Introduction
Ophiobolin A (
Scheme 1) has been identified in the 1960s as a metabolite of the fungus
Helminthsporium oryzeo, toxic to rice seedlings [
1]. A number of related sesterterpenoids have been isolated since then from terrestrial and marine microorganisms, plants and insect secretions. Selected examples of these compounds are shown in
Scheme 1. The main structural feature of ophiobolins is a dicyclopenta[a,d]cyclooctane ring system with methyl groups, or transformed methyl groups, dislocated at positions 3, 7 and 11. A
cis-and
trans -junction of the A/B and B/C rings, respectively, appears typical, however
trans-trans derivatives have also been isolated from natural sources. With regards to biological activity, ophiobolin A induces apoptic cell death in the L1210 cell line, ophiobolin K, 6-epi-N and some other metabolites are active against certain tumor cells [
2]. Potent inhibitory activity of ophiobolin M (16,17-dihydro-ophiobolin K) against nematode worms has been recorded [
3].
Scheme 1.
Ophioboins. Selected structures.
Scheme 1.
Ophioboins. Selected structures.
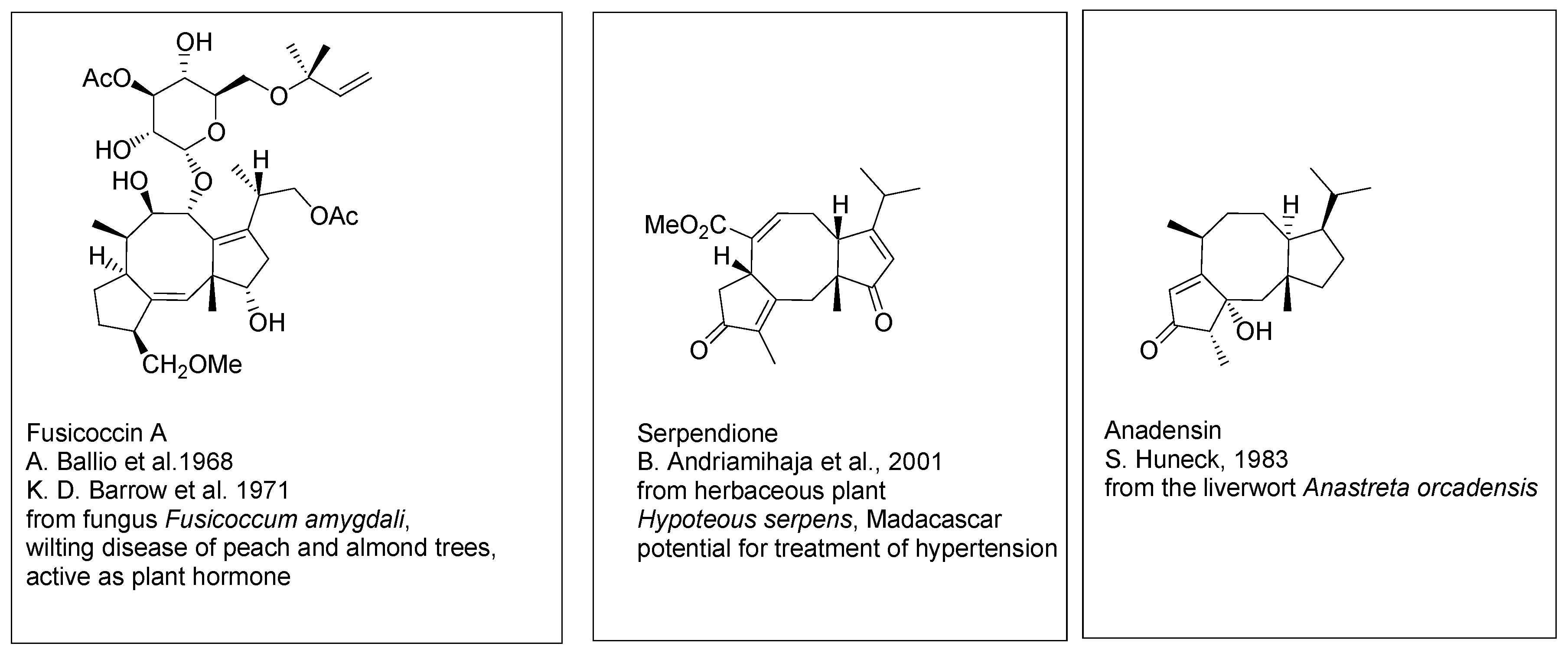
Fusicoccins A (
Scheme 2) and related diterpenoids [
4,
5] embrace the same dicyclopenta[a,d]- cyclooctane ring system, with the side chain shorter by one isoprene unit. The main representatives of this group have been isolated from the fungus
Fusicoccum amygdali causing wilting disease of peach and almond trees. Fusicoccins A and related metabolites occur as glycosides. Several compound of this group are presently known, isolated from fungi, plants, algae and from liverworts. Some of fusicoccins are of interest as potential therapeutic agents. For example, serpendione, isolated from the herbaceous plant
Hypoteous serpens endemic to Madagascar, shows significant blood pressure moderating activity.
Scheme 2.
Fusicoccines. Selected structures.
Scheme 2.
Fusicoccines. Selected structures.
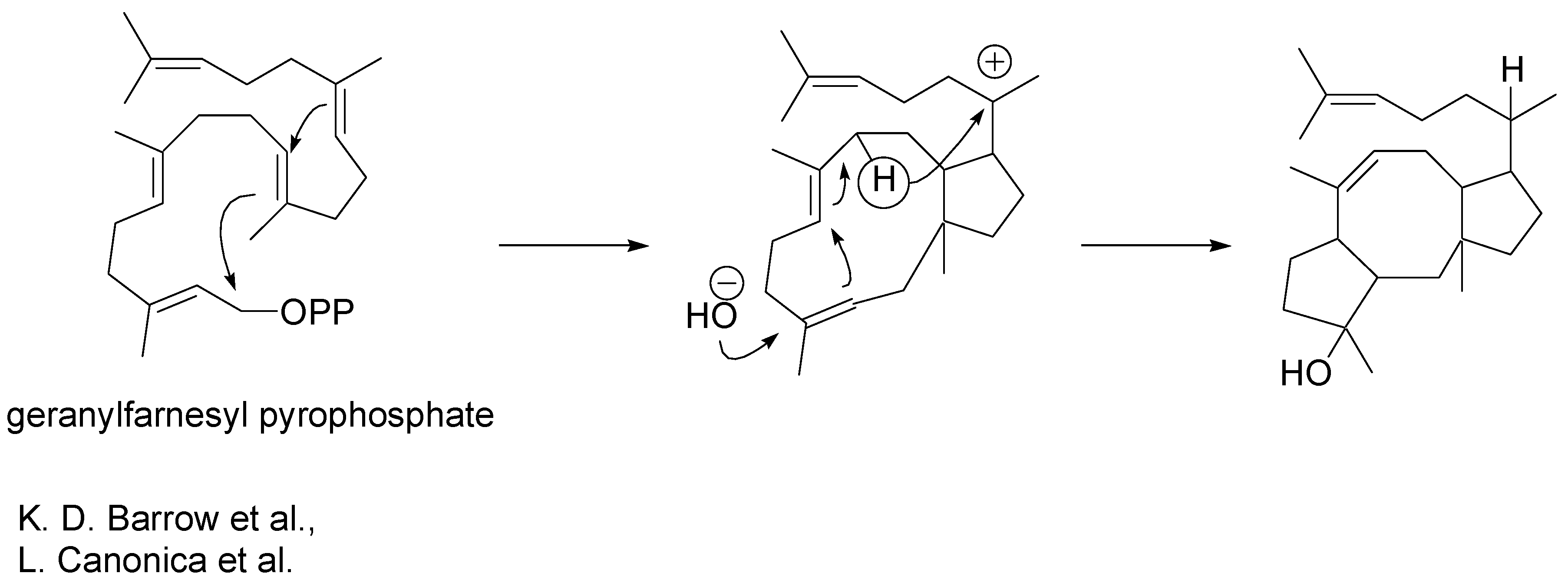
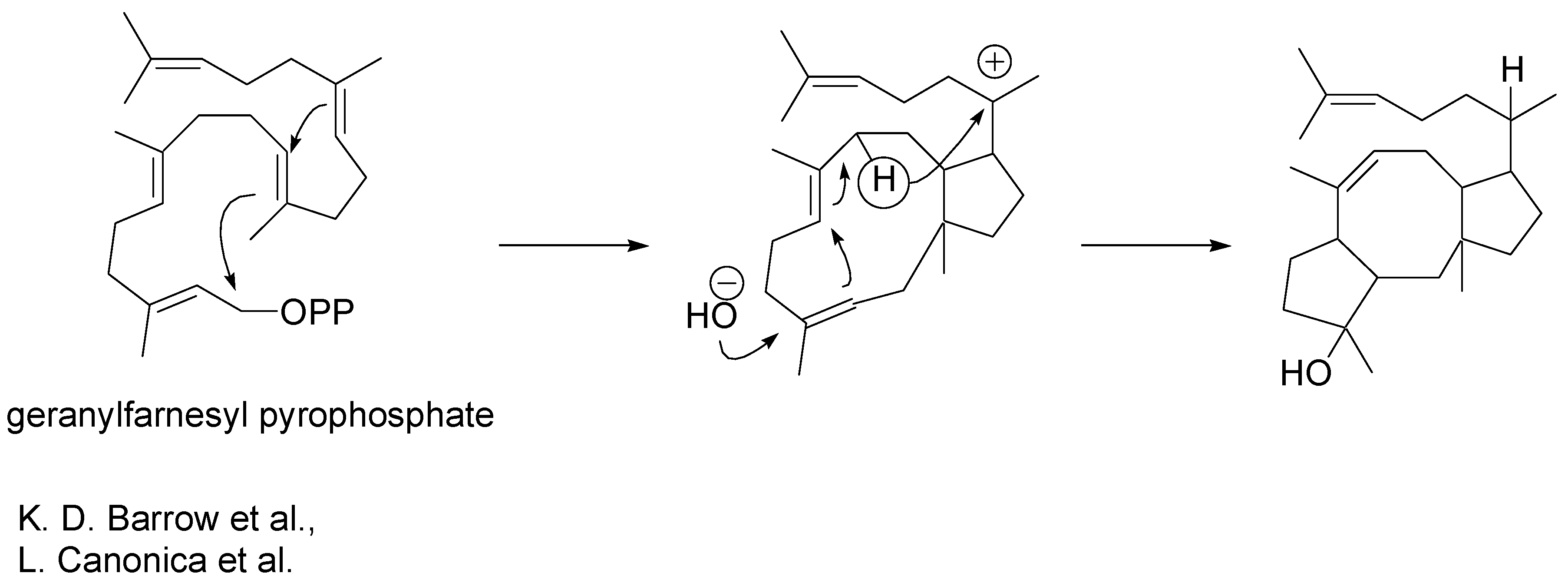
The biosynthetic origins of ophiobolins were revealed some time ago [
5,
6]. Geranylfarnesyl pyrophosphate undergoes macrocyclisation as illustrated in
Scheme 3. Cyclisation is followed by a 1,5- hydride ion shift and trans-annular carbon-carbon bond formation.
Scheme 3.
Biosyntheis of ophiobolins.
Scheme 3.
Biosyntheis of ophiobolins.
The structural complexity and important biological activity of terpenoids with dicyclopenta[a,d]- cyclooctane carbon skeletons attracted a great deal of attention from synthetic chemists [
7,
8,
9,
10]. The majority of work has focused on the methodology for construction of eight-membered ring systems [
11,
12]. Synthesis and elucidation of reactivity of cyclooctane derivatives present a long-standing challenge owing to conformational lability of the cyclooctane ring and, in certain cases, astonishing unpredictability of conformation effects.
In the present paper, a synthesis of a key building block of the ophiobolates and fusicoccins, embracing rings A and B (5/8) will be presented. Special attention will be given to application of the ring closing metathesis (RCM) reaction to annulation of cyclopentane into cyclooctapentane derivatives.
Results and Discussion
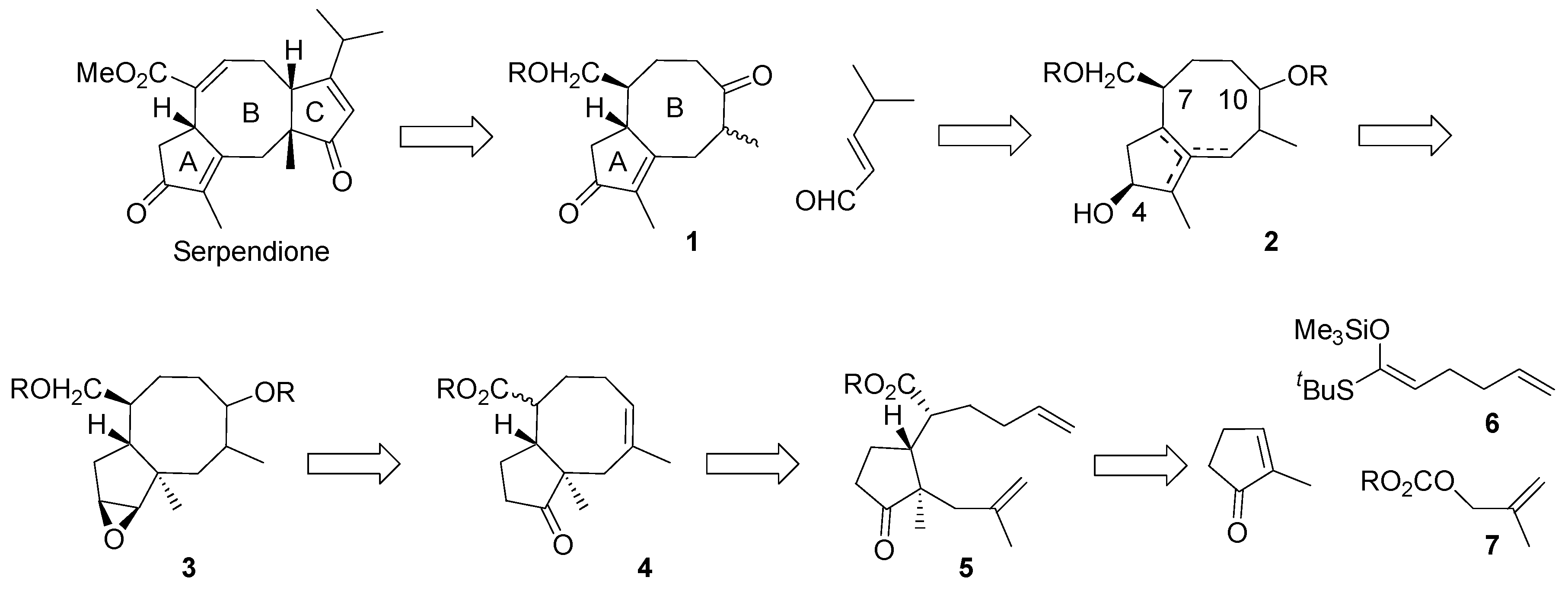
Our synthetic plan is illustrated in
Scheme 4. It was reasoned that cyclopentaannulene derivative
1 with two protected oxo groups, will serve as a versatile key intermediate for ophiobolates and fusicoccanes, for example, for serpendione. The oxo group in the position 10 (ophiobolate numbering) will be used for attachment of the ring C, via aldol reaction with α,β-unsaturated aldehyde or by another means. The carbonyl group in position 4 will allow for controlling of the double bond position in the ring A; the hydroxymethyl group will be used to adjust the double bond position in the ring B. Unsaturated triol
2 is a synthetic equivalent of the diketene
1.
Scheme 4.
The synthetic plan.
Scheme 4.
The synthetic plan.
It was thought that 2 will be available by Wagner-Meerwein – type rearrangement of epoxide 3. The epoxide function in 3 will be established starting from ketone 4 by methodology well documented in steroid chemistry, via the respective hydrazone and vinyl iodide. With respect to the oxygen function in the eight-membered ring, it will be generated from the double bond in 4 in the hydroboration – oxidation reaction sequence. The hydroxymethyl group in 3 originates in the ester group of 4. The key intermediate 4 will be provided by ring closing metathesis reaction of diene 5. It should be noted that transformation of 5 into 4 would generate considerable strain as a consequence of 1,3- pseudo di-axial arrangement of the angular methyl group and the carbon substituent at C7. The intermediate 5 will be accessible via Mukaiyama-Michael reaction of the appropriate ketene acetal and 2-methylcyclopent-2-en-1-one in tandem with alkylation of the intermediate silyl enol ether.
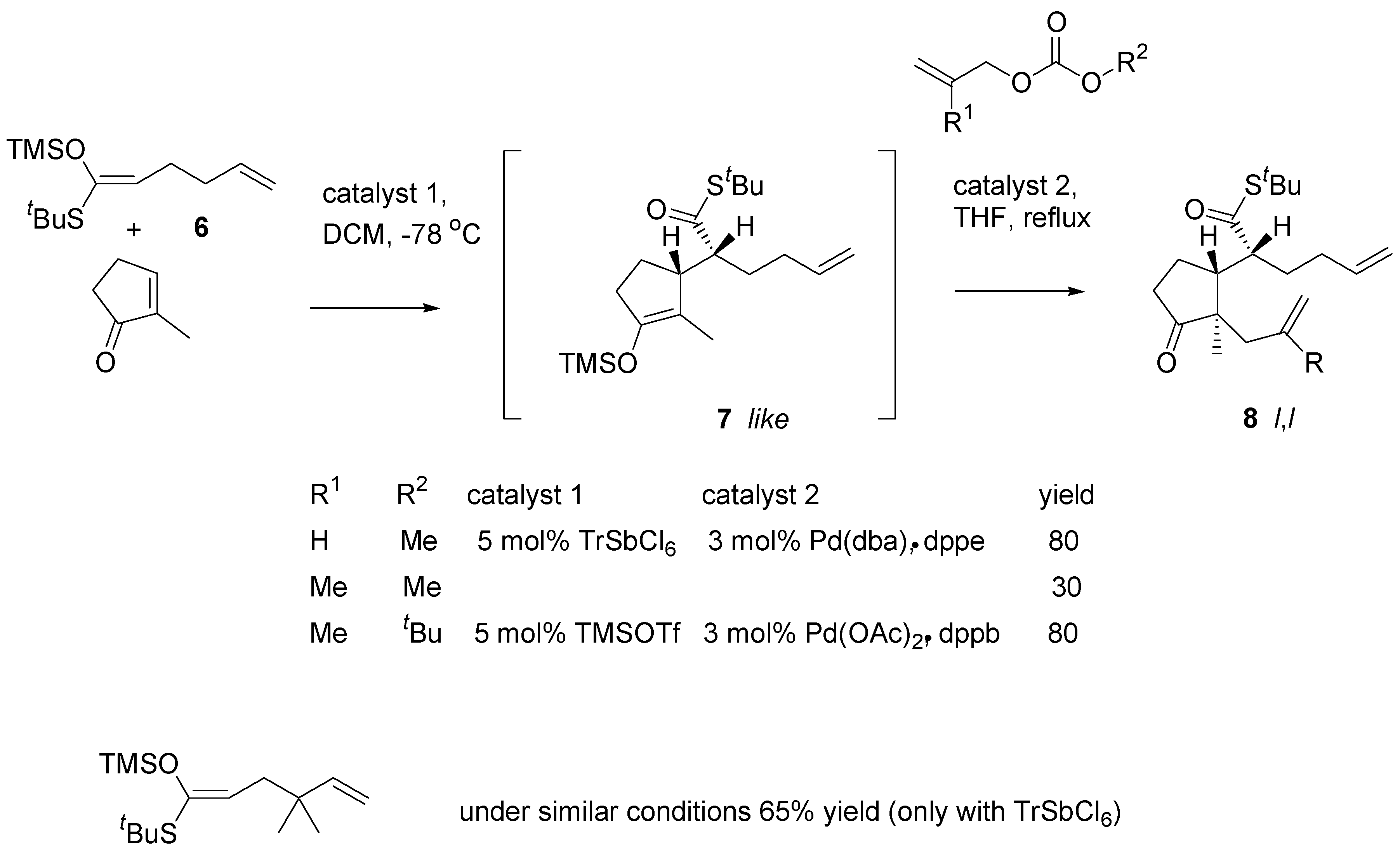
The Mukaiyama-Michael reaction of ketene acetal
6 (prepared from the corresponding thioester) with 2-methylcyclopent-2-en-1-one afforded the adduct
7 (
Scheme 5). The reaction is usually catalyzed with trityl hexachloroantimonate. Alkylation of the silyl enol ether with allyl methyl carbonate according to the Tsuji procedure [
13] affords the product (
8, R = H) in an excellent yield. However, when methallyl methyl carbonate was used for alkylation, the yield of the tandem reactions was low.
Scheme 5.
Study on the conjugate addition – alkylation reaction sequence.
Scheme 5.
Study on the conjugate addition – alkylation reaction sequence.
Two factors were altered in optimalization of the procedure: (1) palladium acetate - bis-(diphenyl-phosphine)butane were found to be the catalyst of choice in the alkylation step and (2) TrSbCl6 was replaced with TMSOTf; the reaction was markedly slower, but the catalyst could be easily removed which was essential for reproducibility of the reaction. The procedure was applied to a number of substrates. As expected, in all cases products of like,like (l,l) relative configuration were obtained.
Scheme 6.
Preparation of starting materials for study of annulation via RCM reactions.
Scheme 6.
Preparation of starting materials for study of annulation via RCM reactions.
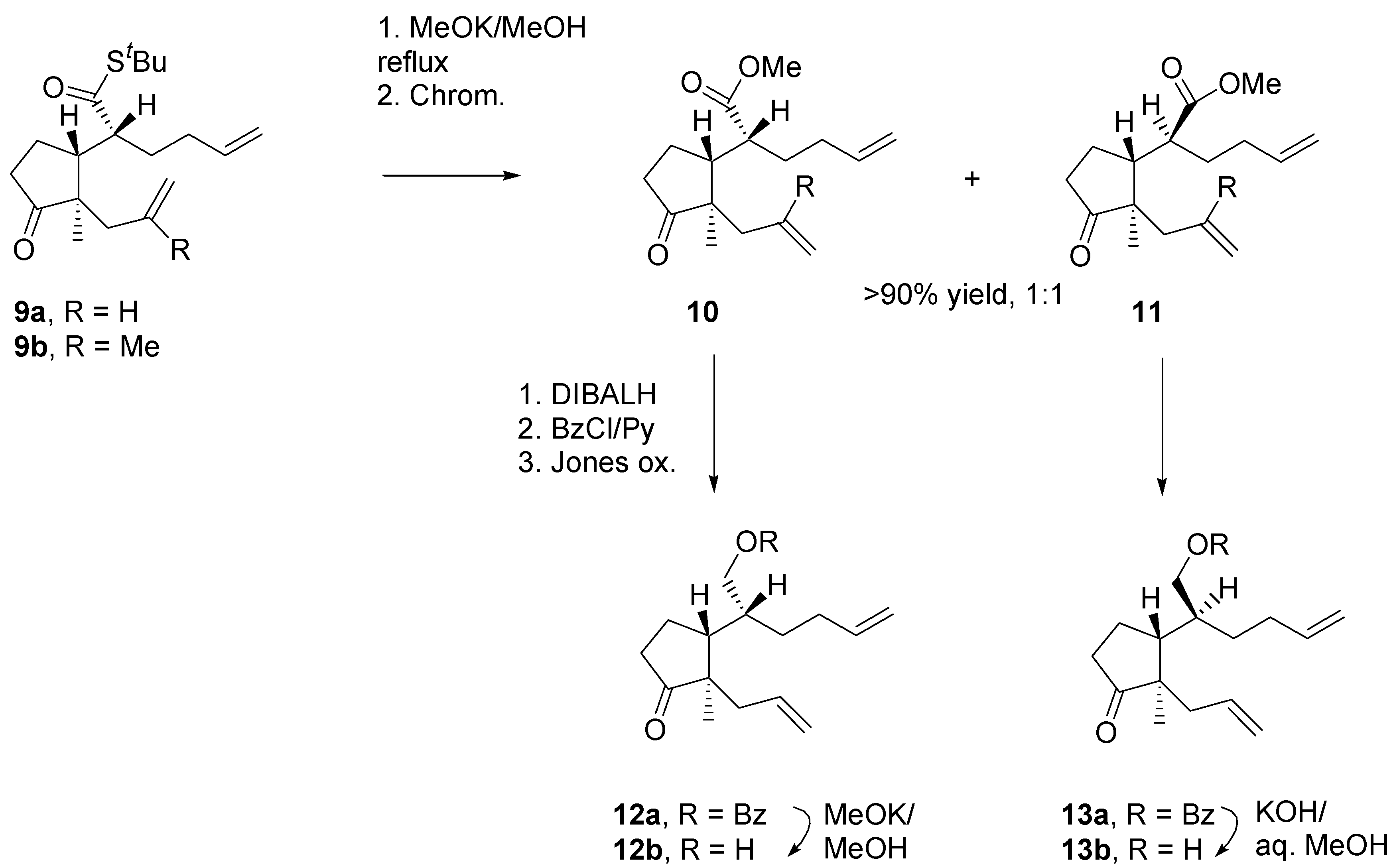
The dienes were transformed straightforwardly into the derivatives that were need for scrutiny of the RCM reaction (
Scheme 6). First, the
tert-butylthio groups were replaced with the methoxy groups. The best results were obtained by carrying out methanolysis reaction using MeOK in MeOH, prepared from potassium and methanol. Methanolysis was accompanied by epimerisation at the stereogenic center in the α-position, to give a mixture of diastereomeric esters (
l,l and
u,l). Differences in steric shielding of
l,l and
l,u diasteromers were obvious: whereas hydrolysis of
l,u benzoate
13a with KOH in aq. MeOH occurred smoothly, for its
l,l–diastereomer
12a only MeOK/MeOH at the reflux temperature was effective in a reasonable time.
Scheme 7.
Preparation of starting materials for study of annulation via RCM reactions, 2.
Scheme 7.
Preparation of starting materials for study of annulation via RCM reactions, 2.
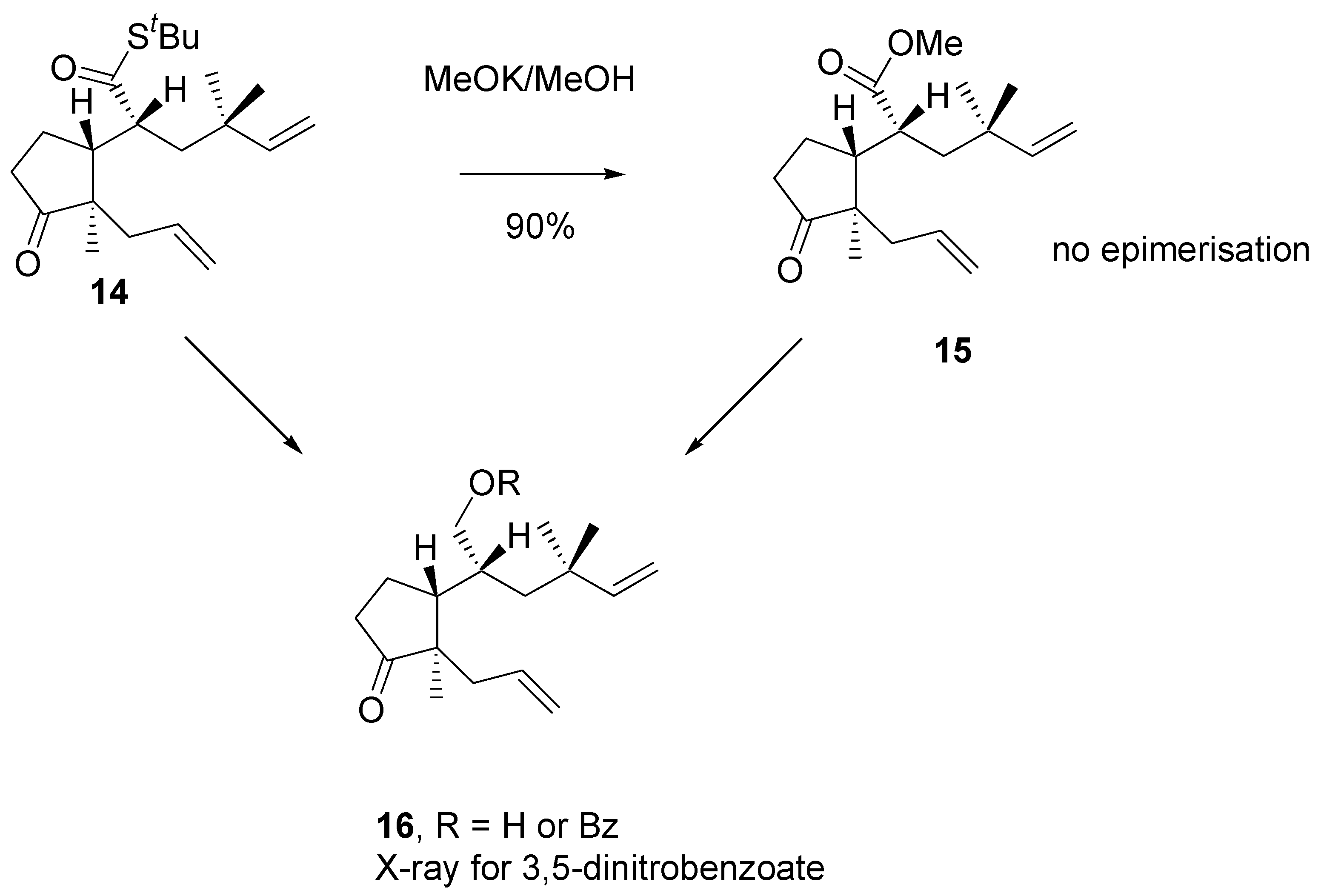
Noteworthly, ester bearing
gem-methyl groups (
Scheme 7) under methanolysis conditions afforded only one product with retained configuration. Correlation of structure of alcohols obtained from methyl ester
15 and thioester
14 was made. The structure of alcohol
16 was confirmed by single crystal x-ray analysis of the respective 3,5-dinitrobenzoate.
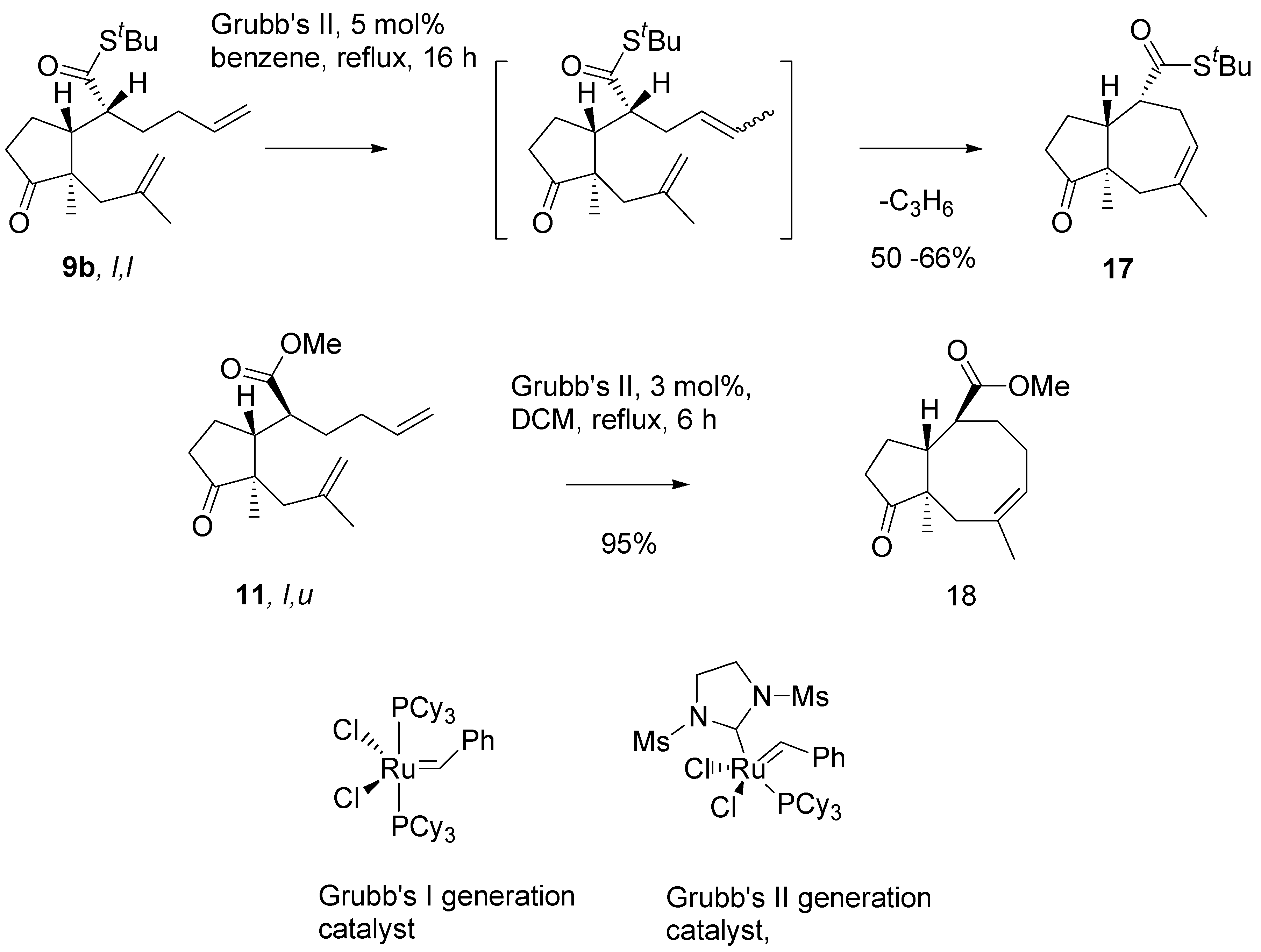
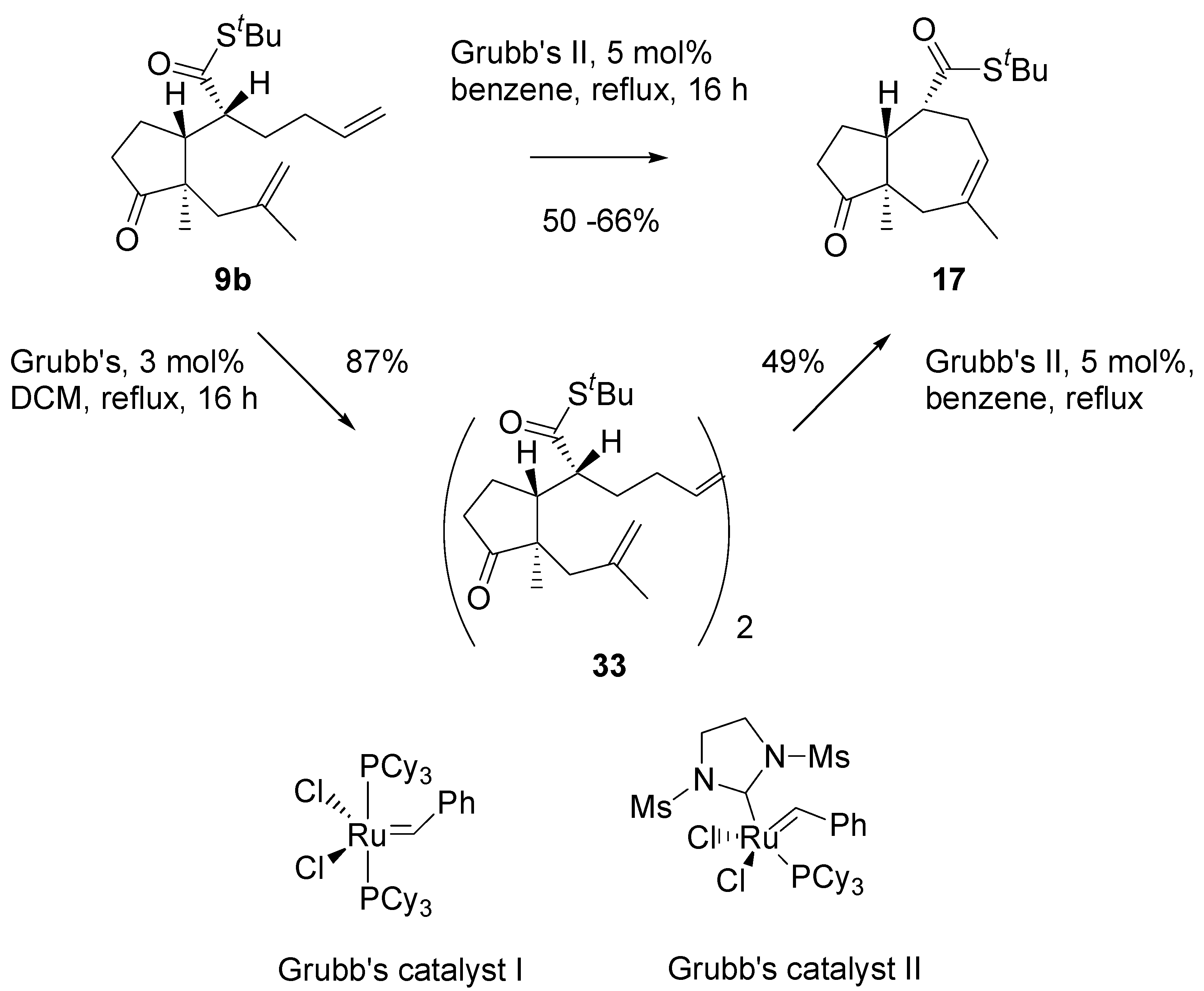
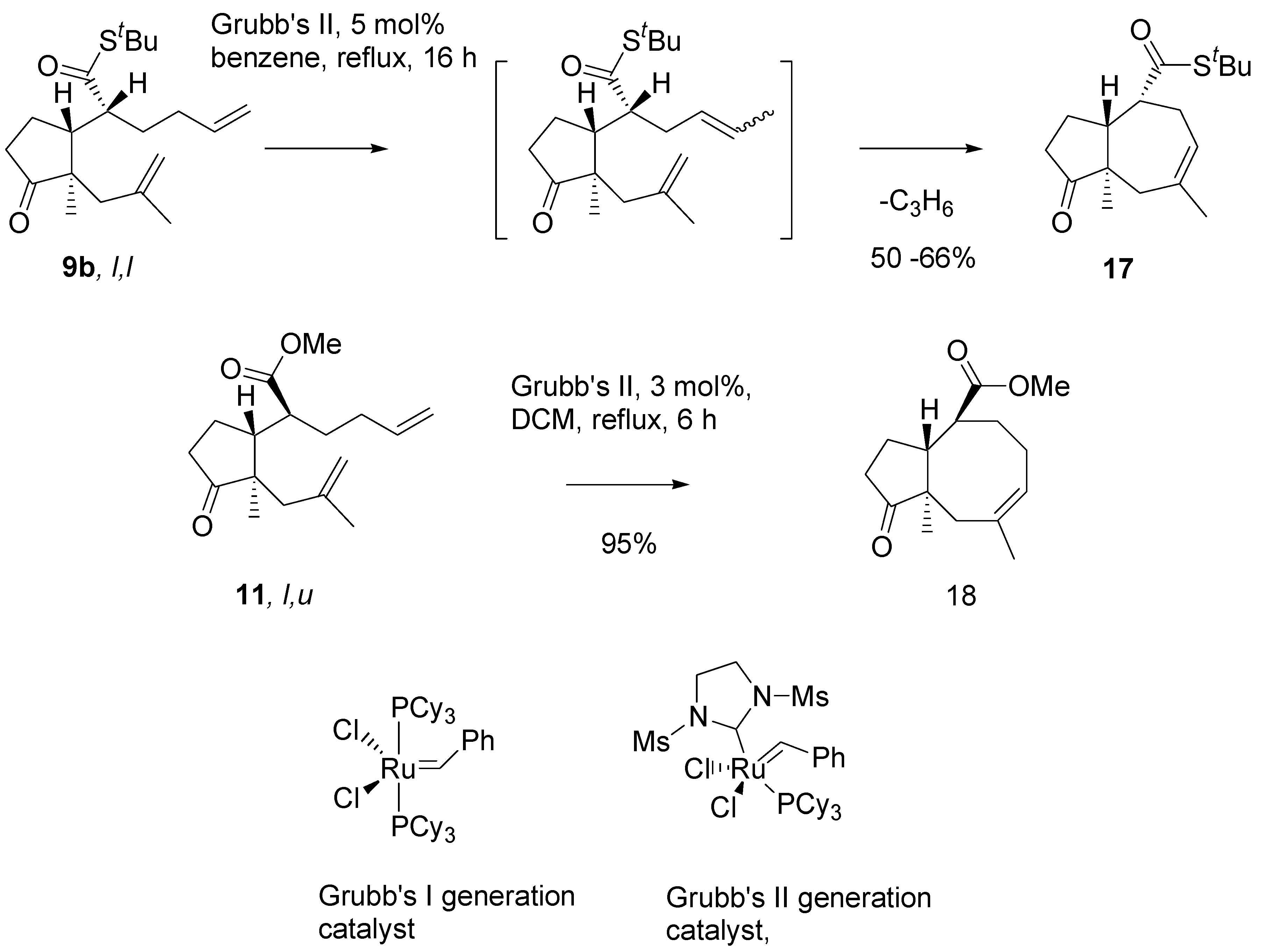
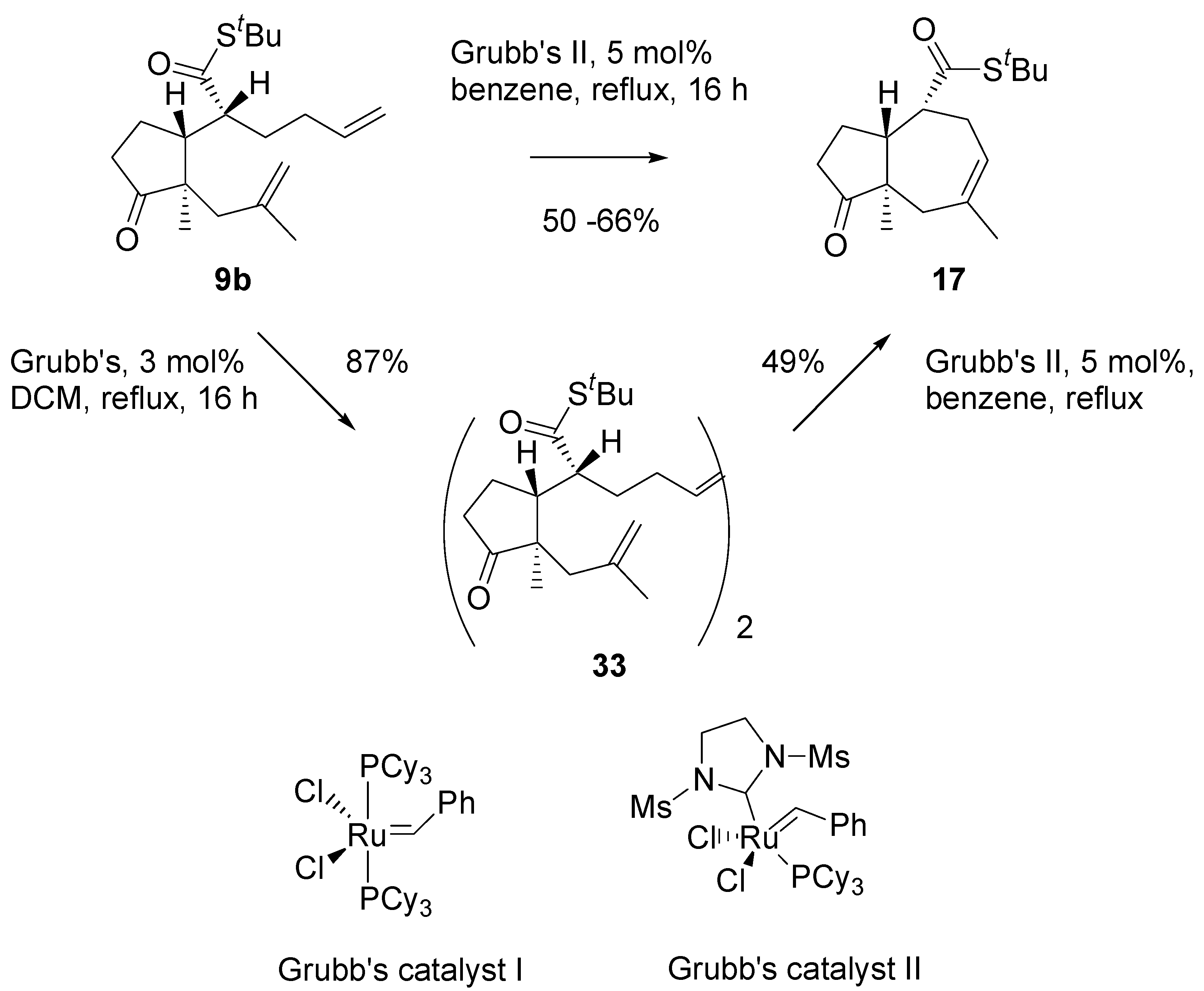
Thioester
9b in metathesis reaction in the presence of the Grubb’s second generation catalyst, in benzene, under reflux, afforded a single product in a moderate yield (
Scheme 8). Analysis of
1H- and
13C-NMR, and mass-spectra of this product indicated the cycloheptane (azulene) derivative
17 has been formed. Evidently, migration of the double bond had occurred prior to ring closure, and propene was generated as the complementary metathesis product.
Scheme 8.
Initial experiments on the RCM reaction.
Scheme 8.
Initial experiments on the RCM reaction.
Migration of a carbon-carbon double bond in the presence of ruthenium catalysts, is a well documented side reaction accompanying RCM reactions [
14,
15,
16,
17]. However, only minor products were formed by this route. Efficient isomerisation – metathesis tandem process has no precedent in the literature, to the best of our knowledge. Our findings on the metathesis reaction will be discussed in the concluding part of the presentation. For the purpose of synthesis, the intermediate with eight membered ring
18 was smoothly obtained by metathesis of methyl ester of
l,
u configurtion
11.
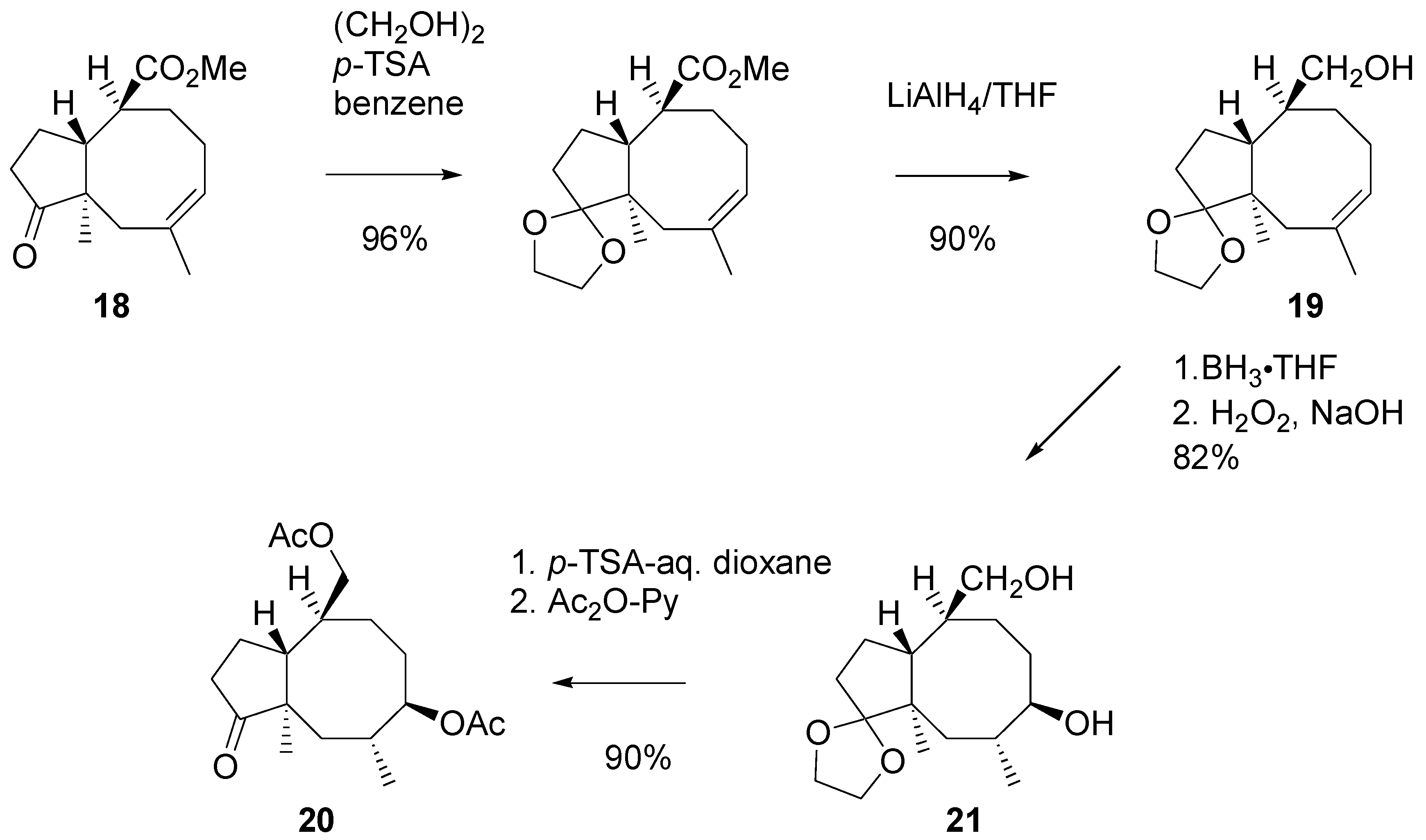
This cyclooctapentane derivative
18 was transformed, into ethylene ketal – alcohol
19 as shown in
Scheme 9. Consecutive hydroboration and oxidation of
19 afforded diol
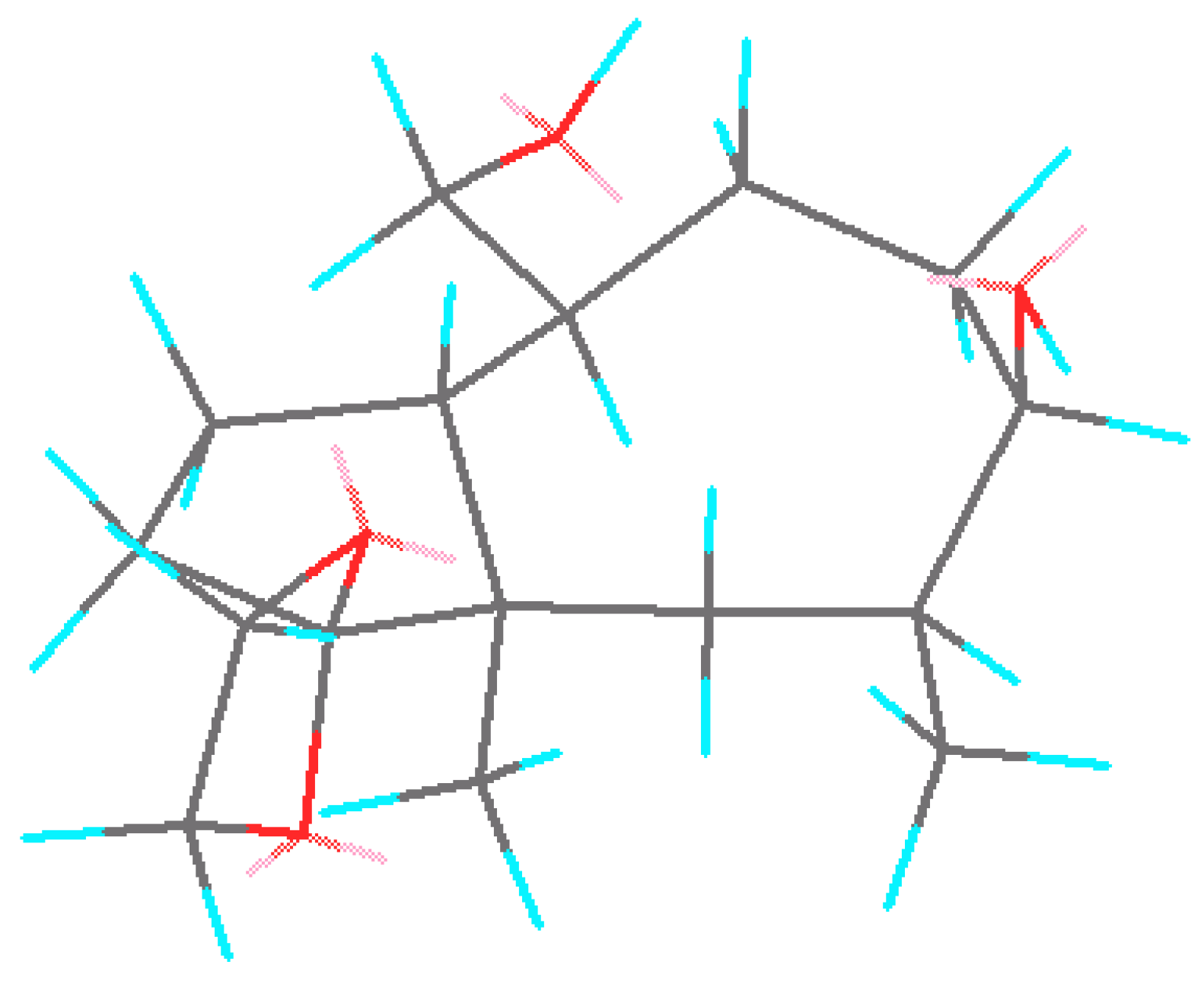
21 as the only product. Attack of the reagent occurred on the outer side of the cyclooctane ring, opposite to the angular methyl group. A low-energy three-dimension structure of
21 is shown in
Figure 1. Standard manipulations with the protective group afforded oxo-diacetate
20.
Scheme 9.
Transformations of cyclopenta[8]annulene derivatives.
Scheme 9.
Transformations of cyclopenta[8]annulene derivatives.
Figure 1.
Projection of three-dimentional structure of intermediate 21.
Figure 1.
Projection of three-dimentional structure of intermediate 21.
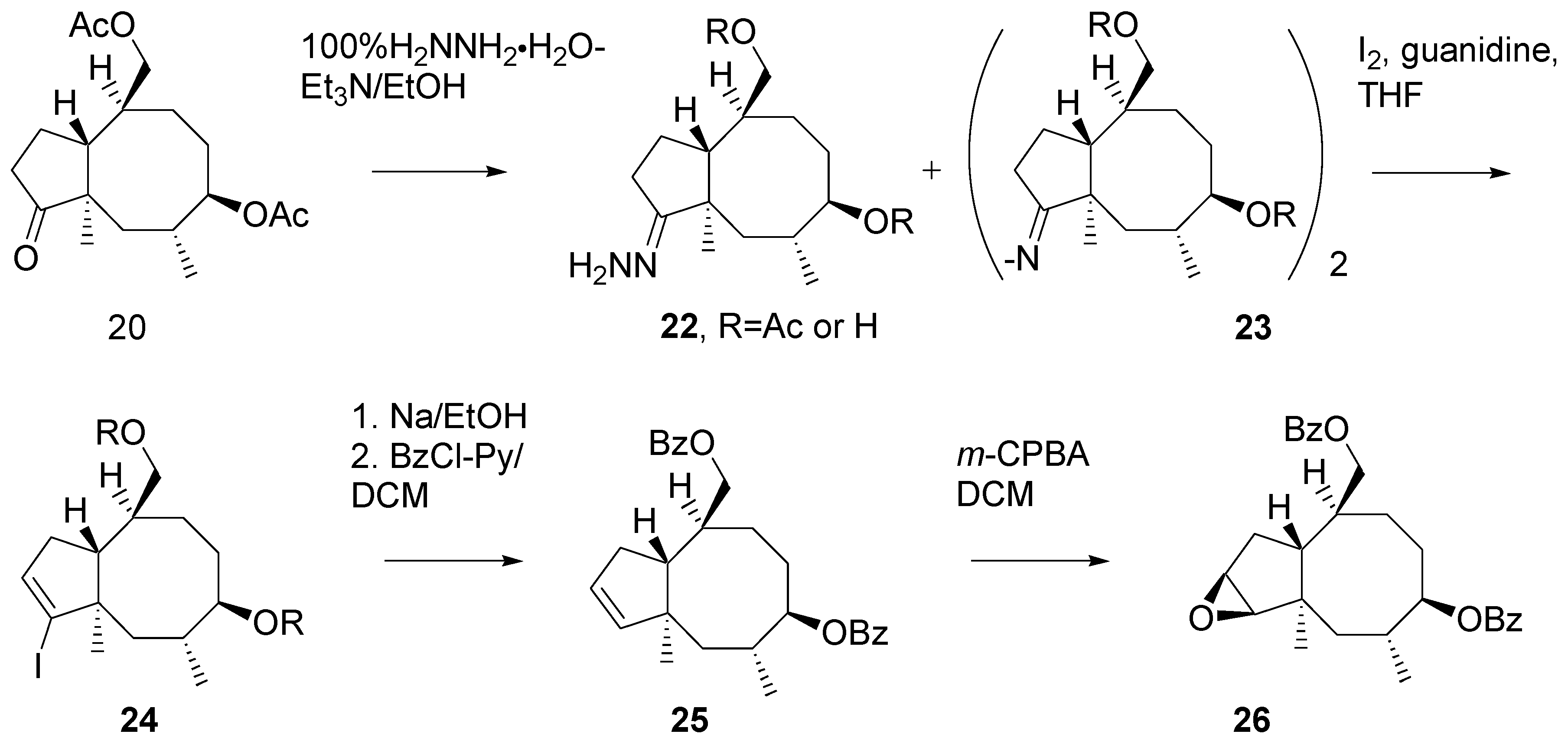
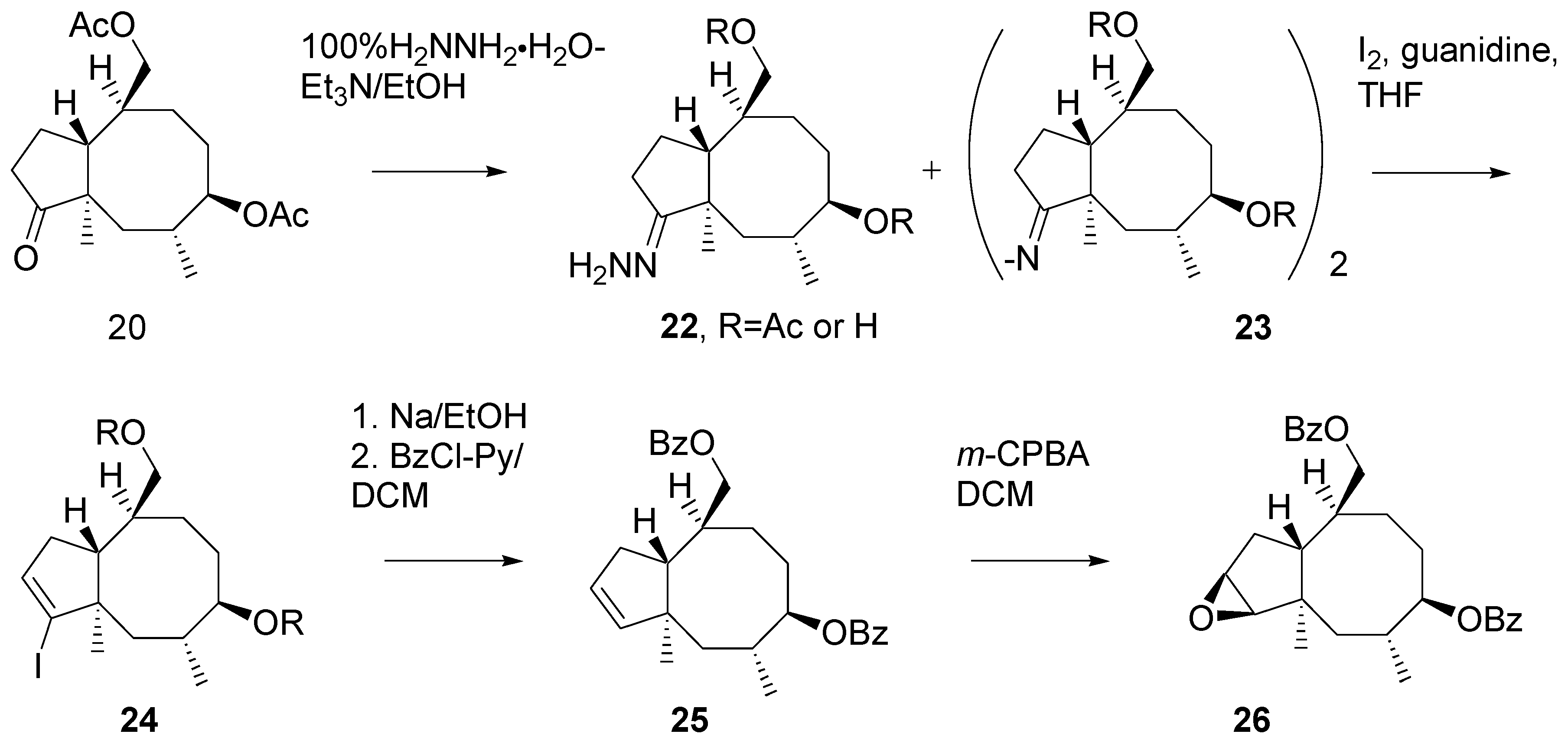
Unexpectedly, transformation of ketone
20 into hydrazone
22 turned out problematic (
Scheme 10). A side product was formed, presumably the diazinene derivative [
18]
23. The crude mixture of hydrazine derivatives was subjected to reaction with iodine [
19]. Vinyl iodide
24 was reduced with sodium in ethanol and then the hydroxy groups in the product were benzoylated. Di-benzoate
25 was obtained in a ca. 50% yield from the ketone
20.
Oxidation of the double bond in 25 with m-CPBA afforded epoxide 26. Analysis of the high-field 1H NMR spectrum of indicated the presence of ca. 5% of some other product, presumably the isomeric epoxide.
Scheme 10.
Preparation of the key intermediate epoxide.
Scheme 10.
Preparation of the key intermediate epoxide.
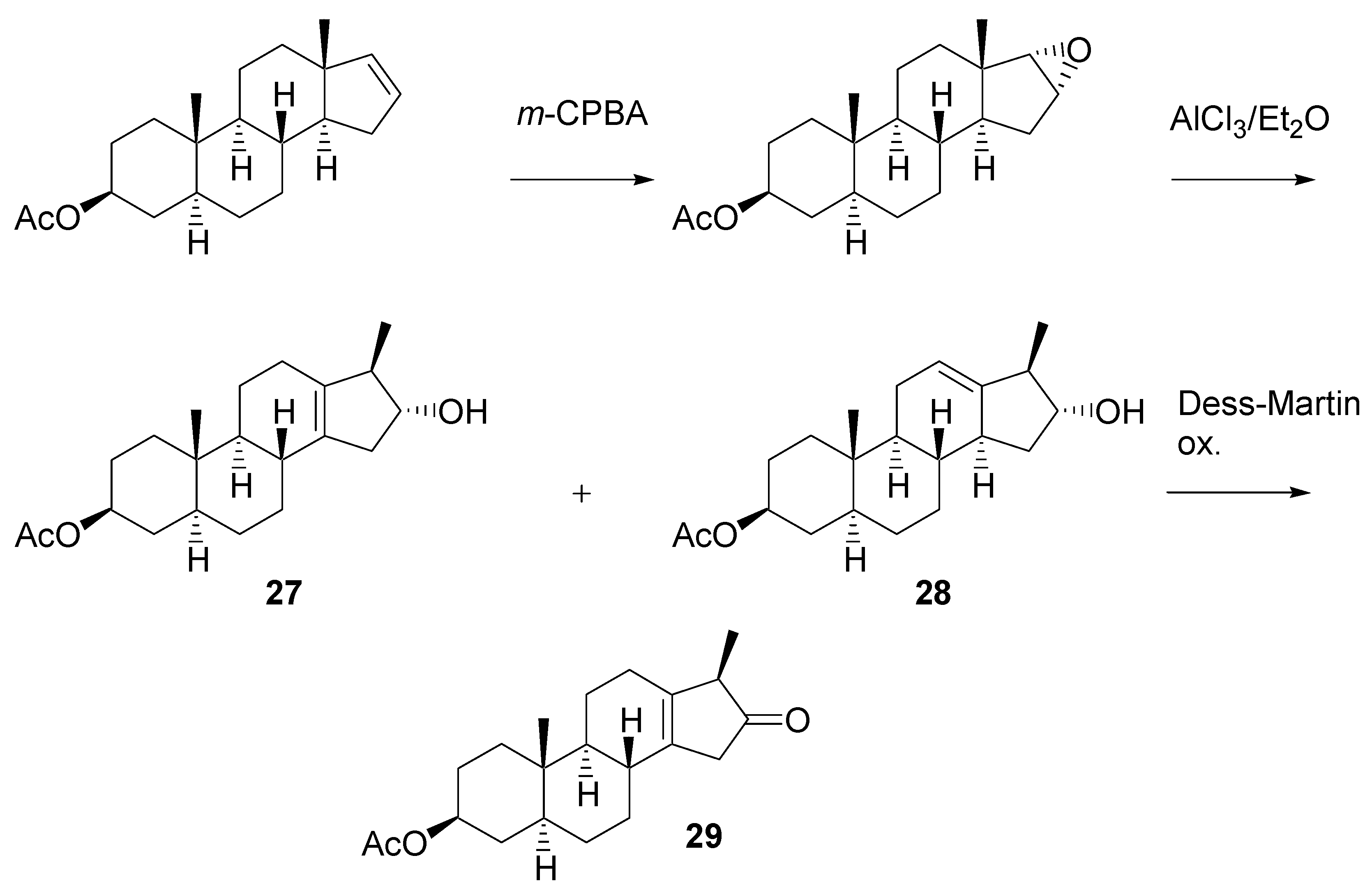
Rearrangement of the epoxide was an important step in the planned synthesis. Model experiments with easily accessible steroid epoxide were carried out (
Scheme 11). It was found that AlCl
3 in ether [
18] gives the best results. A mixture of two products
27 and
28 was formed in ca. 60% yield. Their structure was elucidated from
1H-NMR spectra. After oxidation with the Dess-Martin reagent, pure β,γ-unsaturated ketone
29 was isolated by chromatography.
Scheme 11.
Experiments on rearrangement of the model steroidal epoxide.
Scheme 11.
Experiments on rearrangement of the model steroidal epoxide.
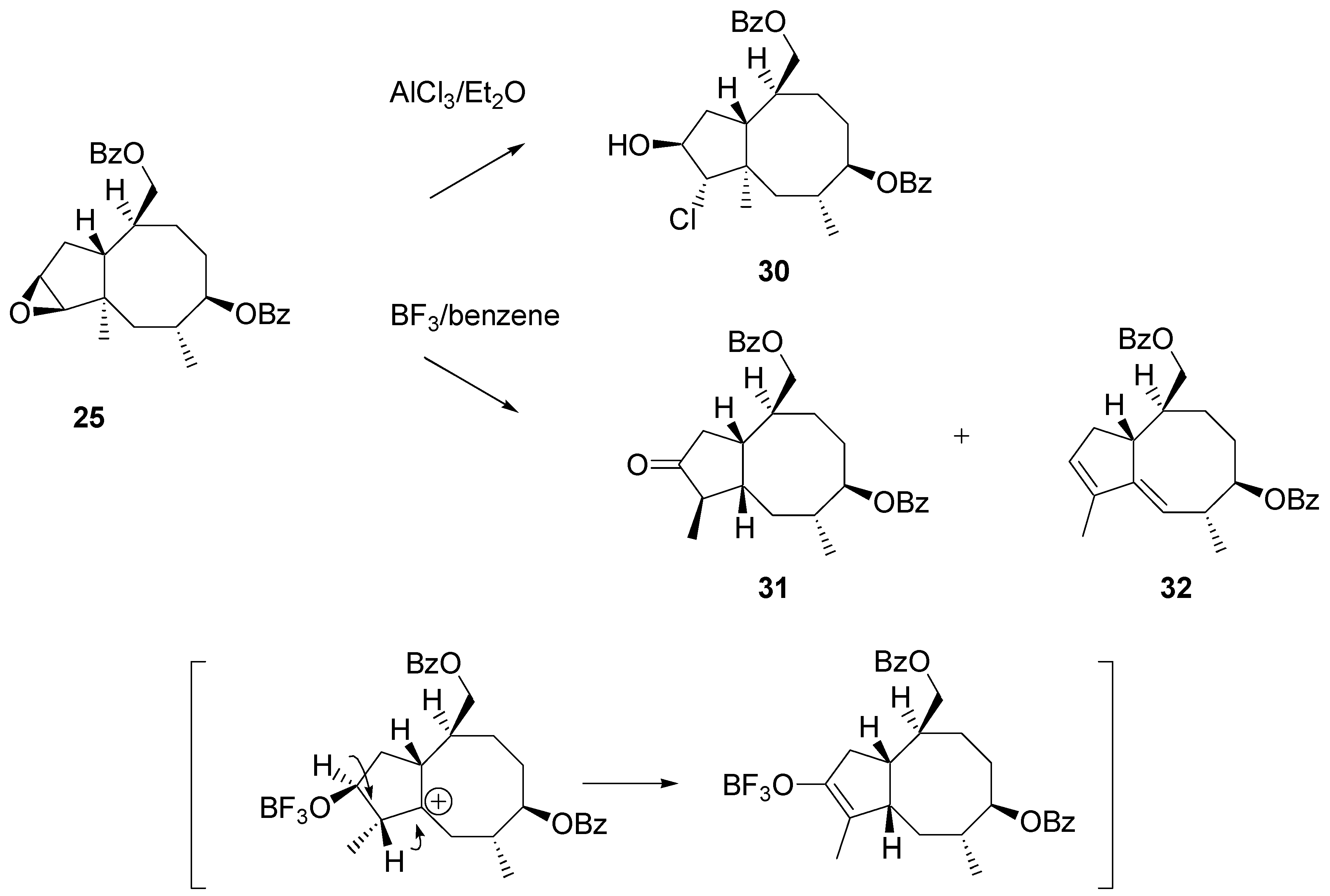
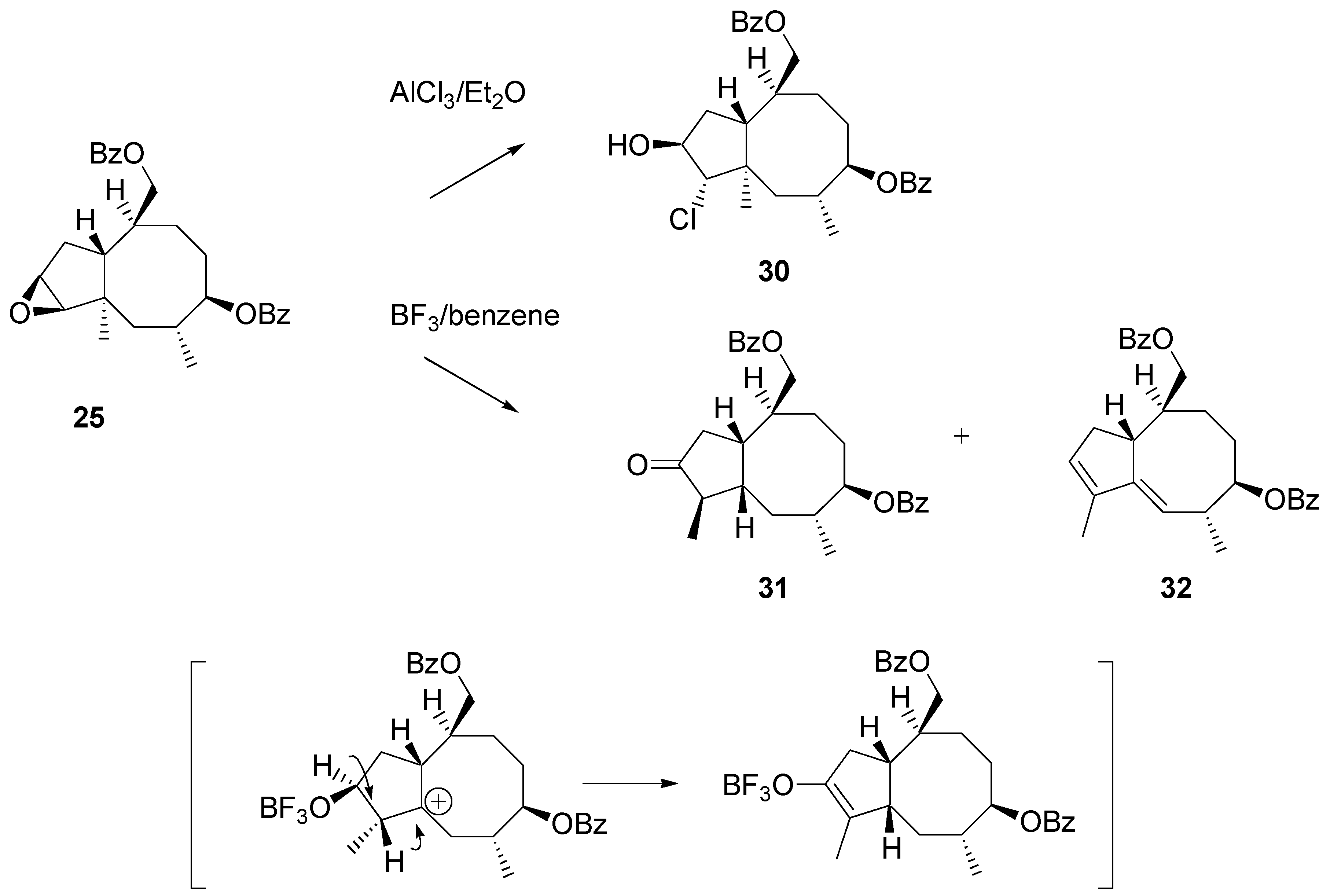
Treatment of epoxide
26 with AlCl
3 in ether afforded a mixture of products. The dominant product was isolated in 60% yield after chromatography. Its NMR spectra clearly showed that the angular methyl group remained unaffected. Instead of rearrangement addition of hydrogen chloride to the epoxide moiety occurred. Surprisingly, the regioselectivity of addition was different to that recorded for the steroidal 16α,17α – epoxide. The chlorine atom was attached to C2 to give structure
30 (
Scheme 12). Fortunately, more cooperative reagents for achieving the synthesis targets were on hand. BF
3·Et
2O-catalyzed rearrangement afforded two products that were separated and identified by NMR, mass and IR spectra. The main product (ca. 40% yield) was identified as ketone
31 and the other isolated product (20%) as diene
32. Stereochemistry of
31 was tentatively assigned from mechanistic considerations: it was assumed that migration of the methyl group and consecutive migration of hydride ion occurred suprafacially, as shown in the scheme.
At this stage our goals in developing total synthesis of ophiobolins and/or fusicoccins have been achieved. Intermediate cyclopentaannulene derivatives with proper dislocation of carbon substituents and functional groups were access on a relatively short synthetic route. Each of the cyclopentaannulenes formed may be used for further synthesis. Application of RCM reaction in the synthetic cycle has allowed us to make some novel and, as we think, interesting observations.
Scheme 12.
Migration of the angular methyl group in the cyclopentaannulene system. Experiments on rearrangement of epoxide 26.
Scheme 12.
Migration of the angular methyl group in the cyclopentaannulene system. Experiments on rearrangement of epoxide 26.
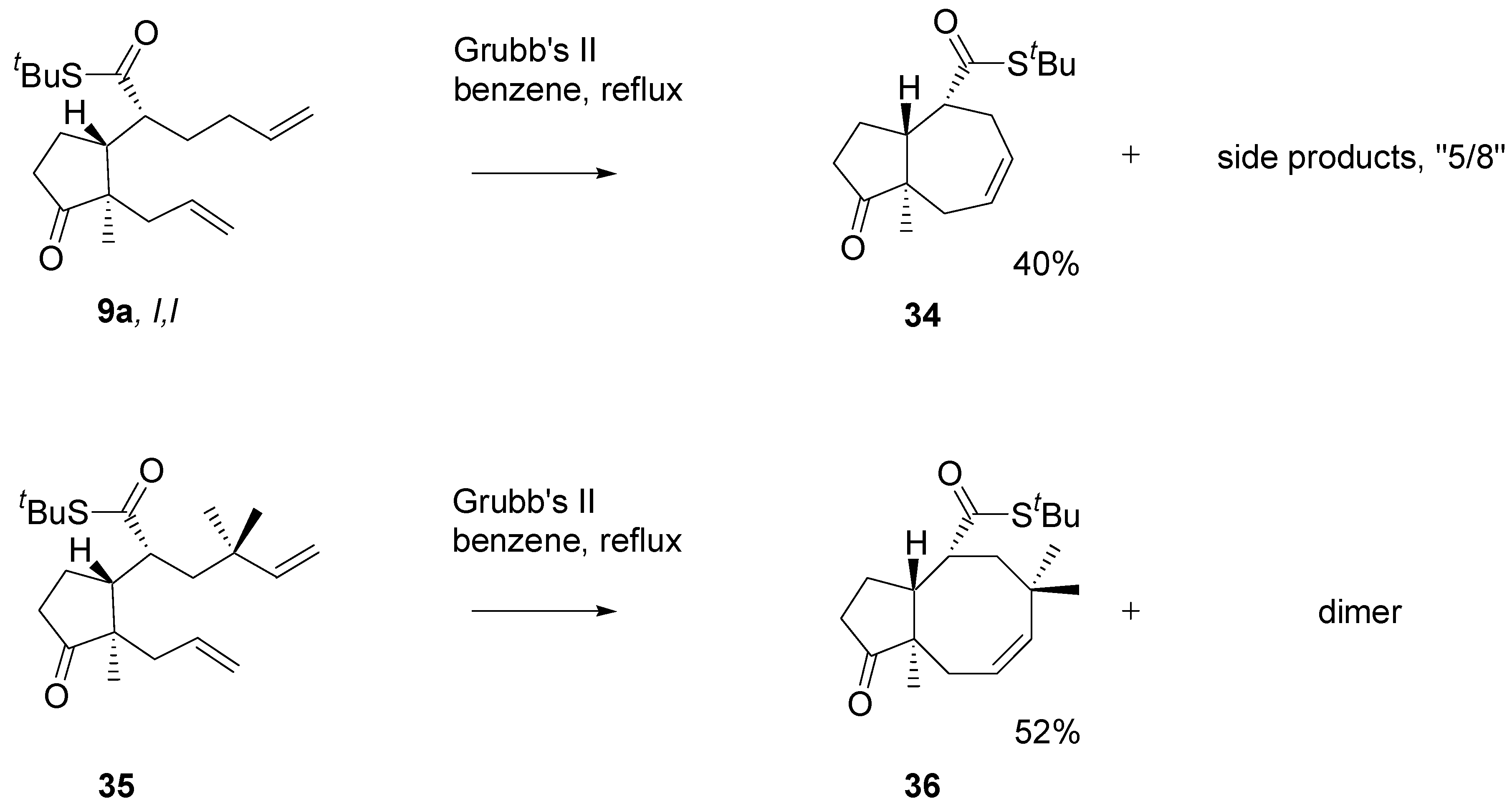
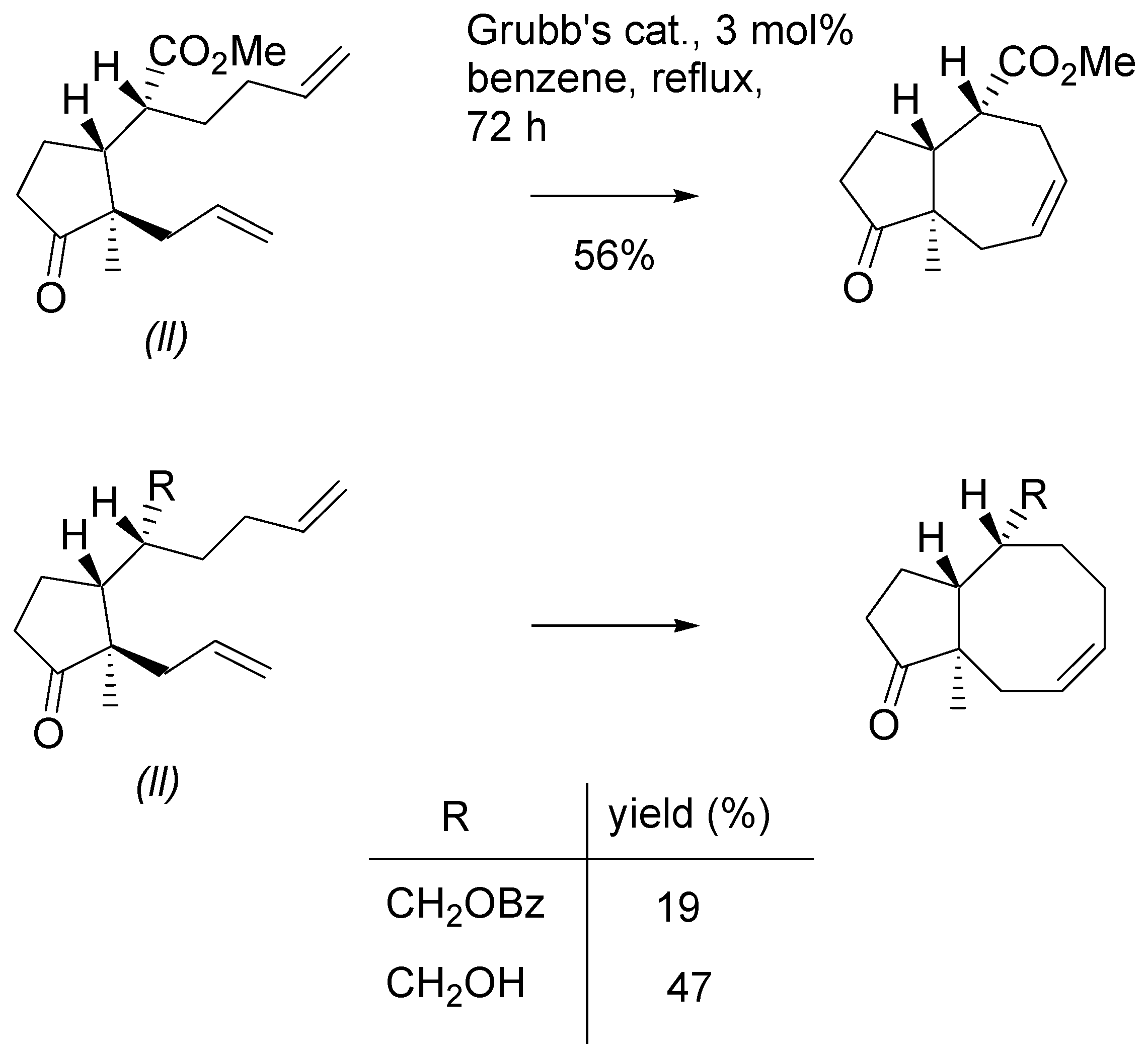
Thioesters turned out bad substrates for the metathesis reaction. RCM occurred only with Grubbs II catalyst and reproducibility of the results was poor. Diene
9b with Grubb’s catalyst, 3 mol%, in DCM, cleanly gave a dimeric product
33 that was isolated and purified by chromatography (
Scheme 13). Submission of the dimer to the action of the Grubb’s II catalyst (5 mol%) in boiling benzene, afforded the 5/7 ring system, similarly as the “direct” metathesis reaction.
Scheme 13.
Annulation of the dienes - thioesters via RCM reaction.
Scheme 13.
Annulation of the dienes - thioesters via RCM reaction.
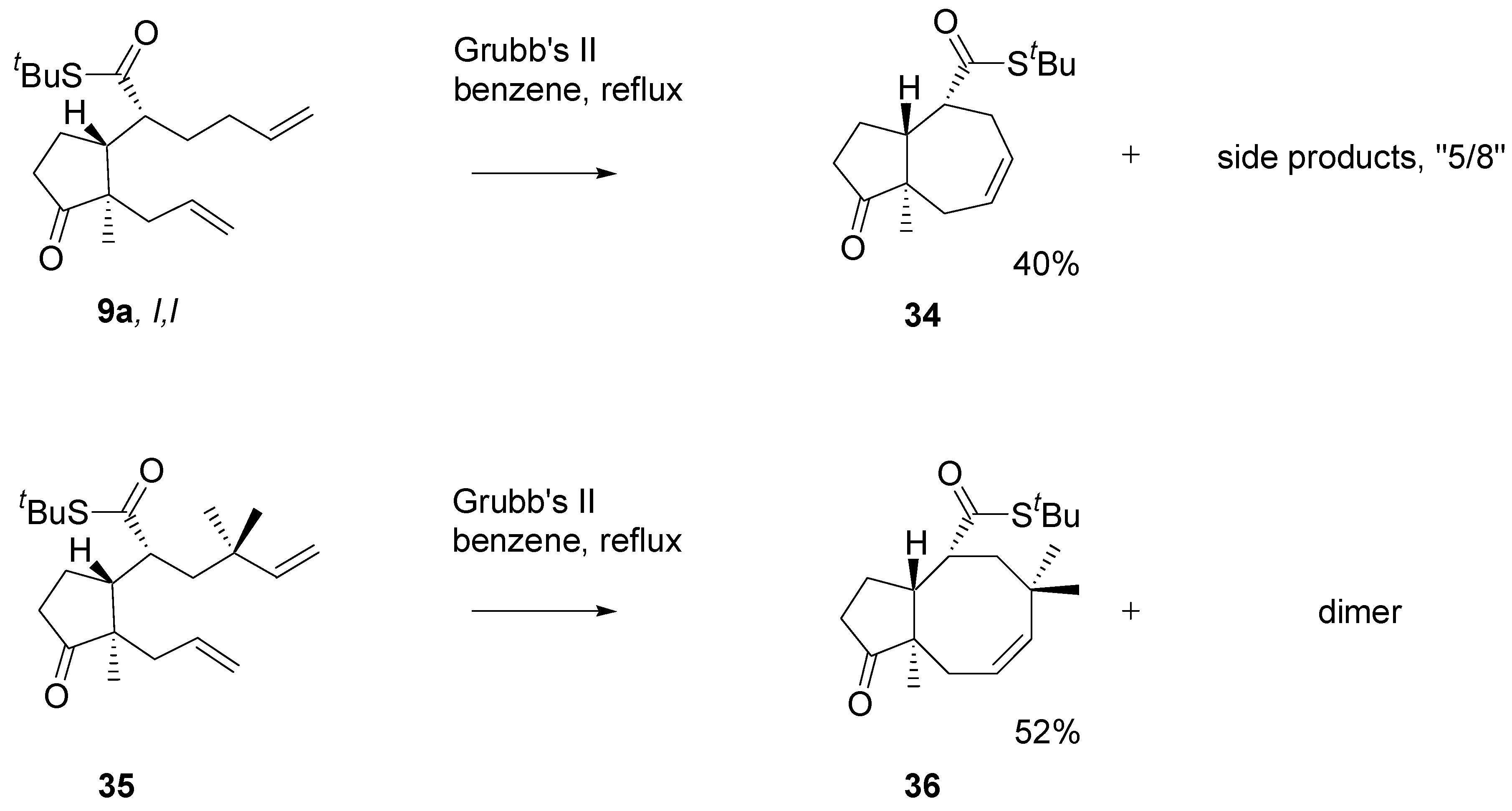
The diene with two mono-substituted double bonds
9a (
Scheme 14) with the Grubb’s II catalyst afforded the 5/7 product
34, isolated in 30-40% yield. Analysis of the crude products using a HPLC – MS instrument indicated the presence of the 8/5 product. The diene
35 carrying
gem-methyl groups under similar conditions afforded the cyclooctane derivative
36 in over 50% yield. The
gem-methyl groups in
35 block migration of one of the double bonds which facilitate formation of the eight-membered ring. On the other hand, non-bonding interactions are markedly higher.
Scheme 14.
Annulation of the dienes - thioesters via RCM reaction. 2.
Scheme 14.
Annulation of the dienes - thioesters via RCM reaction. 2.
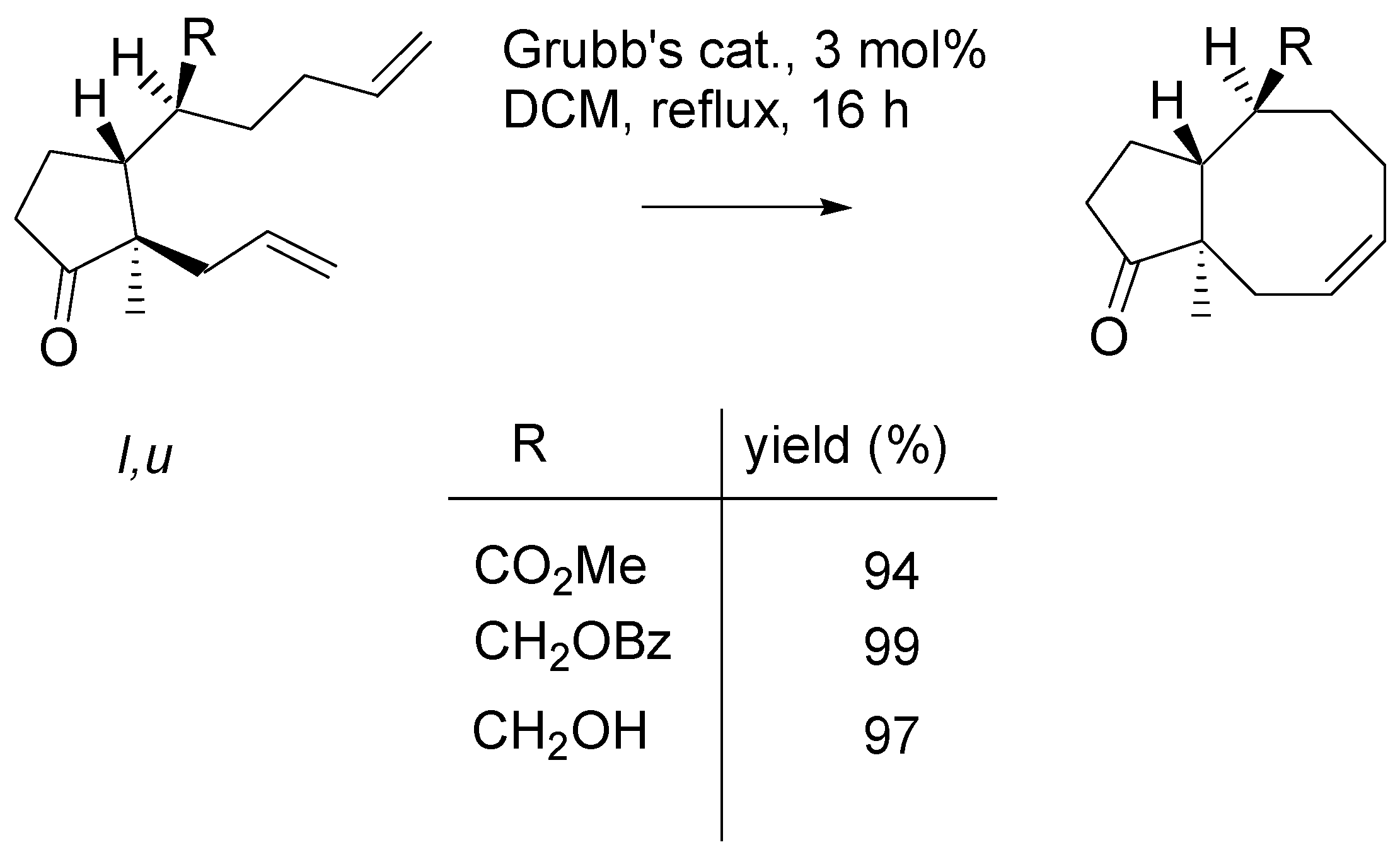
As it was already mentioned, metathesis reaction of methyl esters and their derivatives, with
u,l configuration uniformly gave the products in excellent yields. Some examples involving two mono-substituted double bonds are presented in
Scheme 15. All reactions were carried out using 3 mol% of Grubb’s catalyst in boiling DCM.
Scheme 15.
Metathesis of dienes – esters of u,l configuration.
Scheme 15.
Metathesis of dienes – esters of u,l configuration.
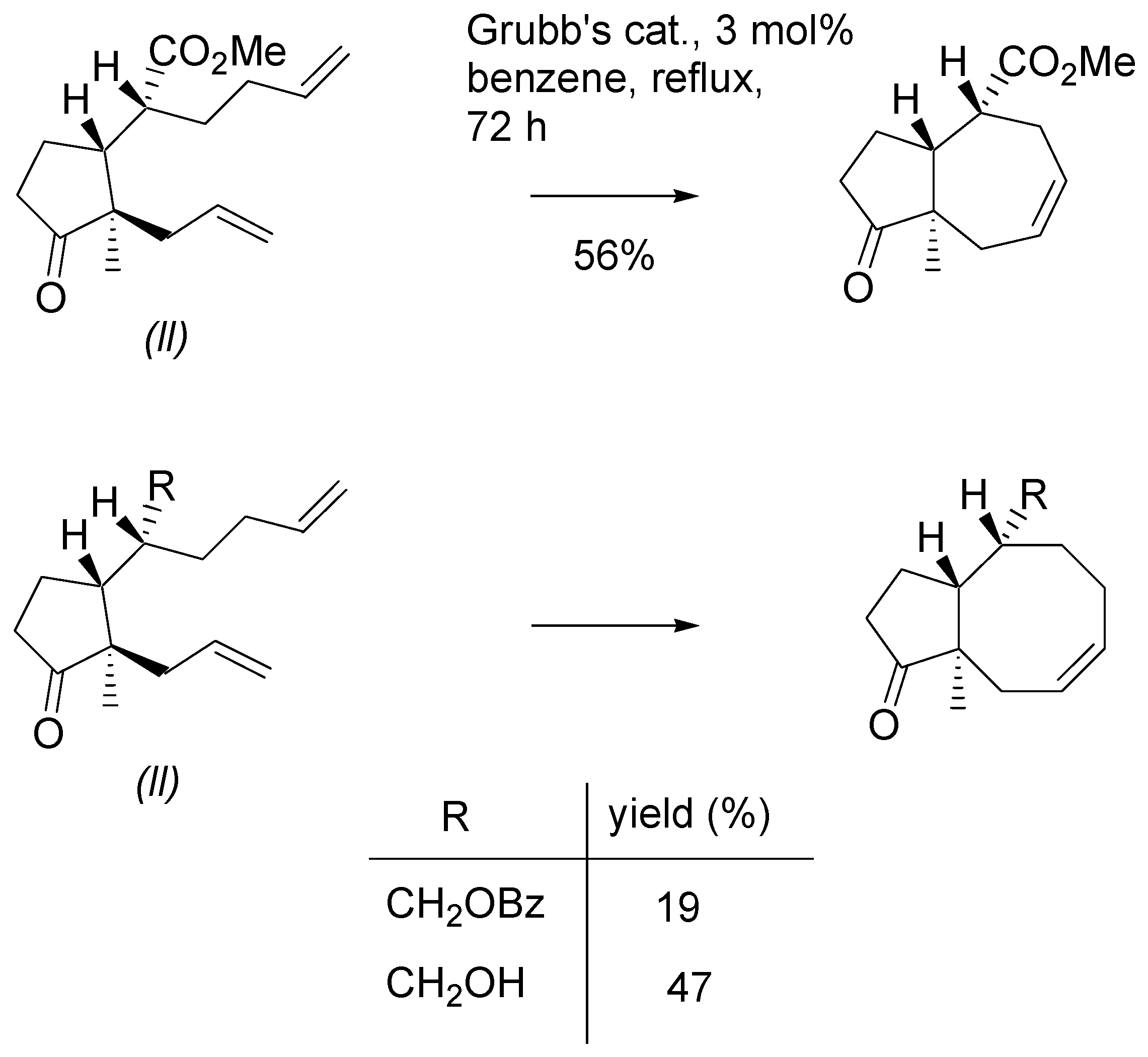
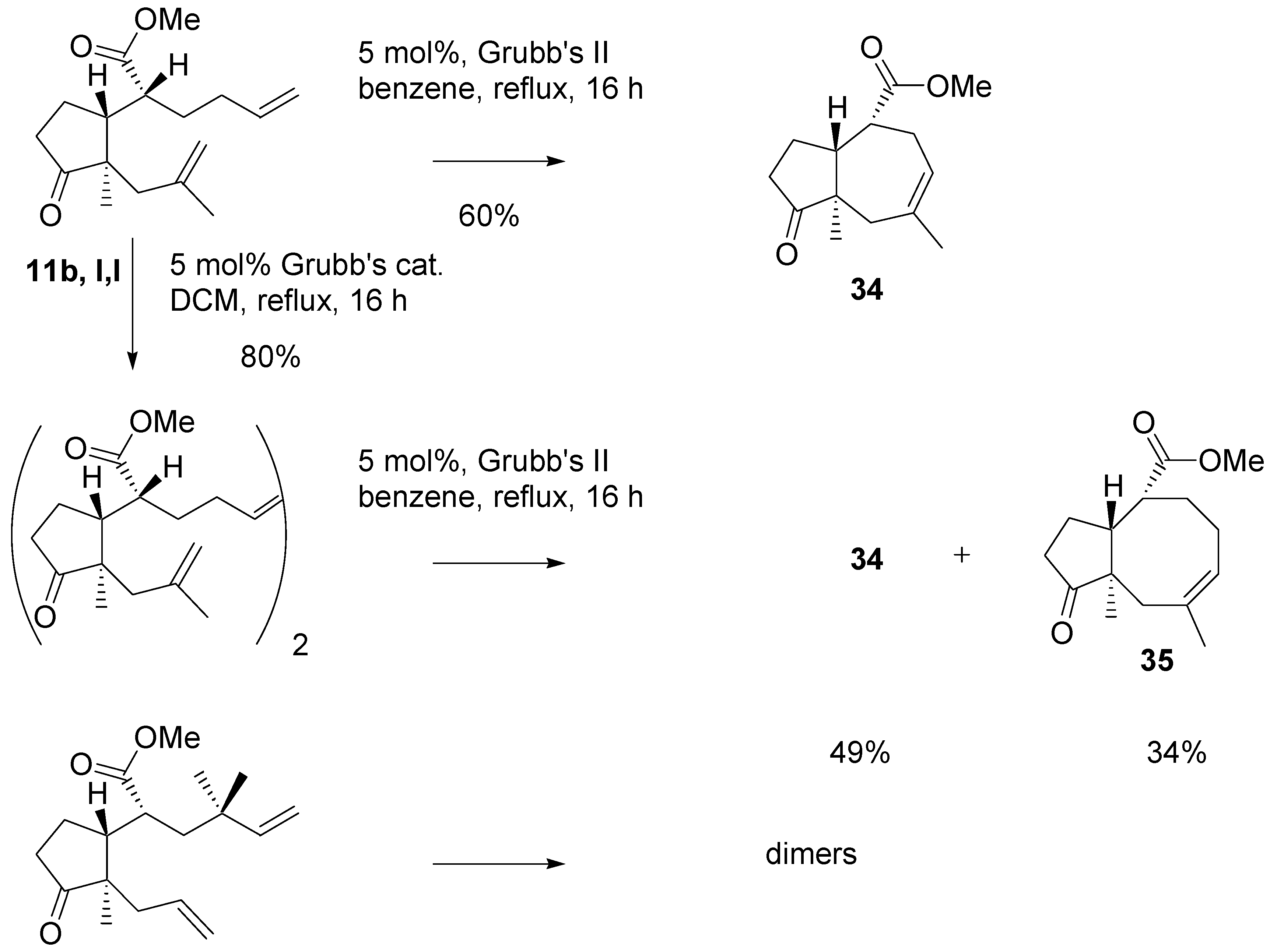
A more complex picture was observed in metathesis reactions of methyl esters of
l,l configuration (
Scheme 16). The diene with one disubstituted double bond with Grubb’s II catalyst cyclized to give the derivative with seven-membered ring in 60% yield. The same substrate was dimerized with Grubb’s catalyst under mild conditions. The dimer was purified by chromatography and then submitted to the action of the Grubb’s II catalyst under more forcing conditions. A mixture of products with seven- and eight-membered rings was obtained in high yield. This experiment shows that potential in employing dimers as intermediates in the metathesis reaction. In contrast to the analogous
tert-butylthio ester, the methyl ester with
gem-methyl groups did not produce the isolable cyclization product. Instead a mixture of dimers was obtained.
Scheme 16.
Metathesis of dienes – esters of l,l configuration.
Scheme 16.
Metathesis of dienes – esters of l,l configuration.
Methyl ester of
l,l configuration with two monosubstituted double bonds with the Grubb’s catalyst in benzene, at the reflux temperature, afforded, again, the product with seven-membered ring (
Scheme 17). The corresponding primary alcohol gave the “normal” product with eight membered ring. Eight- membered ring was form in the RCM reaction of benzoate; however, the product was isolated in low yields.
Scheme 17.
Metathesis of esters of l,l configuration.
Scheme 17.
Metathesis of esters of l,l configuration.
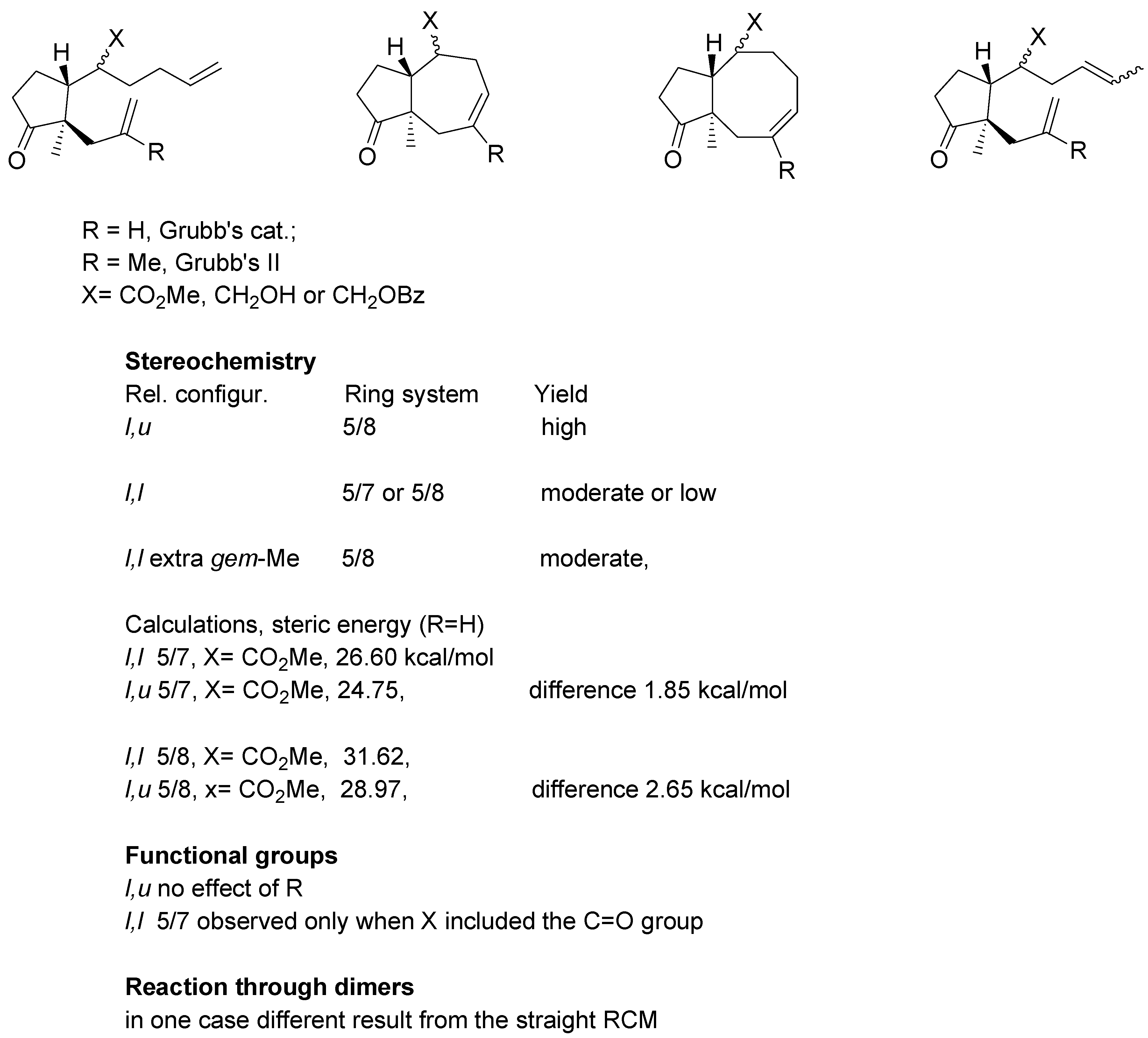
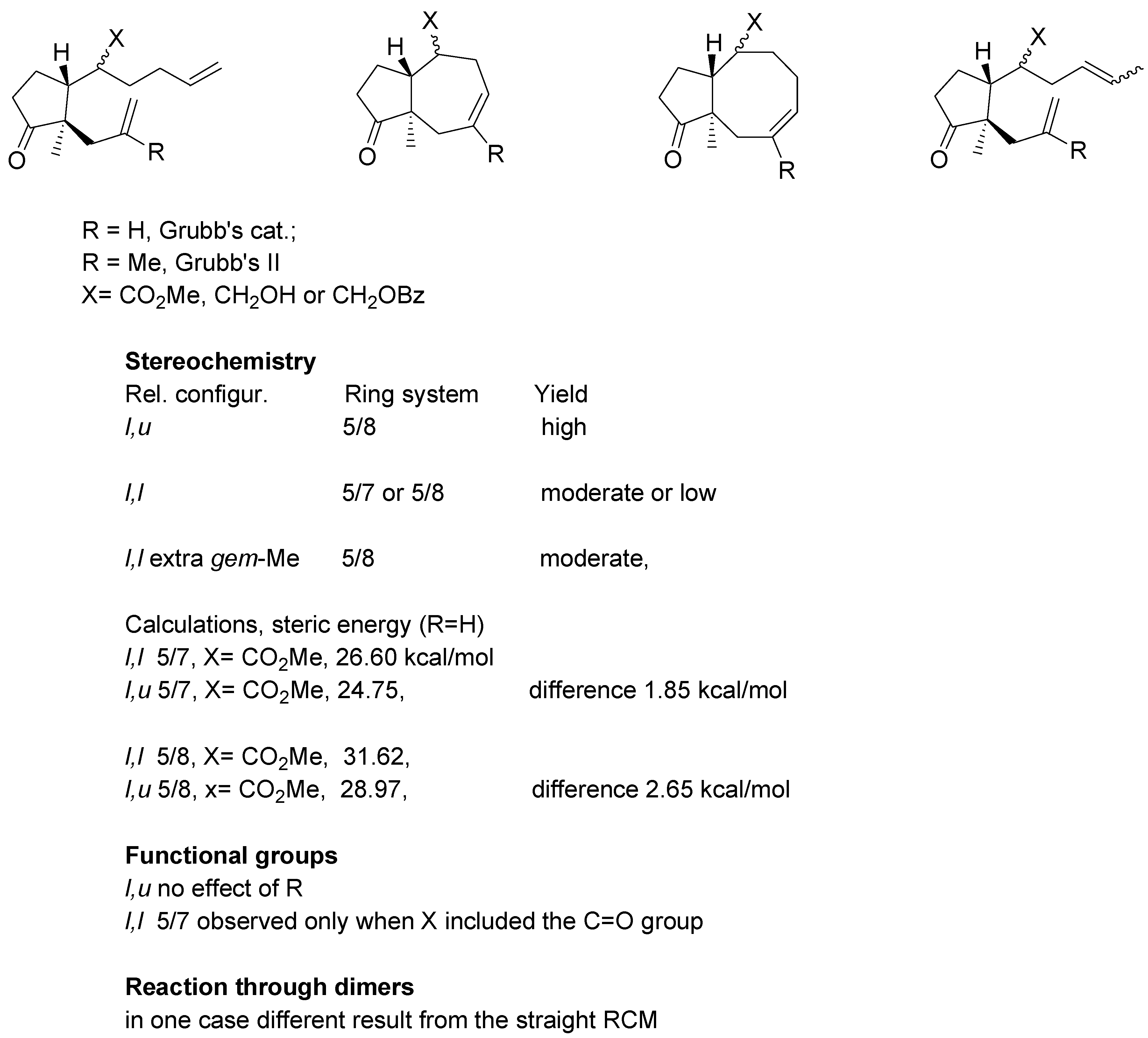
An overview of the experiments on the metathetic annulation with methyl esters and substrates derived from methyl esters, is presented in
Scheme 18. For substrates where R = H Grubb’s first generation catalyst was used, whereas R = Me required the second generation catalyst.
All examined dienes of u,l relative configuration uniformly gave eight-membered ring products in high yields. Their diastereomers of l,l-configuration generated 5/7- or 5/8- system in moderate or low yields. Thioesters gave derivatives (l,l configuration) gave products with 5/7 ring system with one noteworthy exception: the thioester with gem-methyl groups in the side chain afforded 5/8 rings system in a relatively high yield.
For the dienes with two mono-substituted double bonds some calculations were carried out using HyperChem® 5.0 program. For the 5/7 ring system the difference in steric energy between diastereomer with l,l and l,u configuration amounts to 1.85 kcal/mol whereas for the 5/8 system the corresponding difference was 2.65 kcal/mol. The calculation suggest cyclization of the l,l substrates to 5/7 ring system involves somewhat lower strain. It should be noted that calculations are concerned with products of reaction; no estimation of energy intermediates could be done.
With regard to the functional group effects, cyclization with expulsion of propylene was observed only for esters or thioesters (substrates involving the C=O group), but not for their reduction products (X=CH2OR). Finally, in one case a difference between “direct” RCM reaction and reaction carried out via the respective dimer, was noted.
Scheme 18.
Summary of annulation reactions
Scheme 18.
Summary of annulation reactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}