Synthesis of (R)-2-(Benzyloxy)-tetrahydro-5,5-dimethylfuran by a New Oxidative Rearrangement


{kind=link}
{kind=link}
{kind=link}
{kind=link}
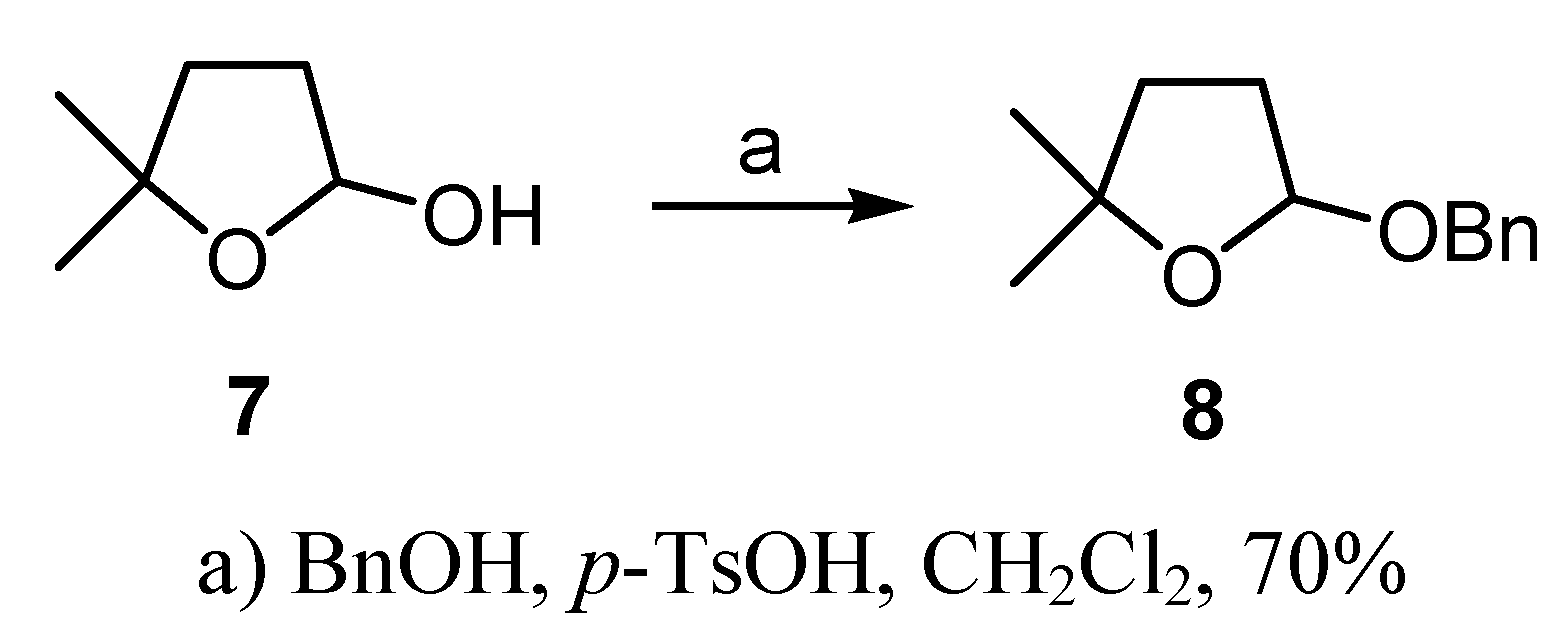{kind=link}
{kind=link}
{kind=link}
Abstract
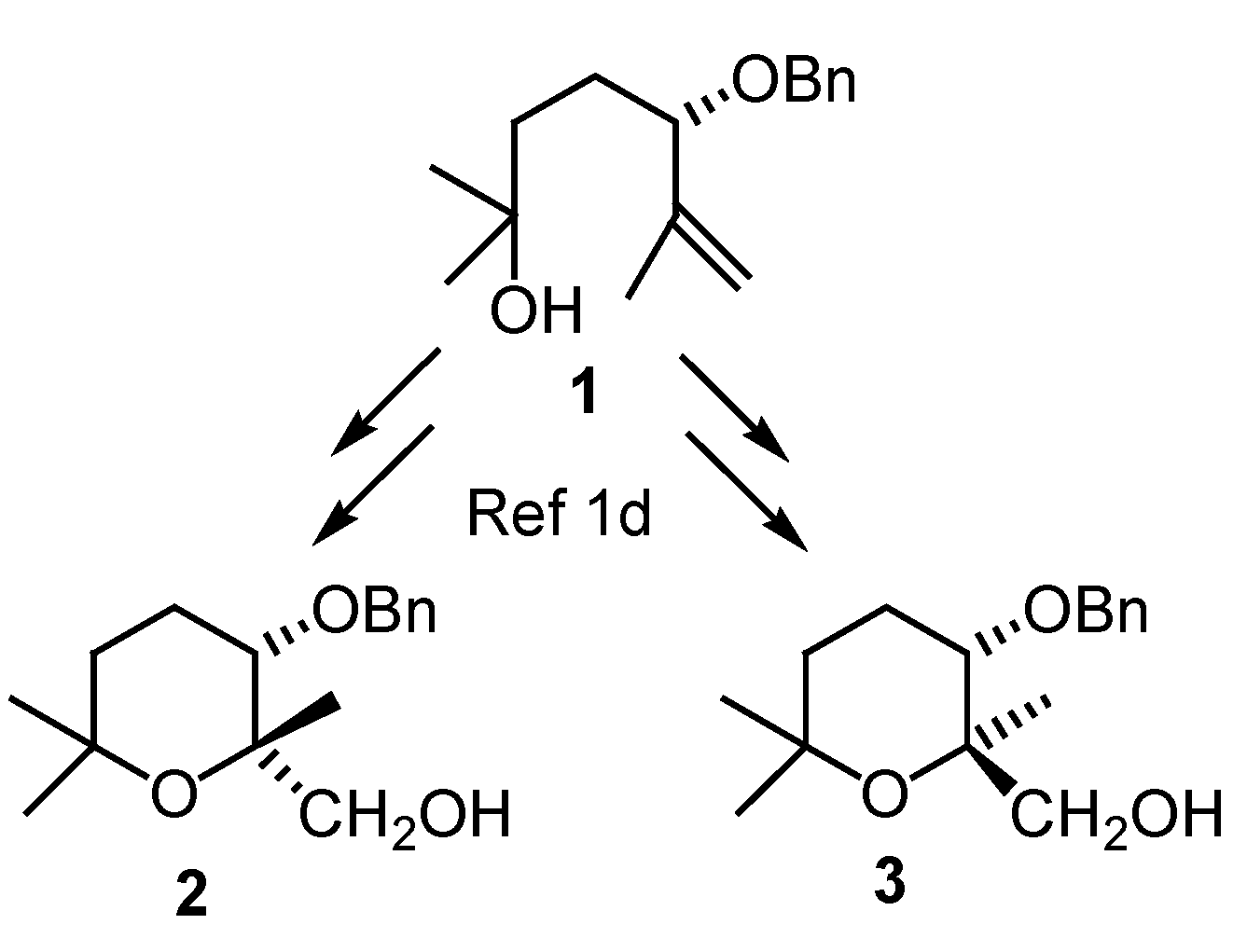
:Introduction
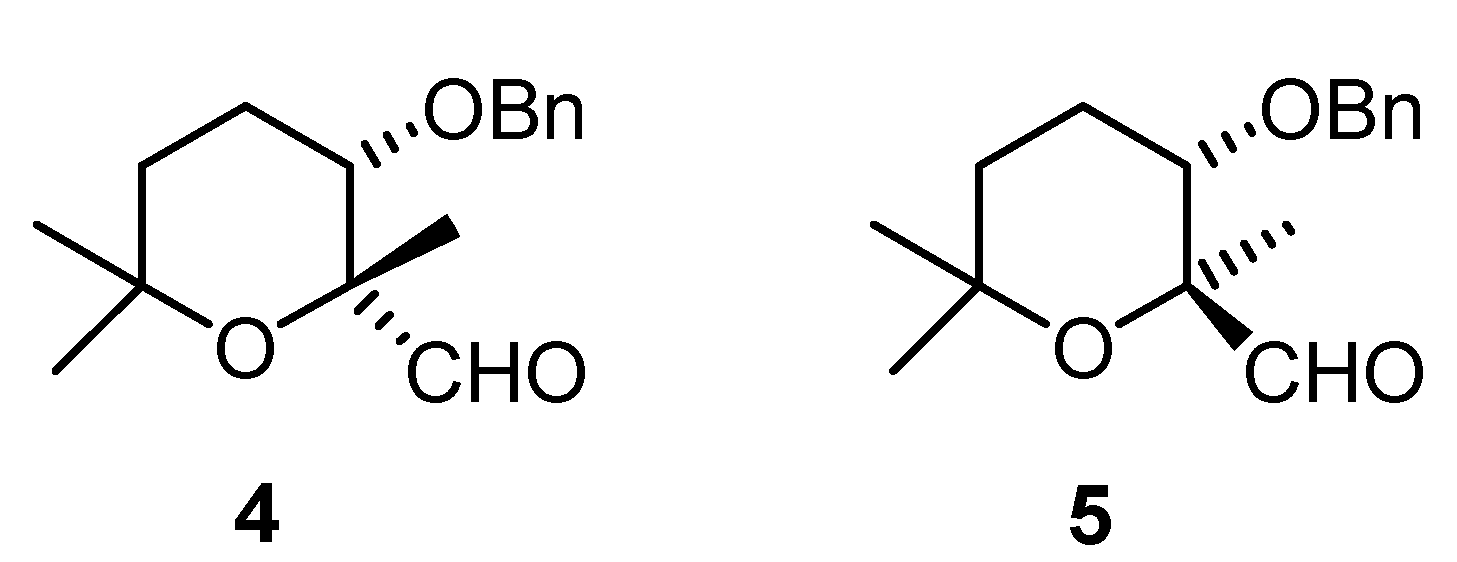
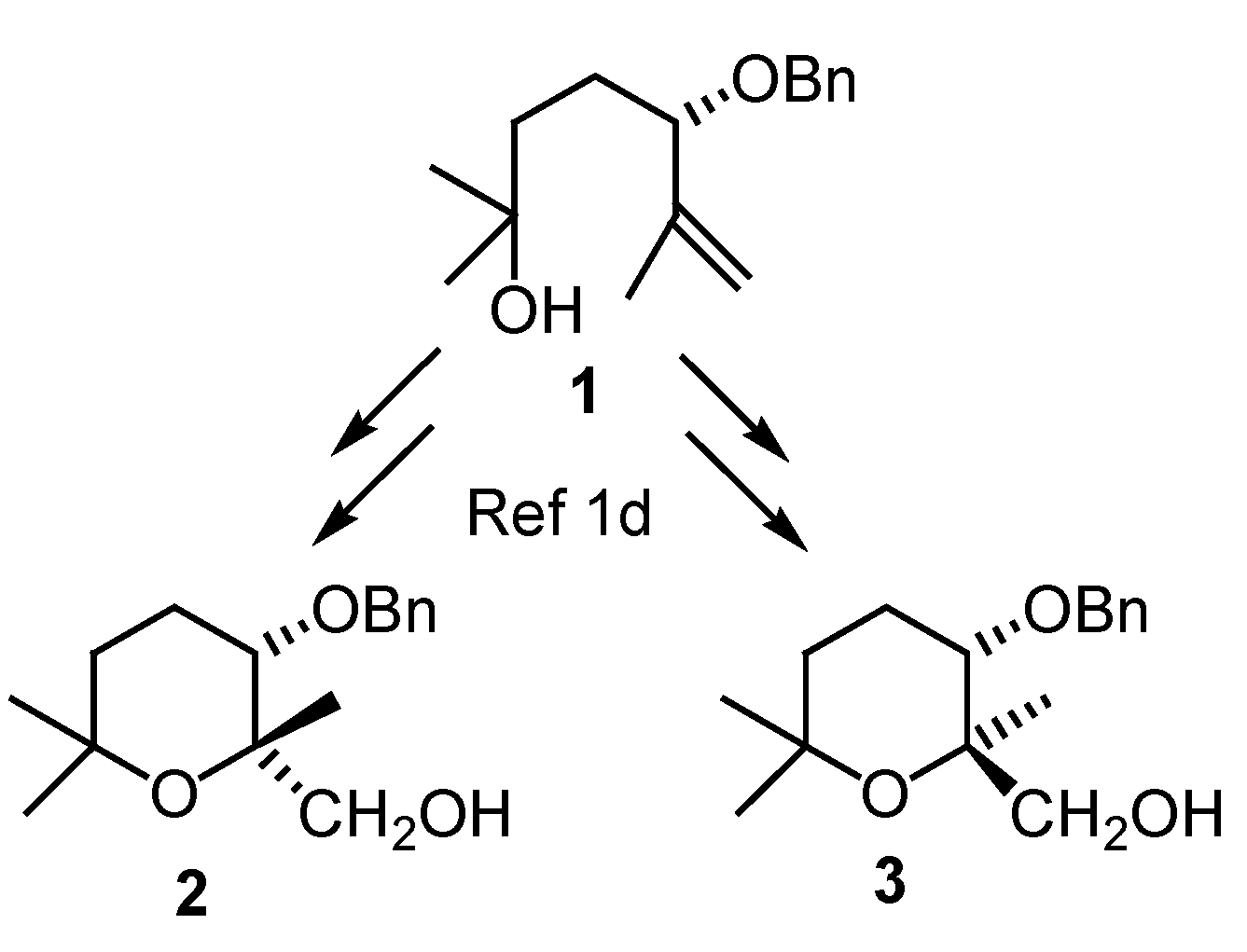
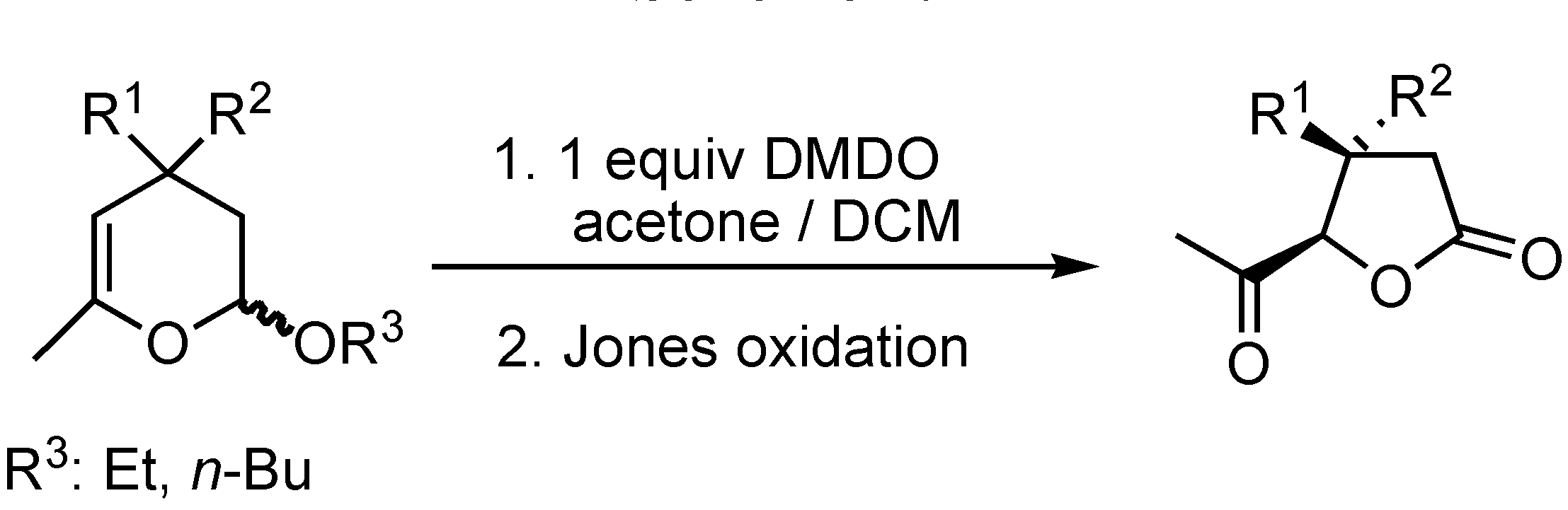
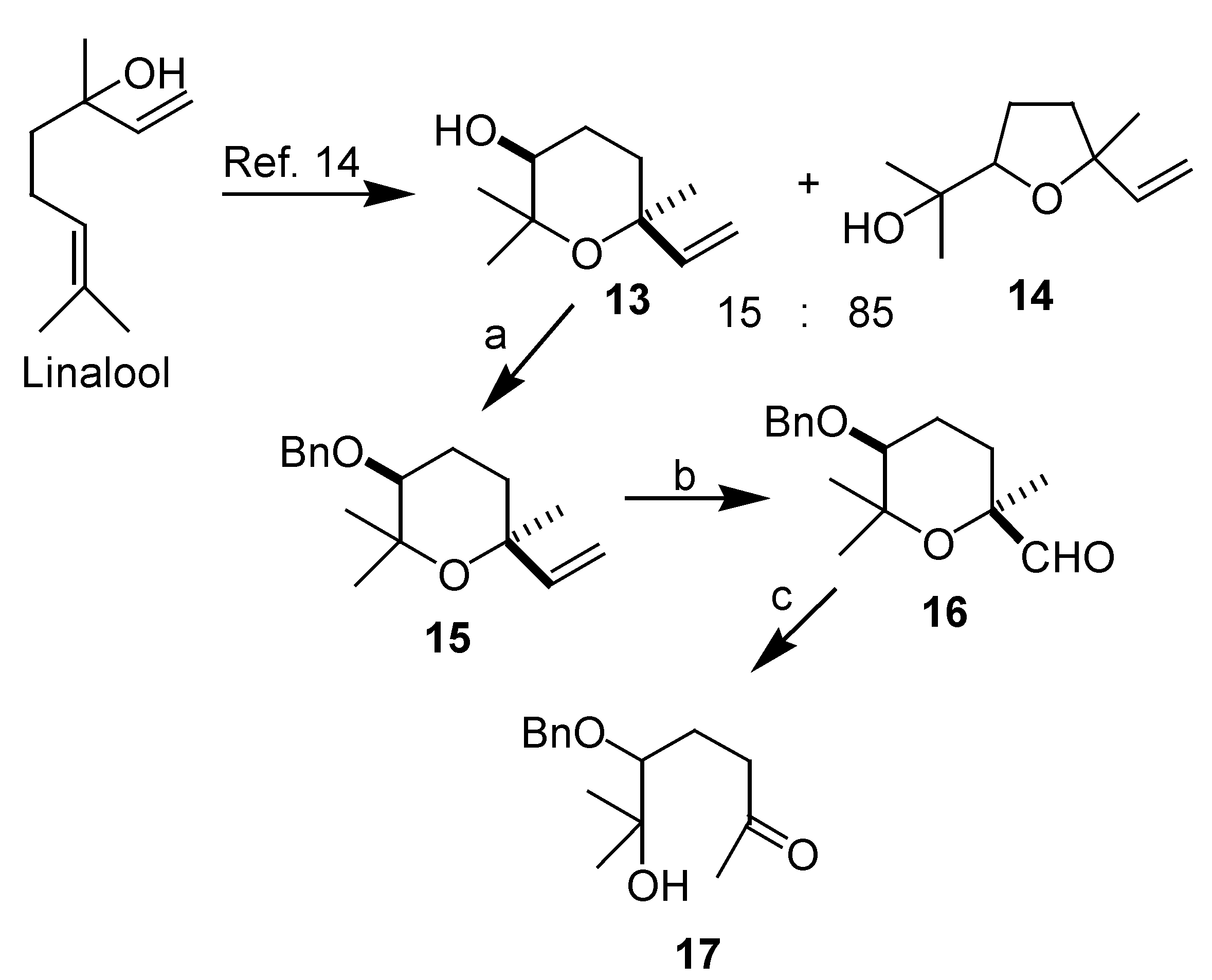
Results and Discussion
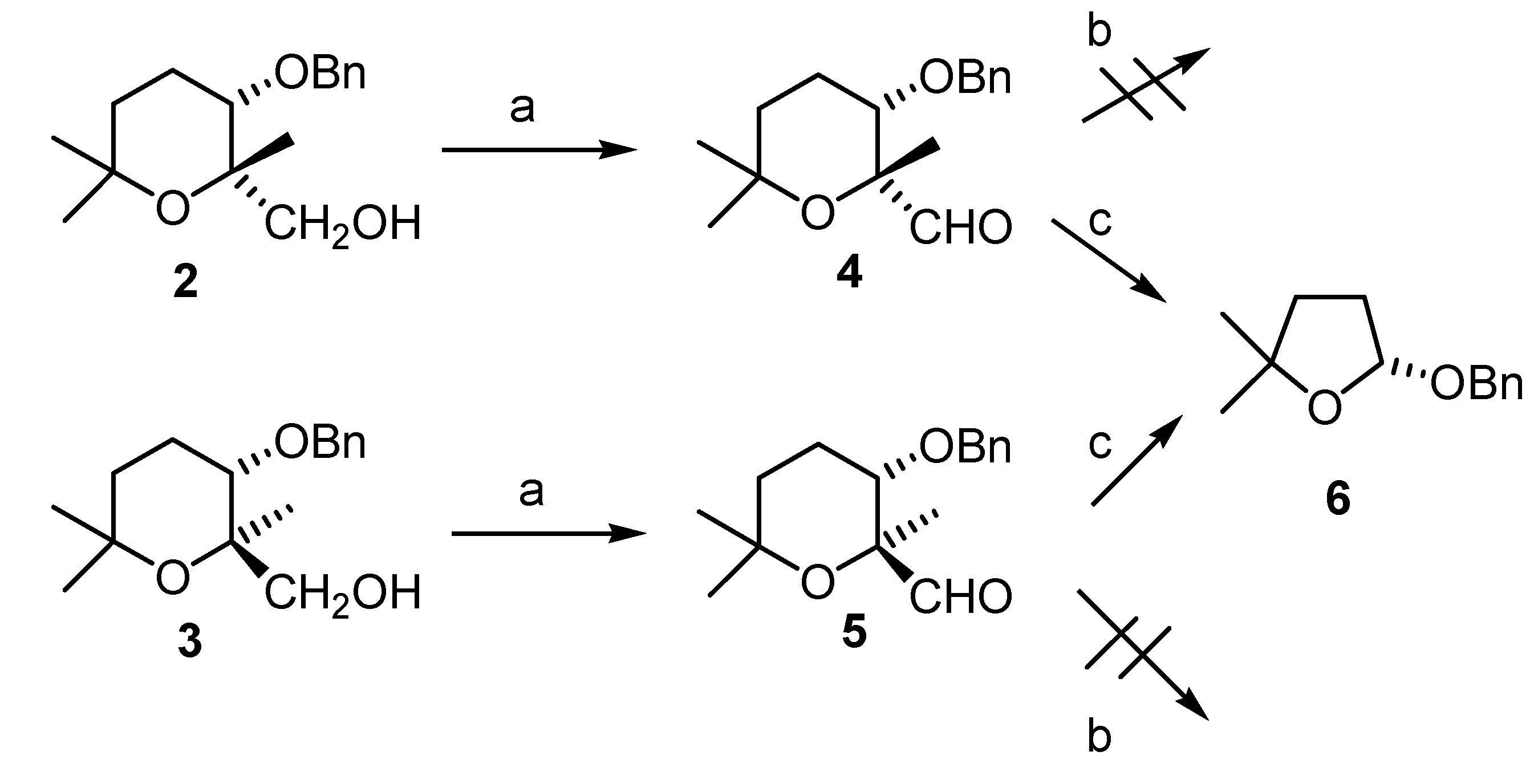
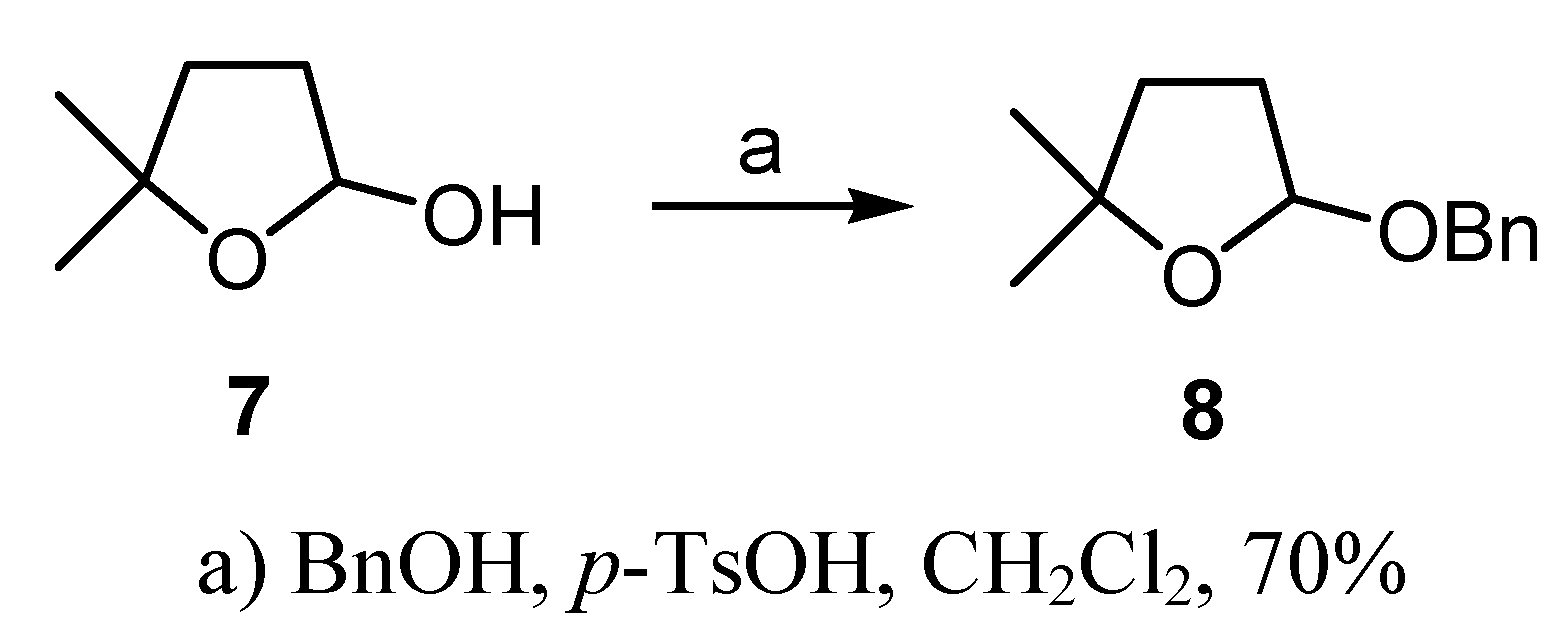
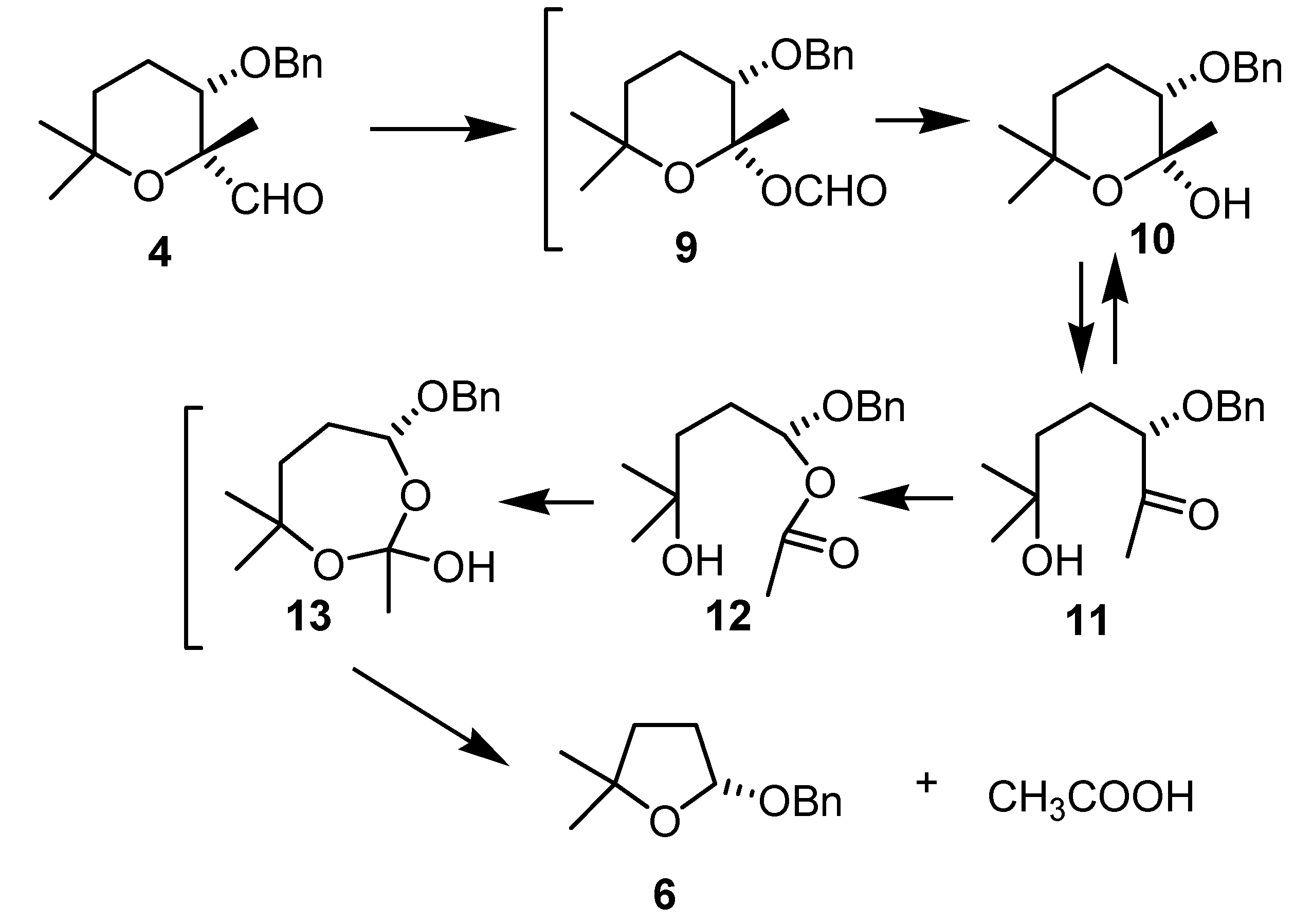
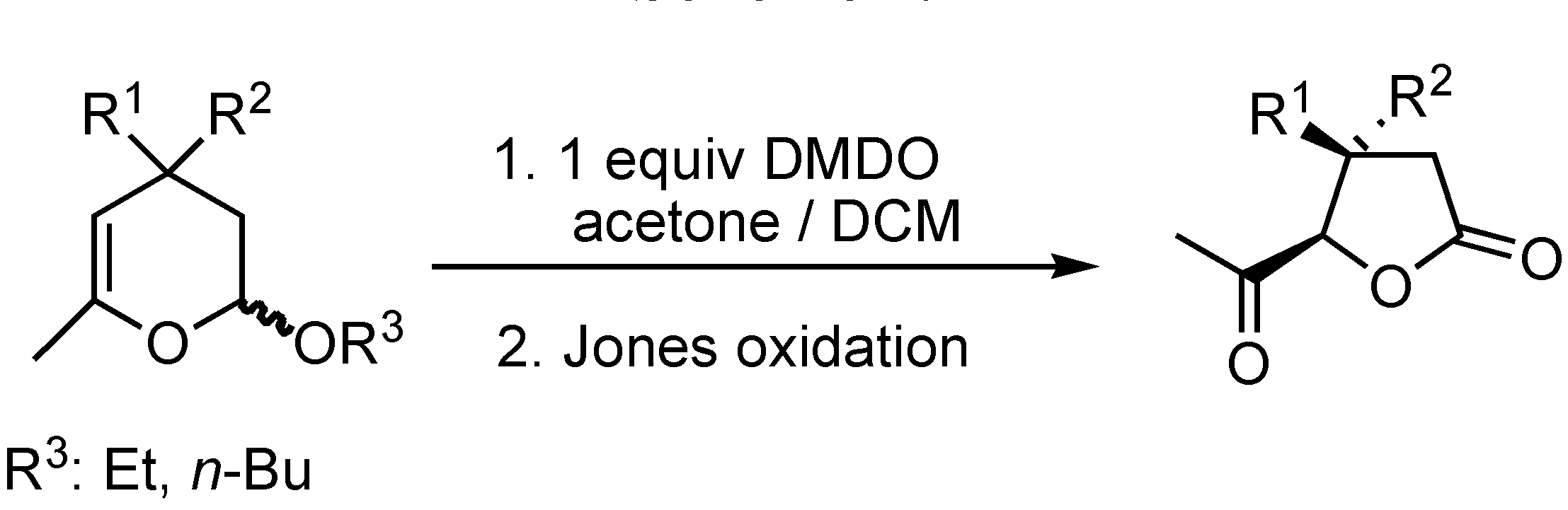
Conclusions
Experimental
General
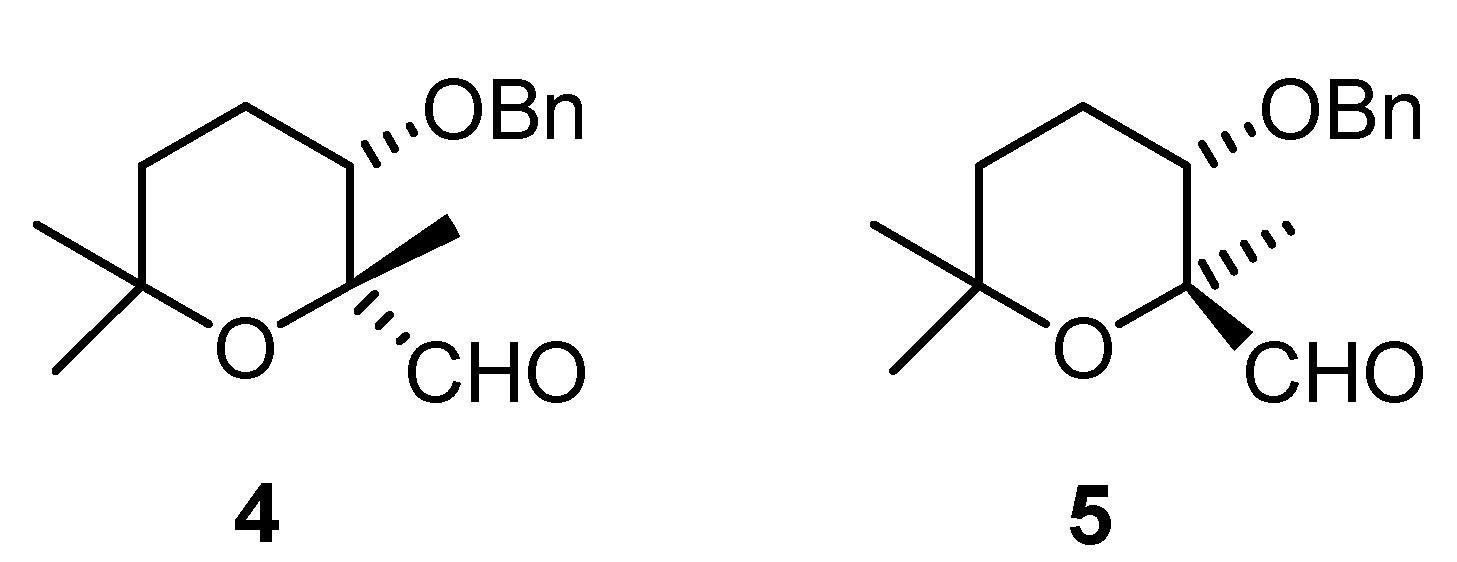
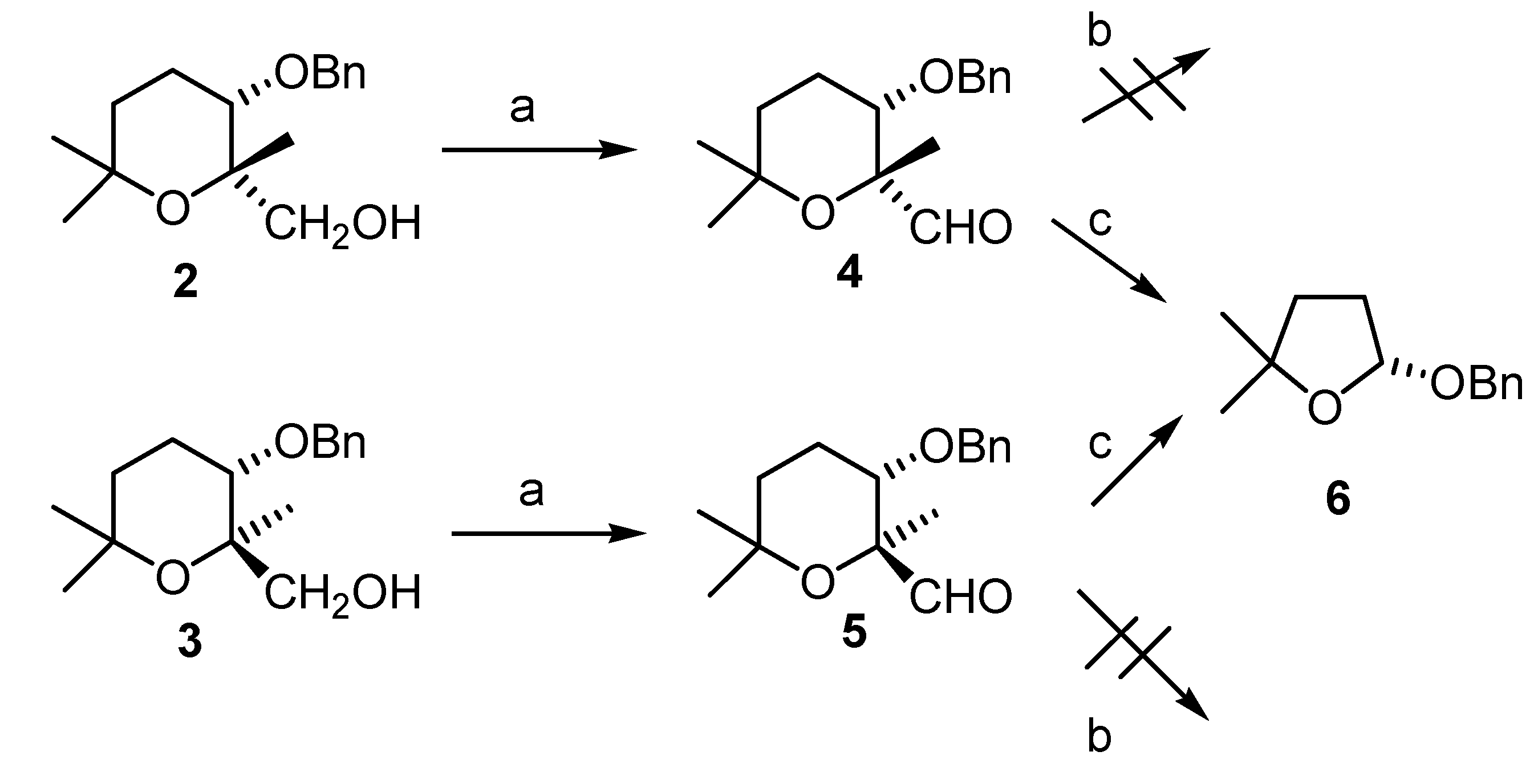
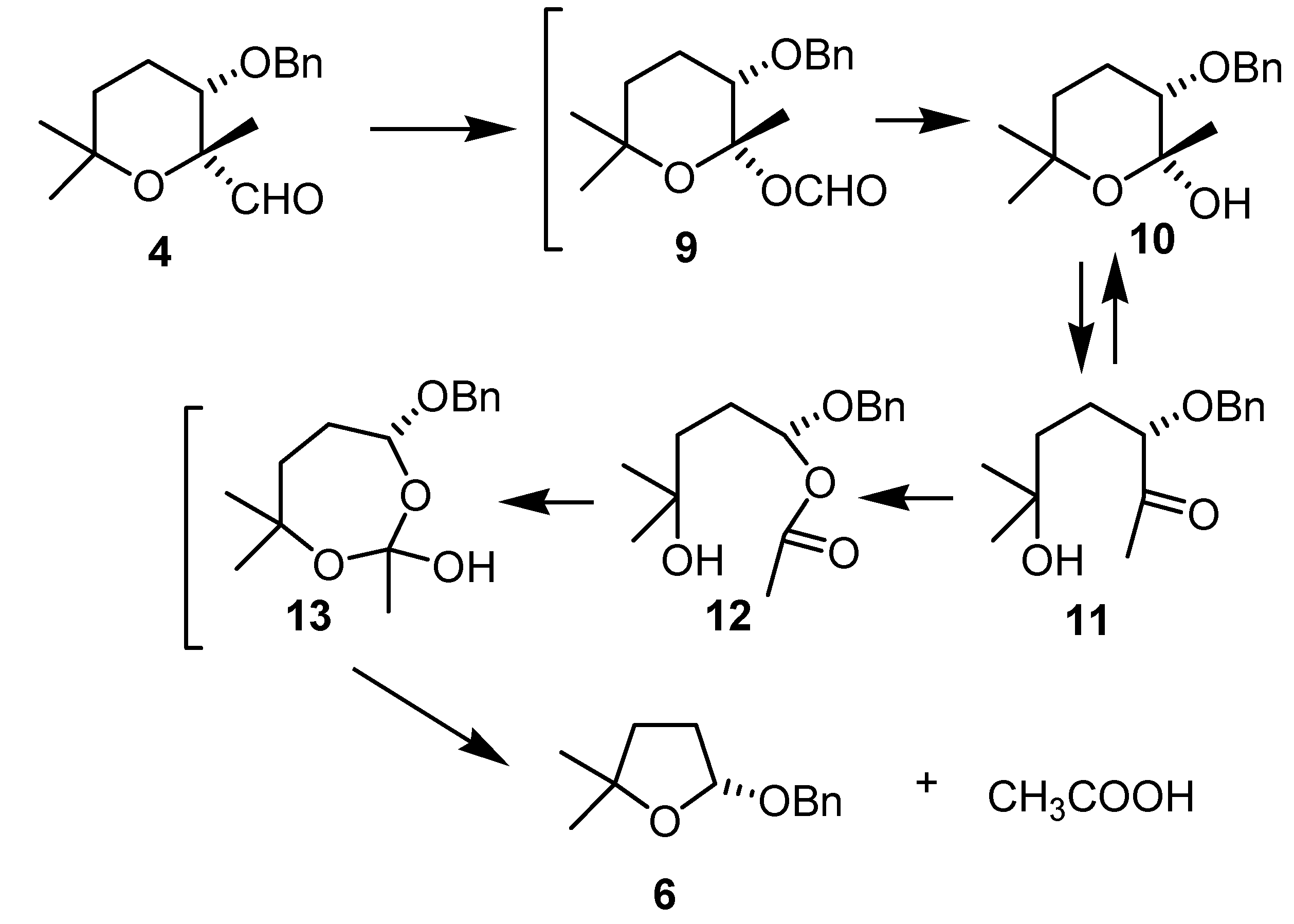
(2R,3S)-3-(benzyloxy)-tetrahydro-2,6,6-trimethyl-2H-pyran-2-carbaldehyde (4)
(2S,3S)-3-(benzyloxy)-tetrahydro-2,6,6-trimethyl-2H-pyran-2-carbaldehyde (5).
(R)-2-(Benzyloxy)-tetrahydro-5,5-dimethylfuran (6)
(±)-2-(Benzyloxy)-tetrahydro-5,5-dimethylfuran (8)
3-(benzyloxy)-tetrahydro-2,2,6-trimethyl-6-vinyl-2H-pyran (15)
5-(benzyloxy)-tetrahydro-2,6,6-trimethyl-2H-pyran-2-carbaldehyde (16)
5-(benzyloxy)-6-hydroxy-6-methylheptan-2-one (17)
Acknowledgements
References and Notes
- Urones, J. G.; Díez, D.; Marcos, I. S.; Basabe, P.; Garrido, N. M.; Escarcena, R.; Lithgow, A. M.; Dominguez, M. F.; Sánchez, J. M. Synlett 1995, 855–856.Díez, D.; Marcos, I. S.; Basabe, P.; Romero, R. E.; Moro, R. F.; Lumeras, W.; Rodríguez, L.; Urones, J. G. Synthesis 2001, 1013–1022.Díez, D.; Moro, R. F.; Lumeras, W.; Rodríguez, L.; Marcos, I. S.; Basabe, P.; Escarcena, R.; Urones, J. G. Synlett 2001, 1335–1337.Díez, D.; Moro, R. F.; Lumeras, W.; Rodríguez, L.; Marcos, I. S.; Basabe, P.; Escarcena, R.; Urones, J. G. Synthesis 2002, 175–184.Díez, D.; Nuñez, M.G.; Moro, R. F.; Lumeras, W.; Marcos, I. S.; Basabe, P.; Urones, J. G. Synlett 2005, 939–941.
- Elliott, M. C. J. Chem. Soc., Perkin Trans. 1 2002, 2301–2340. [CrossRef]Elliott, M. C.; Williams, E. J. Chem. Soc., Perkin Trans. 1 2001, 2303–2323. [CrossRef]Elliott, M. C. J. Chem. Soc., Perkin Trans. 1 2000, 1291–1318. [CrossRef]
- Armstrong, A.; Chung, H. Tetrahedron Lett. 2006, 47, 1617–1619. [Google Scholar]
- Reviews: Frohn, M.; Shi, Y. Synthesis 2000, 1979–2000. [CrossRef]Armstrong, A. Angew. Chem., Int. Ed. 2004, 43, 1460–1462. [CrossRef]Shi, Y. Acc. Chem. Res. 2004, 37, 488–496. [CrossRef]Yang, D. Acc. Chem. Res. 2004, 37, 497–505. [CrossRef]Tsuchiya, T.; Armstrong, A. Tetrahedron 2006, 62, 257–263. [CrossRef]Armstrong, A.; Hayter, B. R. Chem. Commun. 1998, 621–622. [CrossRef]Armstrong, A.; Hayter, B. R.; Moss, W. O.; Reeves, J. R.; Wailes, J. S. Tetrahedron: Asymmetry 2000, 11, 2057–2061. [CrossRef]Armstrong, A.; Moss, W. O.; Reeves, J. R. Tetrahedron: Asymmetry 2001, 12, 2779–2781. [CrossRef]Armstrong, A.; Ahmed, G.; Dominguez-Fernandez, B.; Hayter, B. R.; Wailes, J. S. J. Org. Chem. 2002, 67, 8610–8617. [CrossRef]Shing, T. K. M.; Leung, G. Y. C.; Yeung, K. W. Tetrahedron Lett 2003, 44, 9225–9228.Shing, T. K. M.; Leung, Y. C.; Yeung, K. W. Tetrahedron 2003, 59, 2159–2168. [CrossRef]Shing, T. K. M.; Leung, G. Y. C. Tetrahedron 2002, 58, 7545–7552. [CrossRef]Stearman, C. J.; Behar, V. Tetrahedron Lett. 2002, 43, 1943–1946. [CrossRef]Matsumoto, K.; Tomioka, K. Tetrahedron Lett. 2002, 43, 631–633. [CrossRef]Klein, S.; Roberts, S. M. J. Chem. Soc., Perkin Trans. 1 2002, 2686–2691. [CrossRef]Denmark, S. E.; Matsuhashi, H. J. Org. Chem. 2002, 67, 3479–3486. [CrossRef]Bortolini, O.; Fantin, G.; Fogagnolo, M.; Forlani, R.; Maietti, S.; Pedrini, P. J. Org. Chem. 2002, 67, 5802–5806. [CrossRef]Tian, H. Q.; She, X. G.; Shu, L. H.; Yu, H. W.; Shi, Y. J. Am. Chem. Soc. 2000, 122, 11551–11552.Tian, H. Q.; She, X. G.; Xu, J. X.; Shi, Y. Org. Lett. 2001, 3, 1929–1931. [CrossRef]Tian, H. Q.; She, X. G.; Yu, H. W.; Shu, L. H.; Shi, Y. J. Org. Chem. 2002, 67, 2435–2446. [CrossRef]Shu, L. H.; Wang, P. Z.; Gan, Y. H.; Shi, Y. Org. Lett. 2003, 5, 293–296. [CrossRef]Shu, L. H.; Shi, Y. Tetrahedron Lett. 2004, 45, 8115–8117. [CrossRef]Hickey, M.; Goeddel, D.; Crane, Z.; Shi, Y. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5794–5798. [CrossRef]Armstrong, A.; Dominguez-Fernandez, B.; Tsuchiya, T. Tetrahedron 2006, 62, 6614–6620. [CrossRef]
- Bez, G.; Zhao, C-G. Tetrahedron Lett. 2003, 44, 7403–7406. [CrossRef]Travis, B. R.; Sivakumar, M.; Hollist, G. O.; Borhan, B. Org. Lett. 2003, 5, 1031–1034. [CrossRef]Webb, K. S.; Ruszkay, S. J. Tetrahedron 1998, 54, 401–410. [CrossRef]
- Ley, S. V.; Norman, J.; Griffith, W. P.; Marsden, S. P. Synthesis 1994, 639–666.
- Wang, Z. X.; Tu, Y.; Fhron, M.; Zhang, J. R.; Shi, Y. J. Am. Chem. Soc. 1997, 119, 11224–11235. [CrossRef]
- Yang, D.; Wong, M. K.; Yip, Y. C. J. Org. Chem. 1995, 60, 3887–3889. [CrossRef]
- Clive, D. J.; Ardelean, E-S. J. Org. Chem. 2001, 66, 4841–4844. [CrossRef]
- Measured by chiral HPLC (ChiralPak AD-H, 25 cm x 10 mm, 99:1 hexane/iPrOH), retention times minor enantiomer (4.6 min) and major enantiomer (4.9 min).
- Reviews: Renz, M.; Meunier, B. Eur. J. Org. Chem. 1999, 737–750. [CrossRef]ten Brink, G.J.; Arends, I.W.C.E.; Sheldon, R. A. Chem. Rev. 2004, 104, 4105–4123. [CrossRef]Krow, G. R. Org. React. 1993, 43, 251–798.Crudden, C. M.; Chen, A. C.; Calhoun, L. A. Angew. Chem., Int. Ed. 2004, 43, 2851–2855.Krasutsky, P. A.; Kolomytsin, I. V. Arkivoc 2005, 151–171. [CrossRef]Alvarez-Idaboy, J. R.; Reyes, L.; Cruz, J. Org. Lett. 2006, 8, 1763–1765. [CrossRef]Frison, J-C.; Palazzi, C.; Bolm, C. Tetrahedron 2006, 62, 6700–6706. [CrossRef]Chang, M-Y.; Kung, Y-H.; Chen, S-T. Tetrahedron Lett. 2006, 47, 4865–4870. [CrossRef]
- Dave, V.; Warnhoff, E. W. J. Org. Chem. 1983, 48, 2590–2598, Also see ref. [11c].. [CrossRef]
- Urones, J. G.; Díez, D.; Marcos, I. S.; Basabe, P.; Lithgow, A. M.; Moro, R. F.; Garrido, N. M.; Escarcena, R. Tetrahedron. 1995, 51, 3691–3704. [CrossRef]
- Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Chemistry, 3rd ed.; John Wiley & Sons: New York, 1999. [Google Scholar]
- Hubbs, J. L.; Heathcock, C. H. J. Am. Chem. Soc. 2003, 125, 12836–12843. [CrossRef]
- Sample Availability: Samples of compounds 2-6 and 13-17 are available from the authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Díez, D.; Nuñez, M.G.; Moro, R.F.; Garrido, N.M.; Marcos, I.S.; Basabe, P.; Urones, J.G. Synthesis of (R)-2-(Benzyloxy)-tetrahydro-5,5-dimethylfuran by a New Oxidative Rearrangement. Molecules 2006, 11, 959-967. https://doi.org/10.3390/11120959
Díez D, Nuñez MG, Moro RF, Garrido NM, Marcos IS, Basabe P, Urones JG. Synthesis of (R)-2-(Benzyloxy)-tetrahydro-5,5-dimethylfuran by a New Oxidative Rearrangement. Molecules. 2006; 11(12):959-967. https://doi.org/10.3390/11120959
Chicago/Turabian StyleDíez, D., M. G. Nuñez, R. F. Moro, N. M. Garrido, I. S. Marcos, P. Basabe, and J. G. Urones. 2006. "Synthesis of (R)-2-(Benzyloxy)-tetrahydro-5,5-dimethylfuran by a New Oxidative Rearrangement" Molecules 11, no. 12: 959-967. https://doi.org/10.3390/11120959
APA StyleDíez, D., Nuñez, M. G., Moro, R. F., Garrido, N. M., Marcos, I. S., Basabe, P., & Urones, J. G. (2006). Synthesis of (R)-2-(Benzyloxy)-tetrahydro-5,5-dimethylfuran by a New Oxidative Rearrangement. Molecules, 11(12), 959-967. https://doi.org/10.3390/11120959