluene)sulfonamide, which afterwards was alkylated with allyl bromide under mild conditions. This led to ethyl 2-(
N-tosyl-
N-allylamino)benzoate. In the following step the ester group was reduced with LiAlH
4 and the alcohol thus prepared was finally oxidized with MnO
2 to give the required aldehyde
4 (
Scheme 3) [
6].
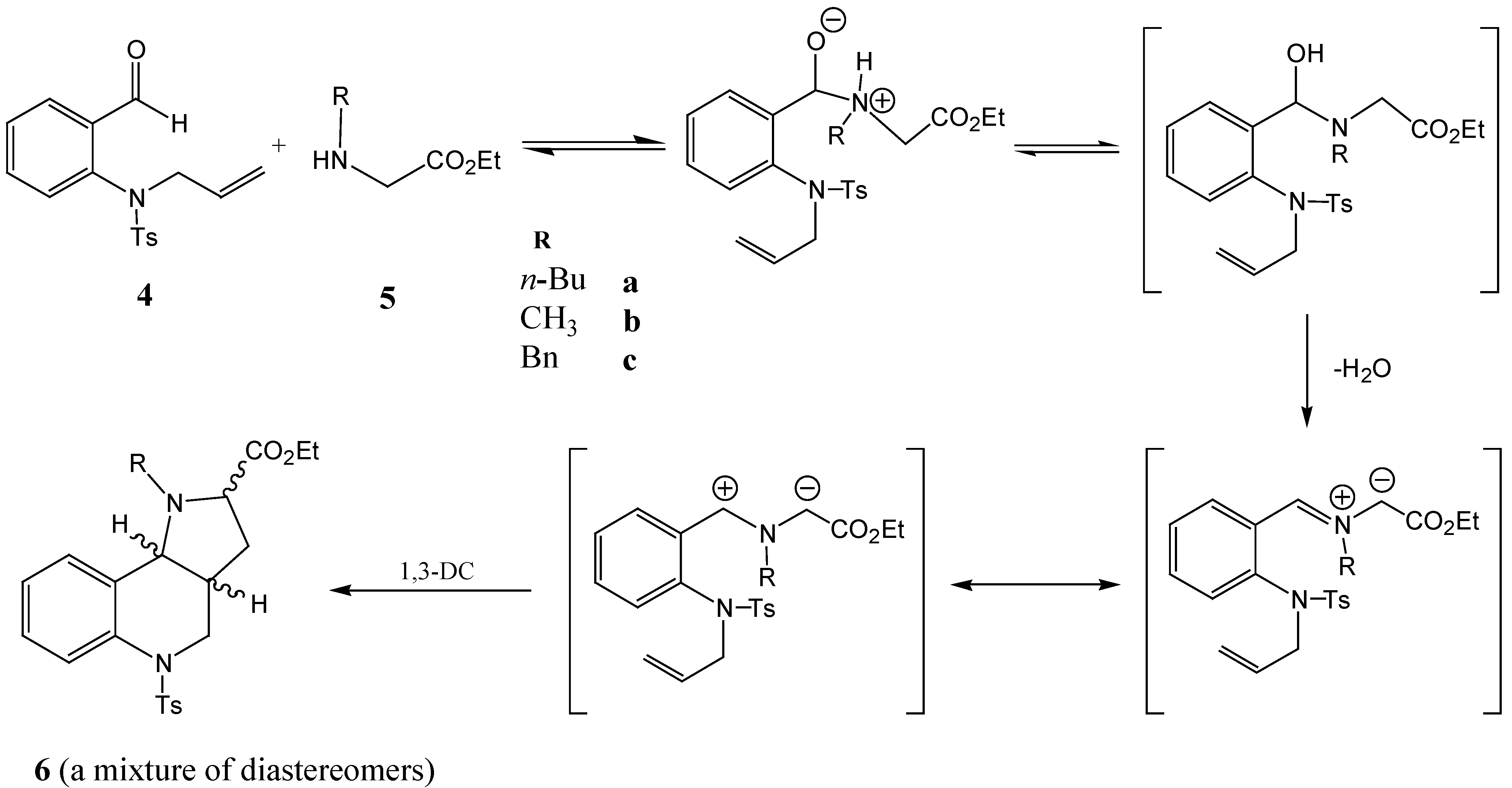
The microwave - assisted reaction of aldehyde
4 with amines
5 was optimized using amine
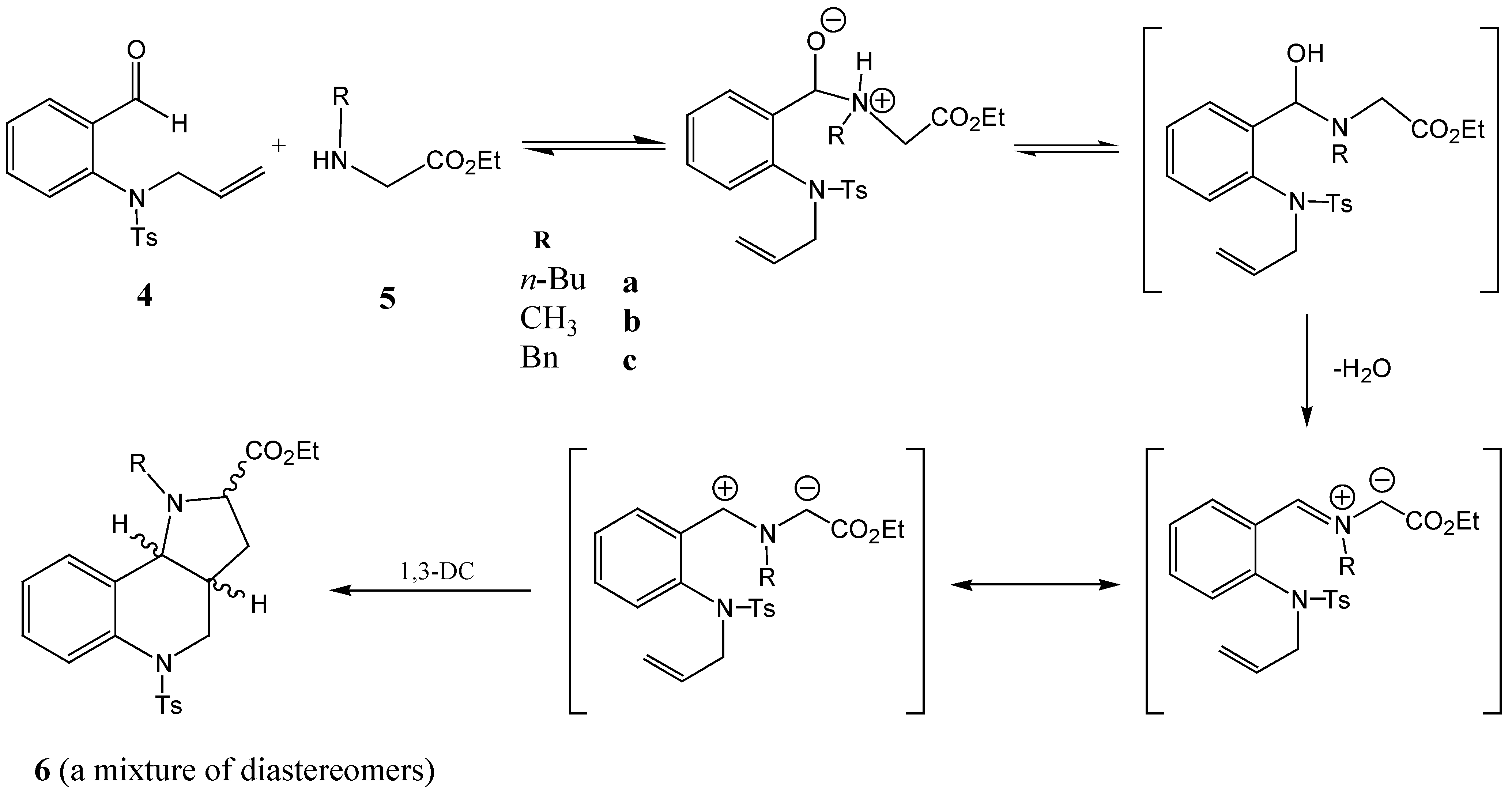
5a as a model substrate. The reactions were carried out in a Prolabo 402 microwave reactor (300 W, 2,450 MHz) and the reaction temperature was controlled by a thermocouple immersed into the reaction mixtures. The proposed mechanism of the reaction is shown in
Scheme 4. Although the mixture of components
4 and
5a was homogenized before its introduction to the reaction vessel, full homogenization did not take place until the melting point of the aldehyde
4 (110 °C) was reached. The reaction temperature was then slowly increased until decomposition of the reaction mixture occurred at a temperature of 235 °C. A temperature of 215 °C was finally found to be optimal. The conversion of aldehyde
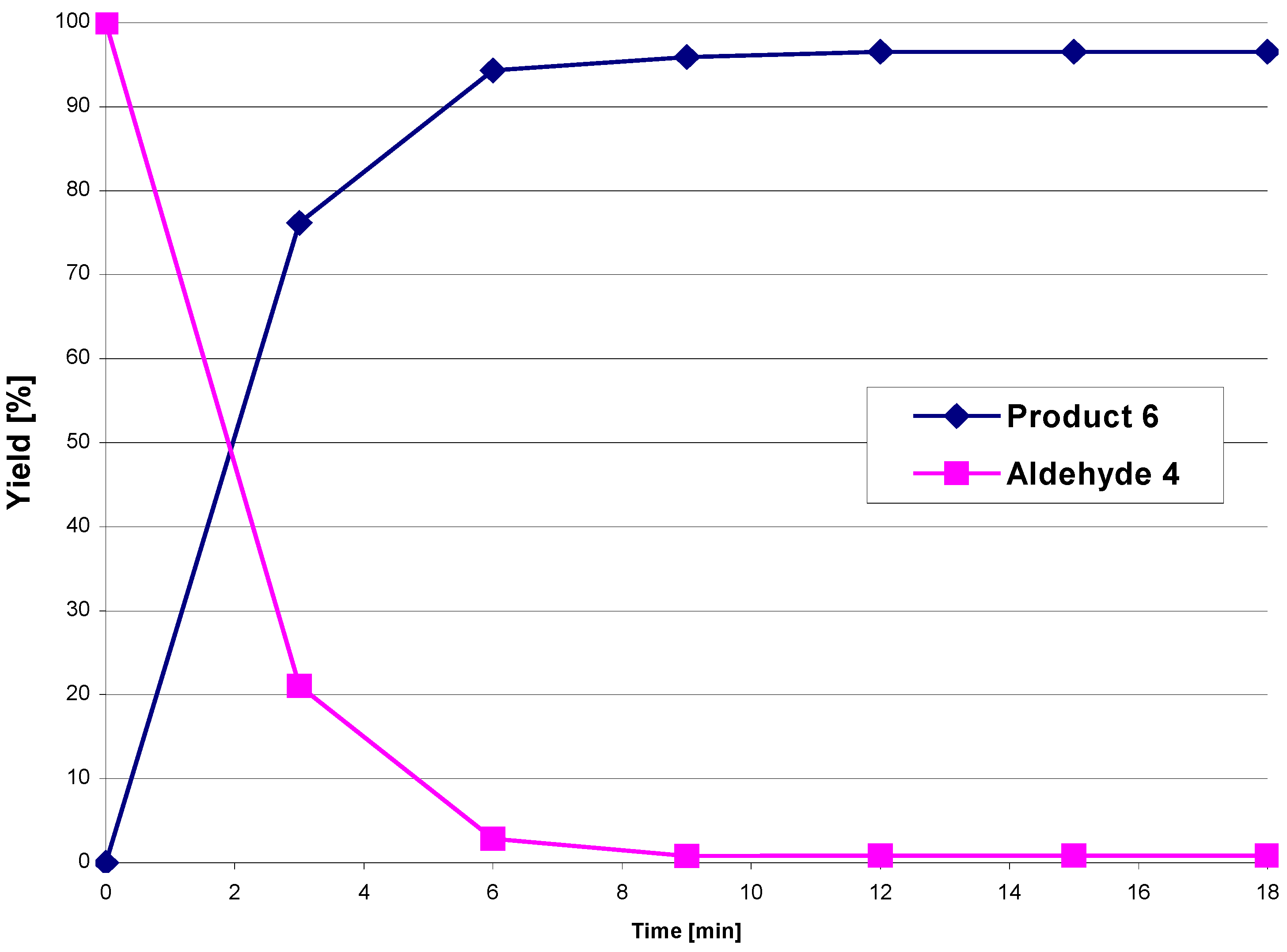
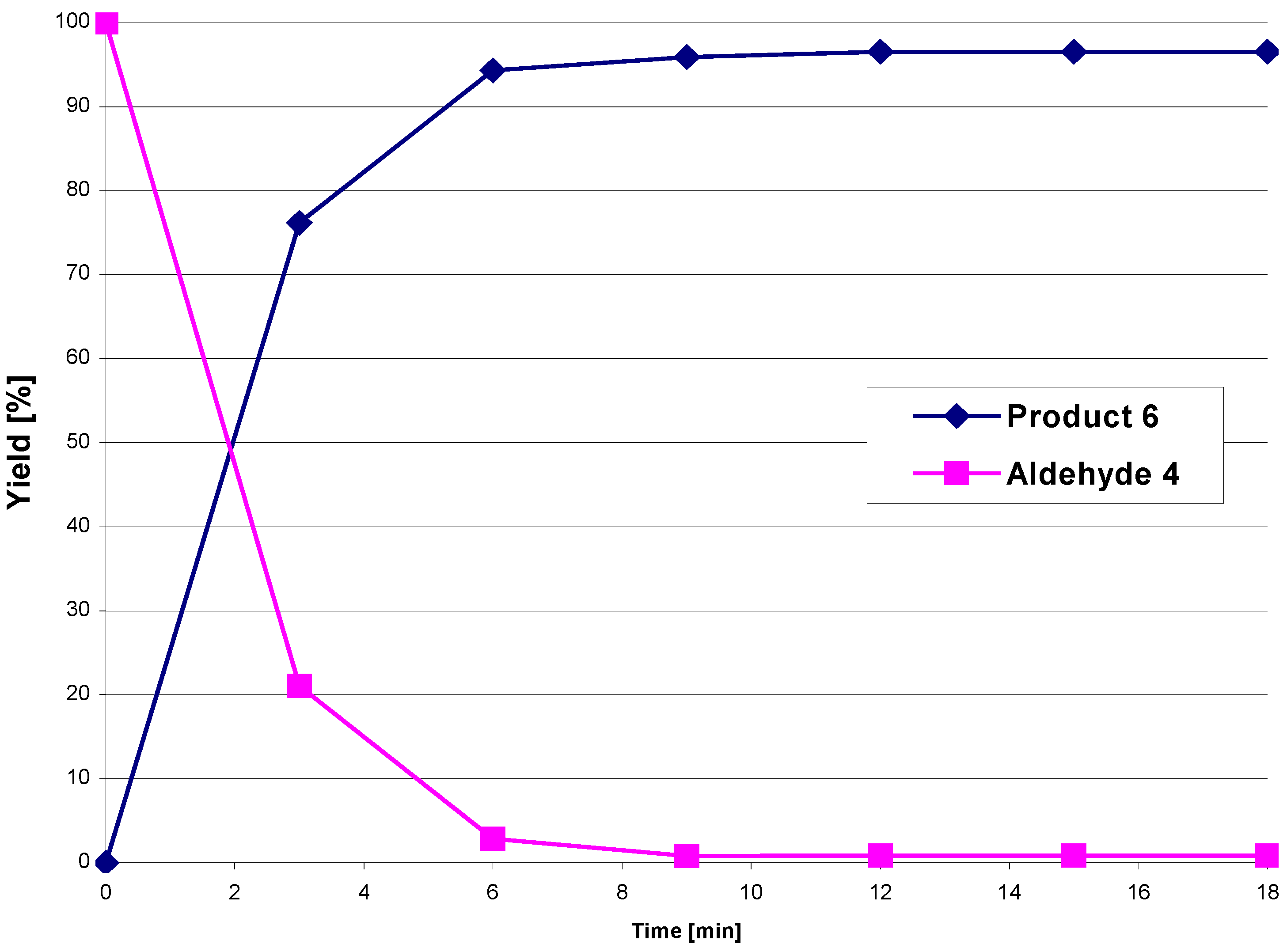
4, as well as the product formation, were followed by HPLC. The reaction was complete (no further increase in the concentrations of the reaction products was observed) within 12 min. The yields ranged between 94 – 97% (
Figure 1).
Scheme 4.
The proposed mechanism of the investigated reaction.
Scheme 4.
The proposed mechanism of the investigated reaction.
Figure 1.
Conversion of aldehyde 4 in the presence of amine 5a to the diastereomeric mixture 6 under microwave irradiation at 215 °C.
Figure 1.
Conversion of aldehyde 4 in the presence of amine 5a to the diastereomeric mixture 6 under microwave irradiation at 215 °C.
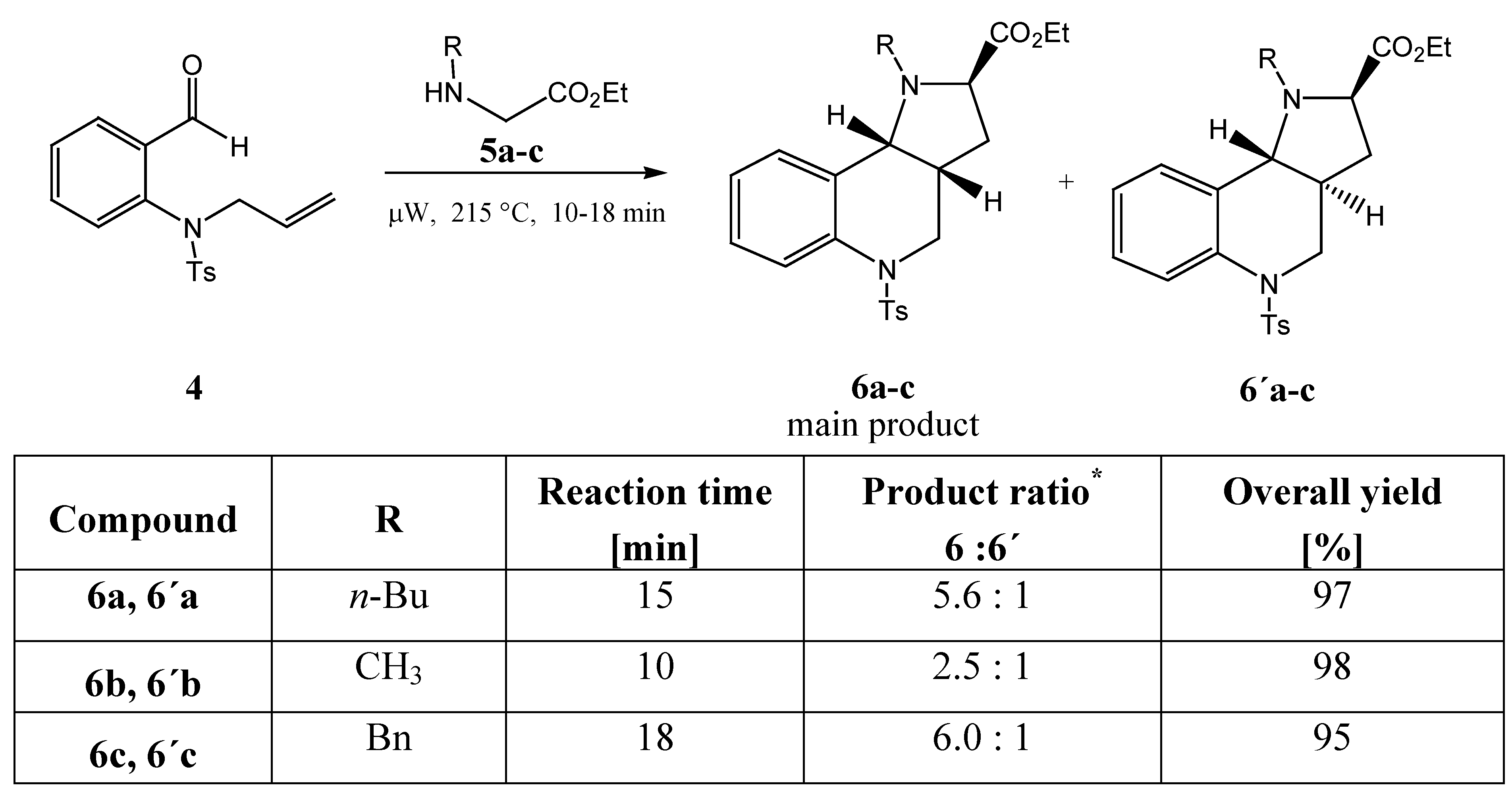
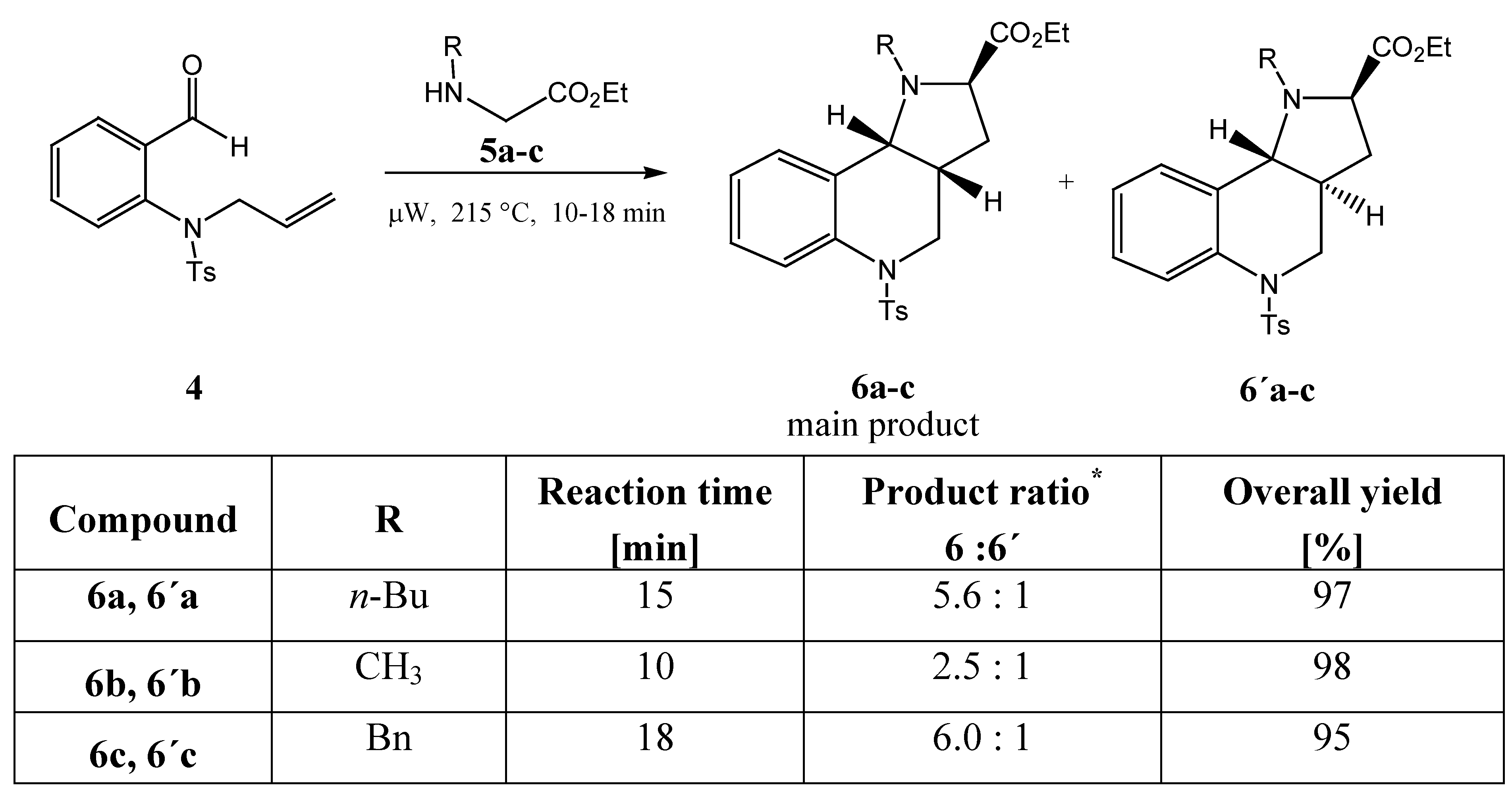
After the reaction conditions were optimized, the relative proportions of diastereomers in the reaction mixtures and the configuration at their stereogenic centres were investigated. Substituents R were found to have an effect on the diastereomeric ratio (as determined by HPLC and shown in
Scheme 5).
The relative configurations at the stereogenic centres were determined by NMR spectroscopy (HSQC, COSY and NOESY methods were applied). Only two of the four possible diastereomers were found in the reaction mixtures. When attempts to isolate them were carried out, we could not achieve their full preparative separation on the column and only the major one was isolated.
Scheme 5.
* determined by HPLC
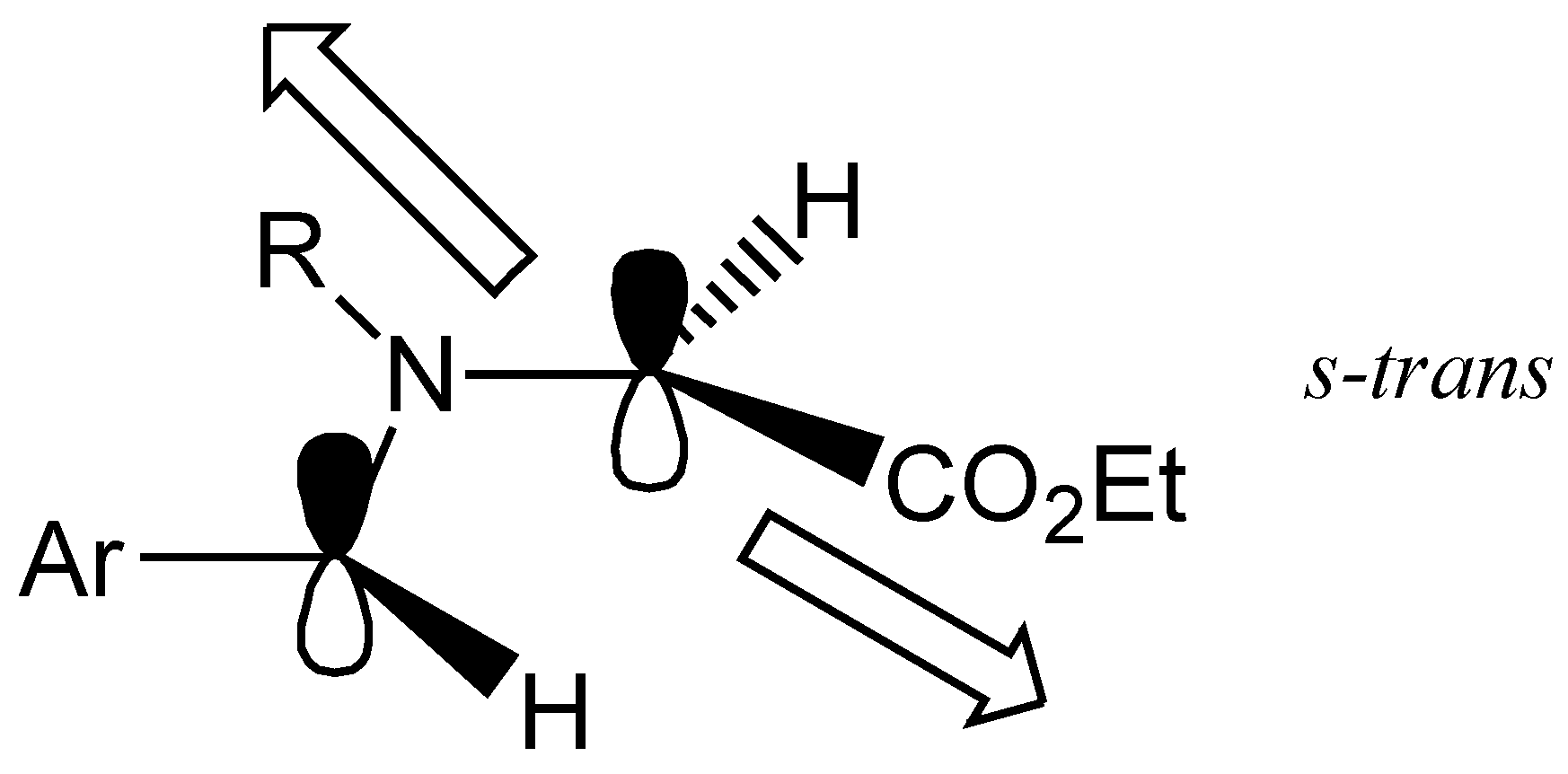
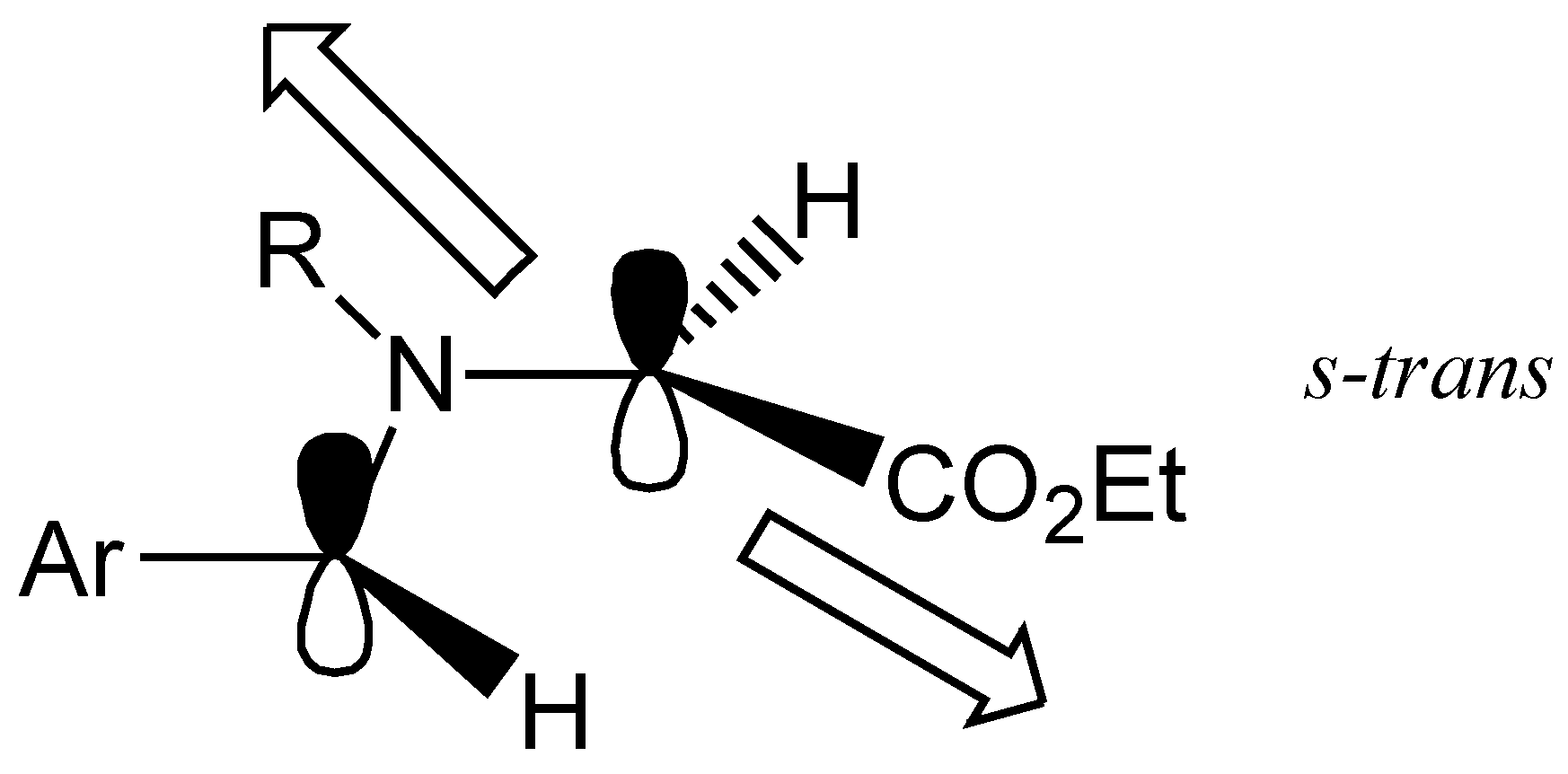
The presence of the only two diastereomers reflects the orientation of the substituents in the 1,3-dipole reacting in the cycloaddition step, which displays a
s-trans orientation of the ethoxycarbonyl with respect to the substituent on the nitrogen atom (
Figure 2).
Figure 2.
Preferred geometry of the ylide.
Figure 2.
Preferred geometry of the ylide.
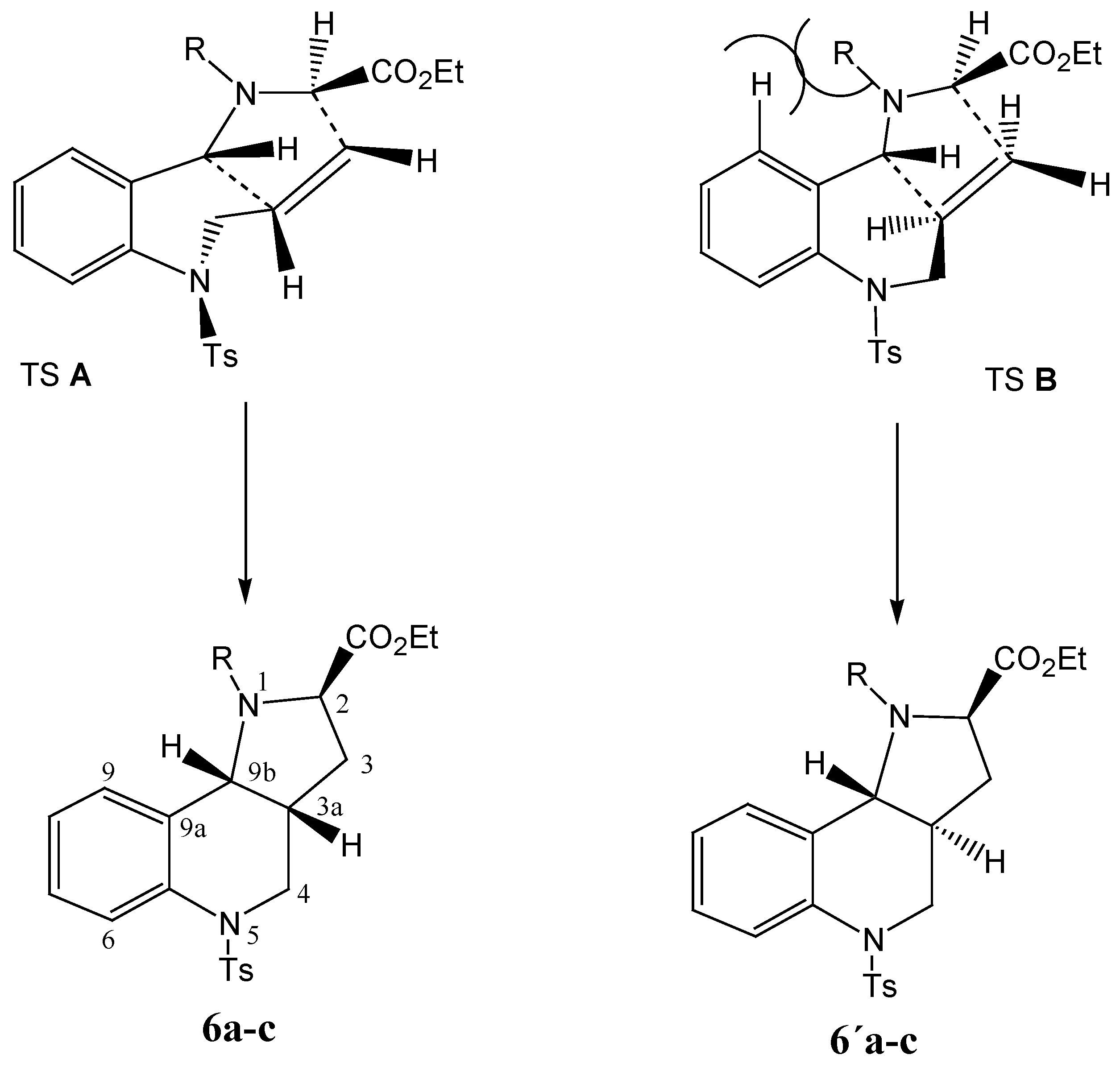
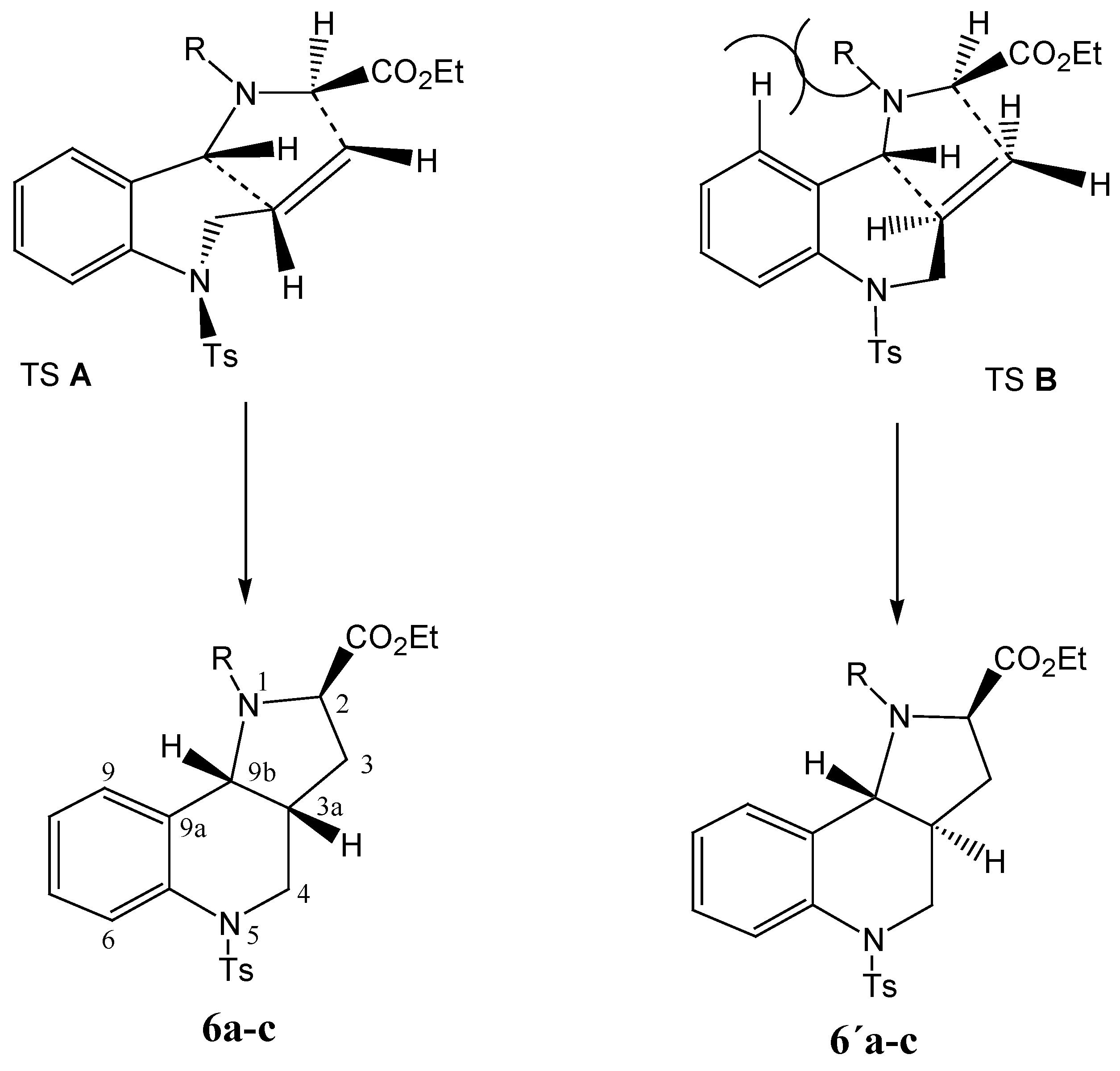
As already stated, the stereoselectivity is rather low, due to the minimal steric effects. In any case, when modelling the stereochemistry of the possible transition states, interaction of the R substitution on the nitrogen atom with the hydrogen atom of the benzene ring can be expected, making the transition state TS
B less probable (cf.
Scheme 6).
Scheme 6.
Schematic depiction of the transition states TS A and TS B during the reaction and configuration at the products 6 and 6´.
Scheme 6.
Schematic depiction of the transition states TS A and TS B during the reaction and configuration at the products 6 and 6´.
As the bulkiness of the substituents increases (methyl <
n-butyl < benzyl), the mentioned steric repulsion is higher, and increasing stereoselectivity is observed in the cycloaddition reaction. For entropy reasons the six-membered ring formed possesses the rather less energetically demanding conformation shown in TS
A. At the same time the product
6, with a
cis orientation of the hydrogen atoms on the C3a and C9b carbon atoms common to both fused rings (both hydrogen atoms are oriented equatorially) has less strain and is more flexible than the product
6´ with a
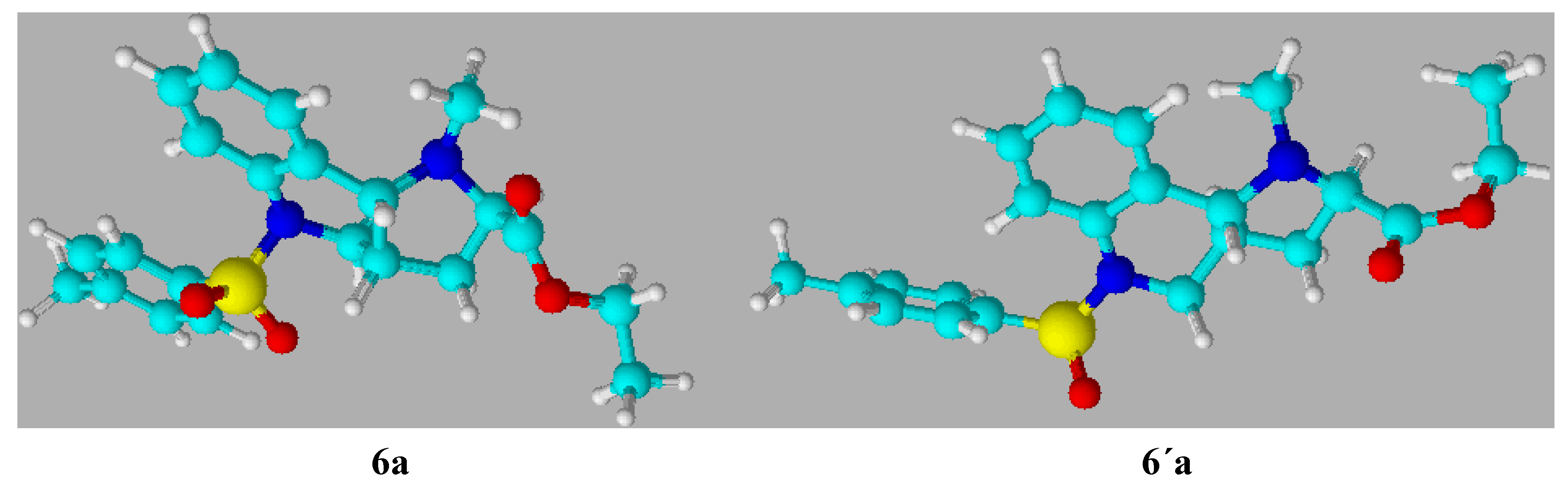
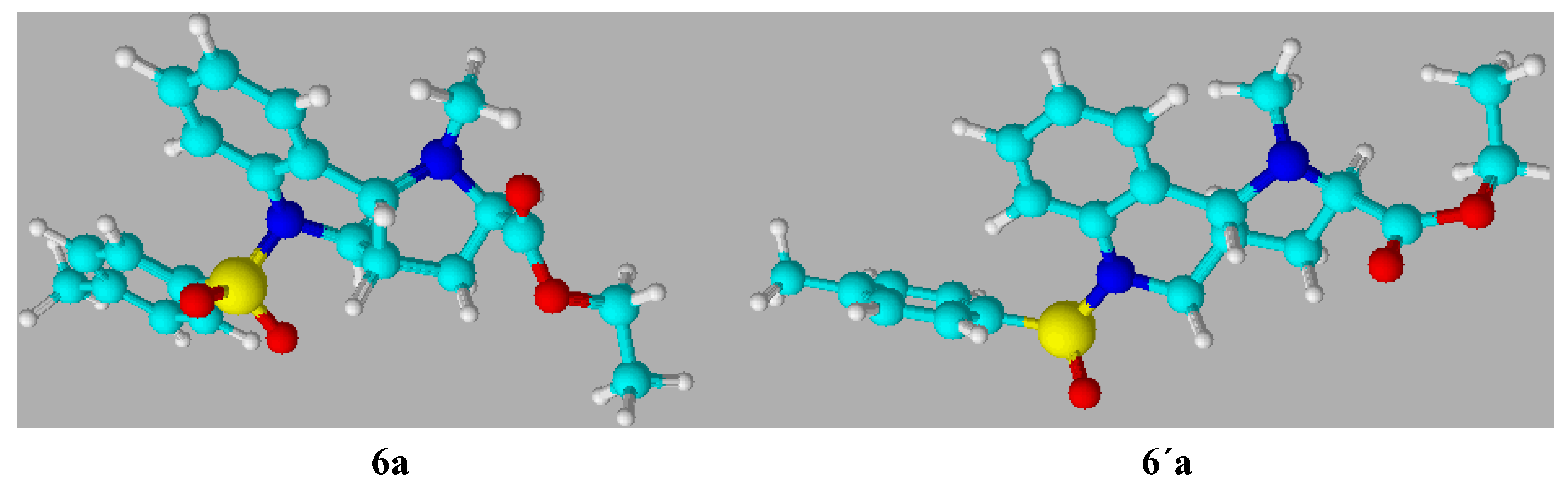
trans orientation of the hydrogen atoms, which to the contrary is more rigid. The configuration on the common atoms C3a and C9b also has an influence on the value of the dihedral angle (C9,C9a,C9b,N1), which in the case of the
cis isomer is about 70
o, while the
trans isomer has an angle of about 30
o and consequently the interaction of the R substitution on the aromatic part with the N1 atom in the
cis isomer is negligible (
Scheme 7).
Scheme 7.
Optimized view of the compounds 6a and 6´a.
Scheme 7.
Optimized view of the compounds 6a and 6´a.
The investigated reaction was compared with the same reaction carried out under the classical conditions of thermal heating in toluene in the presence of triethylamine and a catalytic amount of lithium bromide. The results are presented in
Table 2. The reaction under microwaves is substantially faster and the yield is nearly quantitative after 15 min. The classical reaction (even with the recommended catalyst [
16]) gave only a 44% yield after 18 h. As for the stereoselectivity, it was lower in the classical case. This effect might be partially due to the effect of the lithium used, which is known for its complexing ability.
Table 2.
Comparison of the reaction yields of products 6a and 6’a under different reaction conditions.
Table 2.
Comparison of the reaction yields of products 6a and 6’a under different reaction conditions.
| Products | Reaction conditions | Reaction time [min] | Product ratio | Overall yield [%] |
|---|
| 6a, 6´a | μW | 15 | 5.6 : 1 | 97% |
| 6a, 6´a | Δ, toluene, Et3N, LiBr | 1080 | 1.8 : 1 | 44% |
Experimental
General
Melting points were measured on a Küstner Nachf. KG HMK 66/1565 Kofler apparatus (Dresden, Germany). Infrared spectra were recorded in KBr pellets or as a neat film on NaCl discs on a FTIR ATI Mattson Genesis series spectrometer (wavenumbers are given in cm
-1). NMR spectra were recorded on an Bruker Avance 300 instrument, operating at 300 MHz for
1H and 75 MHz for
13C; the solvent used was CDCl
3 with an internal TMS standard. Chemical shifts (δ) are reported in ppm downfield of TMS and coupling constants (
J) in Hz. For the microwave assisted reactions a PROLABO 402 Synthewave (300 W, 2,450 MHz) instrument was used. The temperature inside the reaction vessel was measured by a thermocouple. Mass spectra were recorded on a Shimadzu GC-MS-QP 2010 in positive mode with EI ionization at 70 eV. HPLC chromatography was carried out using a Tessek SGX C18 column (15 cm length, internal diameter 3 mm) and a mobile phase of acetonitrile–water (65:35) on a Shimadzu LC-10AD VP instrument equipped with a UV–VIS detector. TLC chromatography was carried out on Kieselgel 60 F
254 (Merck) sheets. Column chromatography was carried out on silica (40–63 μm), using a mobile phase containing various ratios of hexane–ethyl acetate. The molecular modelling and the dihedral angles were estimated by the ACD/3D program, version 1.50 (ACD Labs, Toronto, Canada). Ethyl 2-bromoacetate was prepared according to the literature [
17] and used in subsequent reactions without purification. Secondary amines were prepared by the method published in [
11]. Ethyl 2-(4-toluenesulfonylamino)benzoate was prepared according to [
6] and used for preparation of the starting aldehyde
4 via ethyl 2-[
N-allyl-
N-4-(toluenesulfonyl)amino]benzoate and
N-(2-hydroxymethyl-phenyl)-
N-allyl-4-toluenesulfonamide.
General Method for the Synthesis of Secondary Amines 5 [11].
A primary amine (4.0 mmol) was dissolved in dry acetonitrile (2 mL) and the reaction mixture was cooled down to 0 oC. Then, under vigorous agitation, ethyl 2-bromoacetate (0.17 g, 1.0 mmol) was slowly added. During the addition the temperature was kept at 0 °C. The reaction mixture was then left under stirring at room temperature for 4 h. The solvent was evaporated, the residue poured into 2M NaOH solution (2 mL) and the organic layer was extracted by ether (3 x 3 mL), dried over MgSO4 and after filtration the solvent was evaporated. The pure product was obtained by distillation under vacuum.
General Method for the Preparation of Hexahydropyrrolo[3,2-c]quinolines 6: Microwave - Assisted Intramolecular Cycloaddition.
Aldehyde 4 (1.0 mmol) was mixed with secondary amine 5a-c (1.0 mmol), the mixture was homogenized in a reaction tube, placed in the microwave oven and heated for 15 min to a temperature of 200 – 215 °C. The diastereomeric ratio was determined by HPLC and the structure confirmed by 1H‑, 13C-NMR and 2D NMR experiments. Afterwards, the reaction mixture was separated on a silica gel column.
Ethyl (2R*,3aR*,9bR*)-1-(n-butyl)-5-(4-toluenesulfonyl)-2,3,3a,4,5,9b-hexahydro-1H-pyrrolo [3,2-c]-quinoline-2-carboxylate (6a).
Column chromatography (hexane-ethyl acetate = 4:1) gave 0.57 g of 6a (yellow oil) from aldehyde 4 (0.9 g, 2.84 mmol) and amine 5a (0.45 g, 2.84 mmol). 1H-NMR: δ = 0.73 (t, 3H, 3J = 7.1, NCH2(CH2)2CH3), 1.12 (m, 2H, N(CH2)2CH2CH3), 1.18 (m, 2H, NCH2CH2CH2CH3), 1.29 (t, 3H, 3J = 6.9, CO2CH2CH3), 2.08 (dddd, 2H, 2J3,3´ = 3.3, 3J3´,3a = 13.2, 3J3,3a = 8.3, 3J3´,2 = 3.6, H-3 and H-3’), 2.37 (s, 3H, NSO2PhCH3), 2.46 - 2.58 (m, 2H, NCH2(CH2)2CH3), 2.70 (ddd, 1H, 3J3a,3´ = 13.2, 3J3a,9b = 5.6, 3J3a,4 = 3.3, H-3a), 3.69 (ddd, 2H, 2J4,4’ = 5.0, 3J4´,3a = 13.2, 3J4,3a = 3.3, H-4 and H-4’), 3.79 (dd, 1H, 3J2,3 = 8.3, 3J2,3´ = 3.6, H-2), 3.85 (d, 1H, 3J9b,3a = 5.6, H-9b), 4.17 (q, 2H, 3J = 6.9, CO2CH2CH3), 7.07-7.65 (m, 8H, Ar-H); 13C-NMR: δ = 13.2 (NCH2(CH2)2CH3), 13.9 (CO2CH2CH3), 19.7 (N(CH2)2CH2CH3), 20.9 (NSO2PhCH3), 29.6 (NCH2CH2CH2CH3), 31.6 (C-3), 36.9 (C-3a), 46.8 (NCH2(CH2)2CH3), 50.1 (C-4), 59.5 (CO2CH2CH3), 60.3 (C-2), 60.5 (C-9b), 121.6, 123.3, 126.5 (2xC), 127.1, 129.0 (2xC), 129.7, 130.6, 137.1, 138.5, 143.0 (Carom), 173.6 (C=O); IR (neat) νmax/cm‑1: 3437, 2956, 2868, 1726, 1601, 1489, 1456, 1350, 1165, 1032, 813, 760, 679, 577; EI-MS (m/z, %): 457 (51, M+), 383 (100), 227 (27), 171 (21), 144 (8), 130 (19), 91 (37), 41 (14).
Ethyl (2R*,3aR*,9bR*)-1-methyl-5-(4-toluenesulfonyl)-2,3,3a,4,5,9b-hexahydro-1H-pyrrolo[3,2-c]- quinoline-2-carboxylate (6b).
After reaction of aldehyde 4 (0.55 g, 1.72 mmol) and amine 5b (0.20 g, 1,72 mmol) column chromatography (hexane-ethyl acetate = 4:1) gave 0.36 g of 6b (yellow oil). 1H-NMR: δ = 1.25 (t, 3H, 3J = 6.9, CO2CH2CH3), 2.40 (s, 3H, NSO2PhCH3), 2.48 (s, 3H, NCH3), 3.22 (d, 2H, 3J3´,3a = 13.9, 3J3´,2 = 13.8, H-3), 3.67 (dd, 1H, 3J3a,9b = 7.3, 3J3a,3´ = 13.9, H-3a), 3.73 (q, 2H, 2J = 6.9, CO2CH2CH3), 3.83 (d, 2H, 3J2,3´ = 13.8, H-2), 4.04 (d, 2H, 3J9b,3a = 7.3, H-9b), 4.27 (dd, 1H, 3J4´,3a = 11.2, 3J4,3a = 5.6, H4), 7.21 - 7.64 (m, 8H, Ar-H); 13C-NMR: δ = 14.6 (CO2CH2CH3), 20.9 (NSO2PhCH3), 30.4 (C-3), 35.20 (C-3a), 41.7 (NCH3), 58.4 (C-2), 59.5 (CO2CH2CH3), 60.5 (C-9b), 68.3 (C-4), 117.3 - 156.0 (Carom), 174.5 (C=O); IR (neat) νmax/cm-1: 3022, 2944, 2882, 1722, 1609, 1580, 1490, 1451, 1230, 1195, 1048, 761; EI-MS (m/z, %): 415 (47, M+), 343 (100), 189 (27), 144 (8), 130 (28), 91 (25), 57 (19).
Ethyl (2R*,3aR*,9bR*)-1-benzyl-5-(4-toluenesulfonyl)-2,3,3a,4,5,9b-hexahydro-1H-pyrrolo[3,2-c]- quinoline-2-carboxylate (6c).
From aldehyde 4 (0.53 g, 1.69 mmol) and amine 5c (0.32 g, 1.69 mmol) column chromatography (hexane/ethyl acetate = 2.5:1) gave 0.38 g of 6c (yellow oil). 1H-NMR: δ = 1.25 (t, 3H, 3J = 7.6, CO2CH2CH3), 2.42 (s, 3H, NSO2PhCH3), 2.81 (ddd, 1H, 3J3a,3 = 6.3, 3J3a,9b = 5.6, 3J3a,4 = 13.2, H-3a), 3.54 (dd, 1H, 3J2,3 = 5.9, 3J2,3´ = 12.9, H-2), 3.63 (d, 1H, 2J4´,4 = 4.6, H-4´), 3.77 (dddd, 2H, 2J = 4.6, 3J3,2 = 5.9, 3J3,3a = 6.3, 3J3´,2 = 12.9, H-3), 3.62 (d, 1H, 3J4,3a = 13.2, H-4), 4.08 (s, 2H, PhCH2N), 4.13 (q, 2H, 3J = 7.6, CO2CH2CH3), 4.18 (d, 1H, 3J9b,3a = 5.6, H-9b), 7.05 (m, 2H, Ar-H), 7.18 - 7.21 (m, 8H, Ar-H), 7.60 - 7.73 (m, 3H, Ar-H); 13C-NMR: δ = 15.4 (CO2CH2CH3), 25.84 (C-3), 31.2 (NSO2PhCH3), 38.0 (C-3a), 44.9 (NCH2Ph), 45.6 (C-4), 53.9 (CO2CH2CH3), 57.8 (C-2), 58.8 (C-9b), 115.86, 117.74, 120.71, 120.96 (2xC), 121.95, 122.24 (2xC), 122.30 (2xC), 123.6 (2xC), 125.2, 123.4, 131.6, 132.8, 132.9, 137.4 (Carom), 167.9 (C=O); IR (neat) νmax/cm-1: 3058, 3028, 2956, 2876, 1491, 1348, 1164, 773; EI-MS (m/z, %): 491 (10, M+), 417 (86), 399 (5), 335 (4), 261 (14), 171 (56), 91 (100), 65 (20).
Intramolecular 1,3-Dipolar Cycloaddition Under Classical Conditions: Ethyl (2R*,3aR*,9bR*)-1-(n-butyl)-5-(4-toluenesulfonyl)-2,3,3a,4,5,9b-hexahydro-1H-pyrrolo[3,2-c]quinoline-2-carboxylate (6a).
Aldehyde 4 (0.51 g, 1.6 mmol) was dissolved in dry toluene (30 mL) and then amine 5a (0.30 g, 1.9 mmol), triethylamine (0.25 g, 2.5 mmol) and dried LiBr (0.17 g, 2 mmol) were added. The reaction mixture was refluxed for 18 h, until all starting materials had disappeared (as tested by TLC). The solvent was then evaporated and the residue dissolved in water (30 mL) and extracted with CHCl3 (2 x 30 mL). The extract was dried over MgSO4 and, after filtration and removal of the solvent, a yellow oily residue was obtained, which was purified by column chromatography. Yield 0.32 g (44%).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}