Results and Discussion
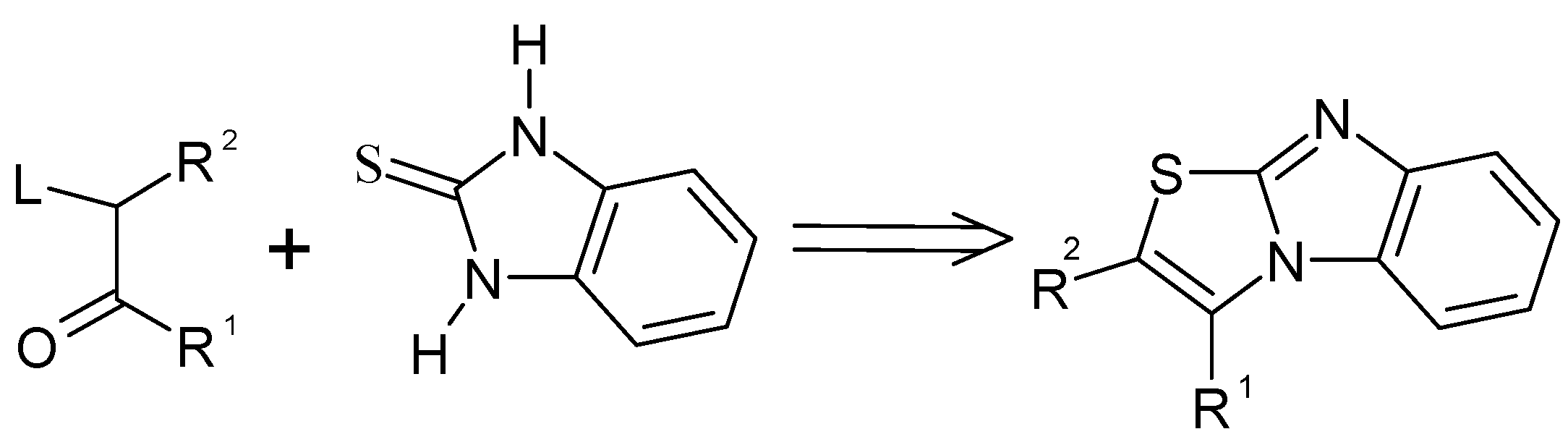
Curiously enough, the reaction sequence in which the C=N bond is formed in the last step from preformed activated thiazole ring has never been attempted to the best of our knowledge. The leaving group in position 2 of thiazolium salt will be expelled during cyclization. This approach is depicted in
Scheme 2.
Among the possible leaving groups in position 2 of the thiazolium salts, the methylthio group was particularly attractive since it could be easily generated by methylation at sulphur of the parent thiazoline-2-thione derivative. (
Scheme 3)
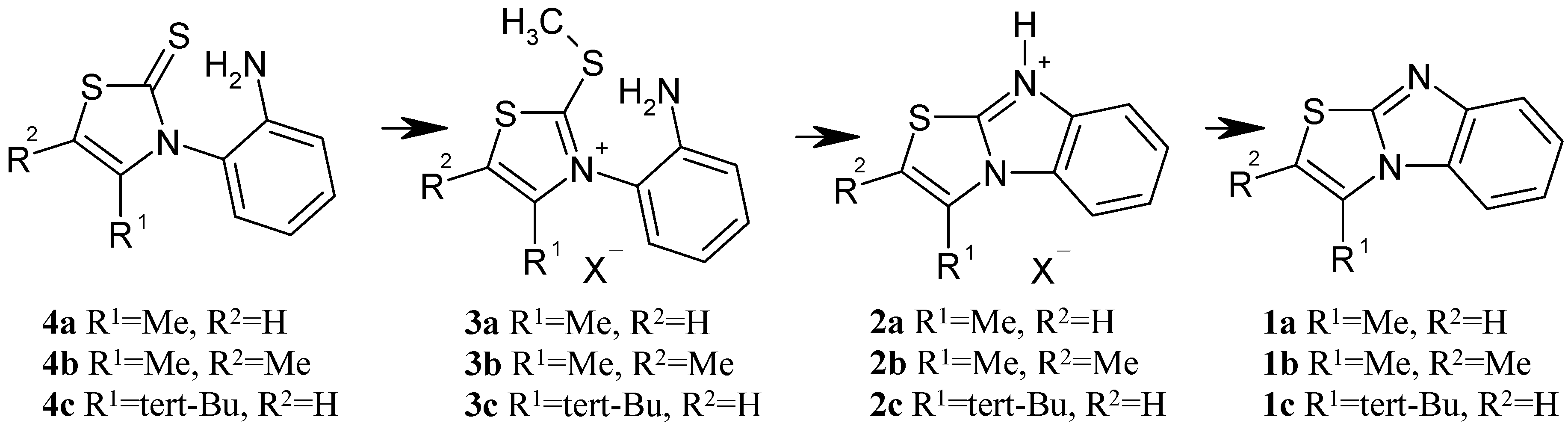
N-(2-Aminophenyl)-4-methyl-thiazoline-2-thione (
4a) has been first described in 1998 [
5]. This compound attracted our attention due to our long standing interest in the atropisomers of N-arylthiazoline-2-thione derivatives in terms of barriers to racemization and chiral recognition mechanisms on chiral stationary phases [
6,
7,
8,
9,
10]. Compound
4a is readily available in high yield by reacting chloroacetone in CH
3CN with the dithiocarbamate obtained quantitatively from 1,2-diamino-benzene and CS
2 in the presence of triethylamine [
5]. In our hands, the isolated yield in recrystallized thiazoline-2-thione derivative was 64%. The thiazolinethione reaction with iodomethane in anhydrous acetone at rt afforded the thiazolium salt
3a (X
-=I
- in
Scheme 2 and
Scheme 3) in quantitative yield. The reaction was monitored by TLC until the total disappearance of the starting thiazoline-thione was confirmed. As expected, due to the very high nucleophilicity of the sulphur atom, no methylation occurred on the amino group. After evaporation of acetone and methyl iodide in excess, NMR data were recorded on the crude reaction medium without any attempts to isolate the thiazolium salts. In the same reactor, the crude thiazolium salt when refluxed in methanol eliminated MeSH to yield 3-methyl-thiazolo[3,2-a]benzimidazolium iodide (
2a) as a single compound after evaporation of the solvent. NMR data of this crude reaction mixture were recorded again to check the disappearance of the S-Me signal and the appearance of the protonated thiazolobenzimidazole
2a. It may prove of interest to obtain directly the iodide salt. More generally, in the reaction sequence we are reporting, the counter anion is coming from the methylating agent and thus a wide variety of 3-methyl-thiazolo[3,2-a]benzimidazolium salts differing by the associated anion could be directly obtained. It must be stressed that obtaining pure isolated and recrystallized iodide salts was not our goal and thus no further analysis were performed on the crude iodide salts. The free 3-methyl-thiazolo[3,2-a]benzimidazole (
1a) is easily obtained in pure state by treatment with aqueous NaHCO
3 followed by careful extraction in CH
2Cl
2. In the starting thiazoline-2-thione derivative
4a the aryl group and the heterocycle are situated in two perpendicular planes giving rise to stable atropisomers [
11], it results from this spatial arrangement that in the
1H-NMR spectrum the 4-methyl group is highly shielded (
δ = 1.93 ppm), whereas in the final product
1a the methyl group is highly deshielded due to the planarity of the structure (
δ = 2.70 ppm). In the two non-isolated thiazolium and thiazolo[3,2-a]benzimidazolium iodide intermediates, the chemical shifts of the proton of that particular methyl group appeared at 2.25 ppm and 2.92 ppm respectively. The reaction sequence was also exemplified with success to yield
1b starting from the already described N-(2-aminophenyl)-4,5-dimethyl-thiazoline-2-thione (
4b)
[12].
It was of interest to check the robustness of the reaction sequence in case of strong steric repulsion between the substituent in position 4 of the thiazolium salts and the aryl group, which shall adopt a coplanar arrangement in the final product. We prepared the corresponding analogues starting from chloropinacolone to obtain the 3-tert-butyl-thiazolo[3,2-a]benzimidazole (1c). The cyclization rate is slower than in the case of the 3-methyl analogues leading to 1a or 1b. After 12 hours reflux in methanol the mixture is composed of 16% of the unchanged thiazolium salt (3c) and 84% of 3-tert-butyl-thiazolo[3,2-a]benzimidazolium iodide 2c. However, the transformation is quantitatively achieved after 41 hrs without the formation of any by-product.
Experimental
General
1H-NMR spectra were recorded at 300 or 200 MHz and 13C-NMR spectra at 75 or 50 MHz on Bruker Avance DPX-300 or 200 instruments. Chemical shifts are reported in ppm with the signal for residual solvent as internal standard. J values are reported in Hz. Melting points were measured using a Kofler hot stage apparatus and are not corrected. Flash column chromatography was performed with silica gel 60 (230-400 mesh). TLC were carried out on Merck 60F254 silica plates.
N-(2-Aminophenyl)-4-methyl-thiazoline-2-thione (4a).
The already reported synthesis of
4a [5], has been used with very slight modifications. Triethylamine (68.6 mL, 492 mmol) was added to a suspension of 1,2-phenylenediamine (28 g, 259 mmol) in CS
2 (600 mL). After stirring for 2 h, the dithiocarbamate salt was filtered off, washed with Et
2O and used without further purification. 1-Chloro-propan-2-one (20 mL, 251 mmol) was added to a suspension of 70 g (245 mmol) of dithiocarbamate salt in 600 mL of acetonitrile. After stirring for 12-48 h at r.t., the solvent was removed
in vacuo and 66 mL of 36% HCl was added and the mixture was left under stirring for 20 min. Water (400 mL) was added and the mixture was extracted with CH
2Cl
2 (3 x 400 mL). The organic phase was washed with water (3 x 200 mL), dried over MgSO
4 and evaporated. Recrystallisation in absolute ethanol afforded (
4a, 35 g, 64 %) as a white powder with m.p. 180°C (lit.[
5]: 184°C); Rf 0.22 (CH
2Cl
2). The
1H-NMR (CDCl
3, 300 MHz) and
13C-NMR (CDCl
3, 75 MHz) were in agreement with literature data for this compound [
5].
N-(2-Aminophenyl)-4, 5-dimethyl-thiazoline-2-thione (4b).
The same procedure as for
4a was used with 3-chloro-butan-2-one (2.45 mL, 24.2 mmol) to afford
4b (5.2 g, 73 %) as a white powder upon recrystallisation in ethanol 96%. In this case, HCl was replaced by H
2SO
4 [
5,
12]. White powder with m.p. 149°C (lit.[
5,
12]: 139-140°C); Rf 0.20 (CH
2Cl
2). The
1H-NMR (CDCl
3, 300 MHz) and
13C-NMR (CDCl
3, 75 MHz) were in agreement with literature data for this compound [
5,
12].
N-(2-aminophenyl)-4-tert-butyl-thiazoline-2-thione (4c).
The same procedure as for 4a was used with 1,2-phenylenediamine (1 g, 9.2 mmol) and 1-chloro-pinacolone (1.21 mL, 9.2 mmol) to afford, after column chromatography on silica gel with CH2Cl2 elution, 4c (0.8 g, 44%) as a crystalline white powder with m.p. 190°C; Rf 0.42 (CH2Cl2); 1H-NMR (CDCl3, 200 MHz): δ = 1.17 (s, 9H, CH3), 3.62 (s, 2H, NH2), 6.41 (s, 1H, =CH), 6.80-7.40 (m, 4H, Ar); 13C-NMR (CDCl3, 50 MHz): δ = 29.48 (CH3), 34.74 (C-(CH3)3), 106.08 (=CH), 117.37, 118.83, 125.68, 130.55, 131.00, 144.04, 153.45, 190.91 (C=S); Anal. For C13H16N2S2: calcd. C 59.05, H 6.10, N 10.59, S, 24.25; found C 59.04, H 6.23, N 10.58, S 24.50.
General Procedure for the synthesis of the thiazolo[3,2-a]benzimidazole (1a, 1b, 1c).
N-(2-Aminophenyl)-thiazoline-2-thione (2.25 mmol) and CH3I (1.4 mL, 10 eq) were stirred in 10 mL of acetone at r.t. for 3 h. Evaporation of the excess of CH3I and acetone afforded quantitatively the thiazolium iodide. NMR and TLC were taken on the crude reaction medium to check the completeness of the methylation. (NMR data shown). In the same vessel, crude 2-methylthio-N-(2-aminophenyl)-thiazolium iodides (2.25 mmol) were refluxed in 20 mL of methanol for 12 h (41 h for the tert-Bu-derivative). Evaporation of the methanol afforded quantitatively the thiazolo[3,2-a]benzimidazolium iodides (NMR data shown). To these crude solids, 20 mL of a saturated NaHCO3 solution was added and the solution was extracted with CH2Cl2 (3 x 10 mL). The organic phase was dried over MgSO4 and evaporated to afford quantitatively the thiazolo[3,2-a]benzimidazole derivatives.
3-Methyl-thiazolo[3,2-a]benzimidazole (1a).
Pale yellow powder m.p. 162°C (lit.[
1]160-165°C); Rf 0.45 (CH
2Cl
2/AcOEt 9/1). NMR data are consistent with literature data [
1].
1H-NMR (CDCl
3, 200 MHz):
δ = 2.70 (d, 3H, CH
3, J = 1.2), 6.31 (q, 1H, =CH, J = 1.2), 7.10-7.40 (m, 2H, Ar), 7.70-7.85 (m, 2H, Ar);
13C-NMR (CDCl
3, 50 MHz):
δ = 14.33 (CH
3), 104.46 (=CH), 110.31, 119.04, 120.47, 123.06, 129.75, 130.23, 157.15(C=N).
2,3-Dimethyl-thiazolo[3,2-a]benzimidazole (1b).
White powder m.p. 151°C (lit. [
1c] 154-156°C); Rf 0.45 (CH
2Cl
2/AcOEt 9/1). NMR data are consistent with literature data [
1c].
1H-NMR (CDCl
3, 200 MHz):
δ = 2.19 (q, 3H, CH
3, J = 6.0), 2.43 (q, 3H, CH
3, J = 6.0), 7.00-7.30 (m, 2H, Ar), 7.50-7.70 (m, 2H, Ar);
13C-NMR (CDCl
3, 50 MHz):
δ = 11.52, 12.36, 110.15, 116.03, 118.88, 120.27, 122.67, 124.47, 130.31, 147.82, 155.00.
3-tert-Butyl-thiazolo[3,2-a]benzimidazole (1c).
Colorless oil; Rf 0.70 (CH2Cl2/AcOEt 9/1); 1H-NMR (CDCl3, 200 MHz): δ = 1.61 (s, 9H, CH3), 6.41 (s, 1H, =CH), 7.20-7.50 (m, 2H, Ar), 7.80-7.90 (m, 1H, Ar), 7.95-8.05 (m, 1H, Ar); 13C-NMR (CDCl3, 50 MHz): δ = 28.40, 33.44, 102.79, 113.90, 119.24, 120.28, 122.95, 130.06, 143.74, 148.92, 158.65; Anal. For C13H14N2S: Calcd. C 67.79, H 6.13, N 12.16, S 13.92; found C 67.76, H 6.31, N 11.85, S 14.17.
3-Methyl-thiazolo[3,2-a]benzimidazolium iodide (2a)
1H-NMR (CD3OD, 300 MHz): δ = 2.92 (d, 3H, CH3, J = 1.2), 7.26 (q, 1H, =CH, J = 1.2), 7.55-7.75 (m, 2H, Ar) , 7.80-7.90 (m, 1H, Ar) , 8.15-8.25 (m, 1H, Ar); 13C-NMR (CD3OD, 75 MHz): δ = 14.21, 112.02, 114.37, 115.50, 125.58, 128.03, 129.20, 134.32, 138.14, 155.85.
2,3-Dimethyl-thiazolo[3,2-a]benzimidazolium iodide (2b)
1H-NMR (CD3OD, 200 MHz): δ = 2.56 (q, 3H, CH3, J = 1.0), 2.84 (q, 3H, CH3, J = 1.0), 7.50-7.95 (m, 3H, Ar), 8.20-8.30 (m, 1H, Ar); 13C-NMR (CD3OD, 50 MHz): δ = 12.18 (CH3), 12.38 (CH3), 114.06, 115.61, 123.30, 125.21, 127.54, 129.26, 138.23, 153.25 (C=N).
3-tert-Butyl-thiazolo[3,2-a]benzimidazolium iodide (2c)
1H-NMR (CD3OD, 200 MHz): δ = 1.67 (s, 9H, CH3), 7.25 (s, H, =CH), 7.50-7.70 (m, 2H, Ar), 7.80-7.95 (m, 1H, Ar), 8.25-8.35 (m, 1H, Ar); 13C-NMR (CD3OD, 50 MHz): δ = 28.98, 34.75, 110.16, 116.26, 117.19, 124.93, 127.45, 129.43, 139.99, 146.94, 157.96.

{kind=link}
{kind=link}
{kind=link}