Introduction
The development of new methods to obtain indans is a particularly active area in synthetic organic chemistry [
1] because these molecules are very useful building blocks for the synthesis of bioactive compounds, such as the well-known Indinavir
® [
2] and Aricept
® [
3]. Examples of important strategies to construct indans are cycloaddition and Friedel-Crafts reactions [
4]. Another approach is the ring contraction of a naphthalene derivative, which can be performed by a thallium(III)-mediated oxidative rearrangement [
5,
6,
7,
8,
9,
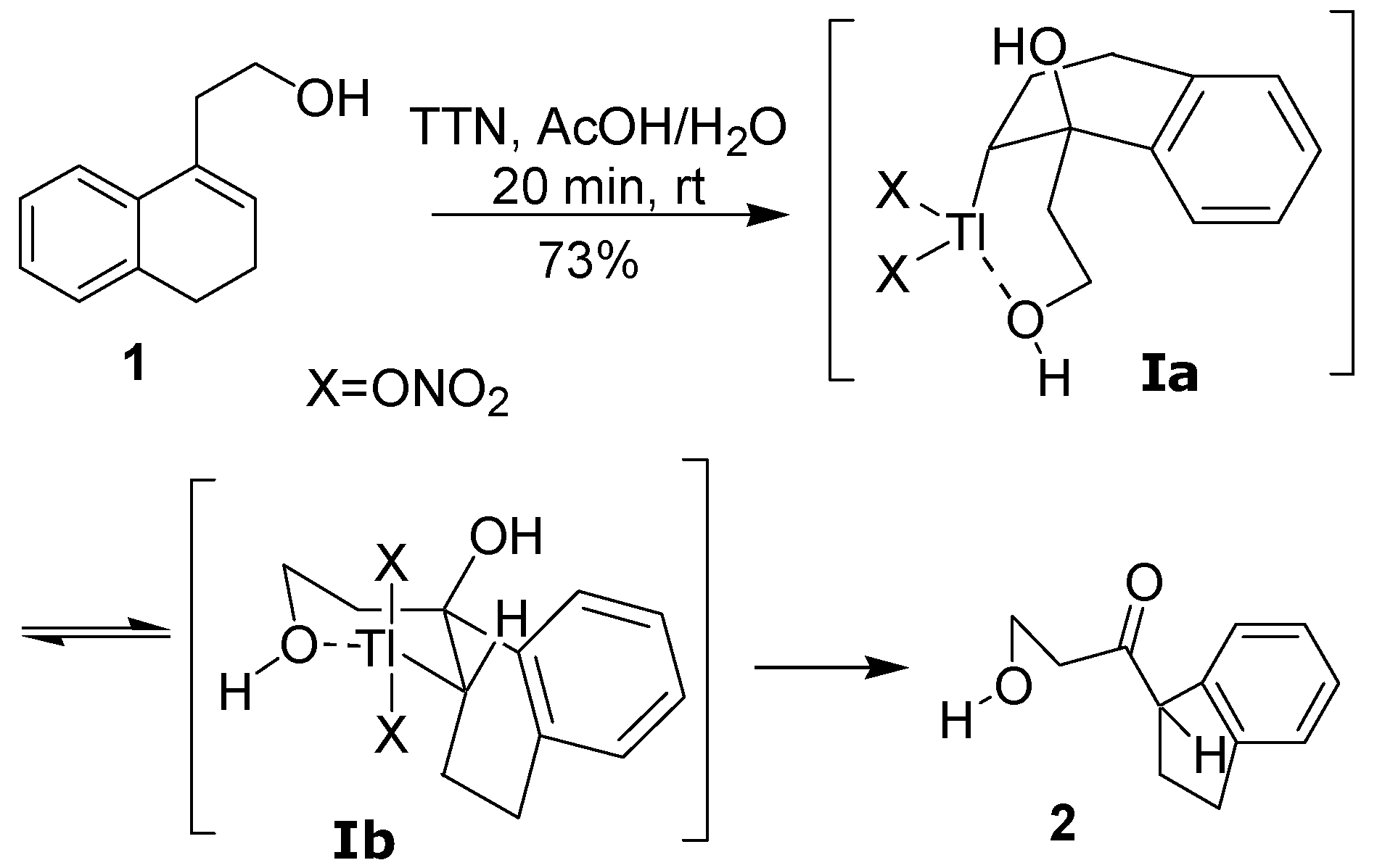
10]. An example of this reaction is the treatment of a homoallylic alcohol, such as
1, with thallium trinitrate (TTN) in a mixture of AcOH and H
2O, leading to the indan
2 in good isolated yield. The hydroxyl group of the side chain plays an active role in this ring contraction, coordinating with the thallium atom in the addition step [
5] (
Scheme 1). Although the behavior of several homoallylic alcohols analogous to
1 has been investigated, the effect of the reaction conditions was not studied. Considering that thallium(III)-mediated oxidations can be highly sensitive to the counter-ion of the thallium salt, as well as to the reaction conditions [
11], we decided to perform a detailed study of the oxidation of
1 with several different thallium(III) salts, including the new thallium tris-[
(S)-2-acetoxypropionate] (TTAP) and thallium tripropionate (TTP), to show that the indan
2 can be obtained under a variety of different conditions. During this work, we also discovered that a double bond dimethoxylation can take place when the oxidation of
1 is performed with thallium triacetate (TTA) in methanol. Although the selective dihydroxylation of double bonds is a well-established reaction [
12], there are only a few methods for the corresponding dimethoxylation reaction [
13,
14,
15].
Results and Discussion
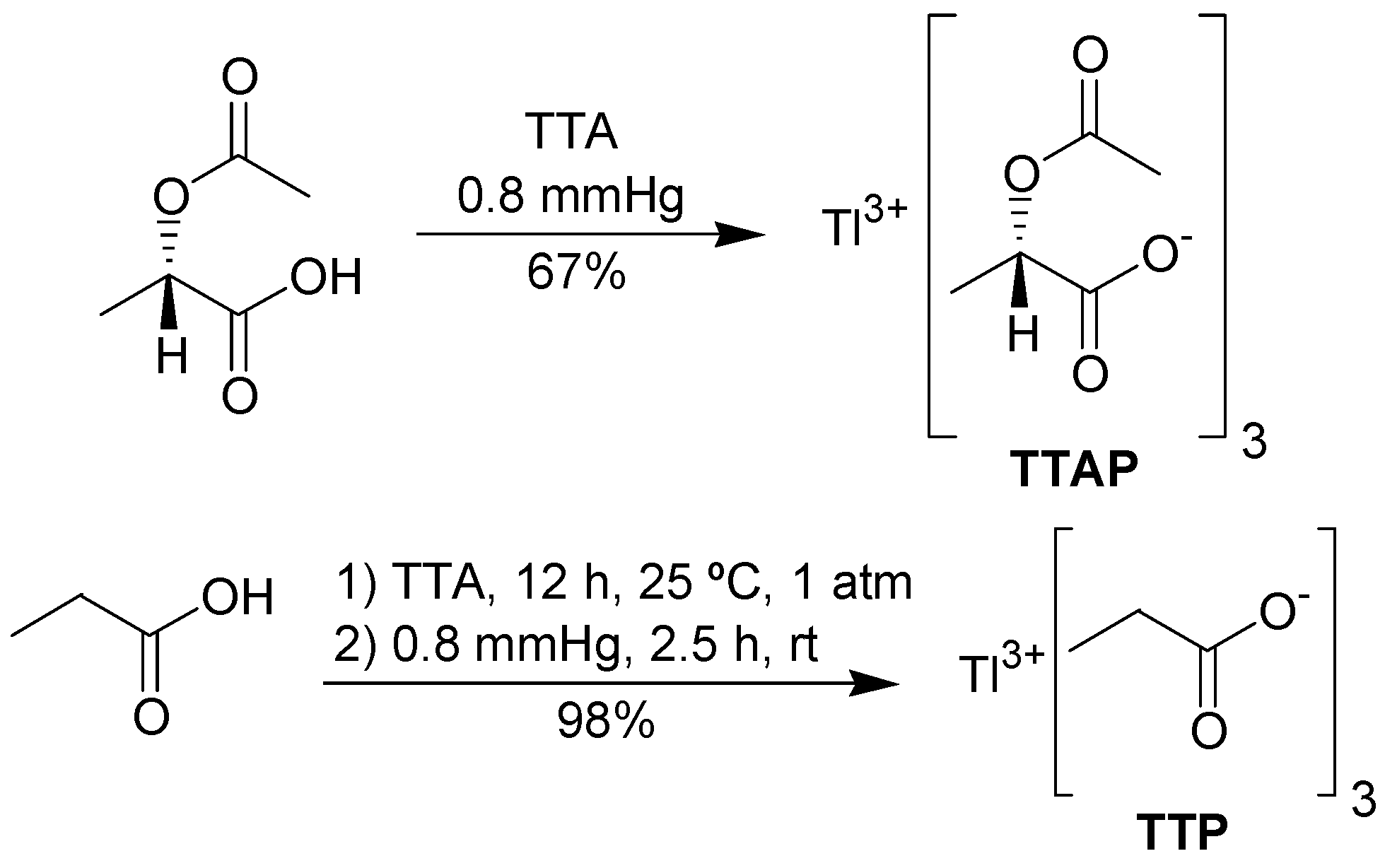
In addition to the commercially available TTA and thallium tris-trifluoroacetate (TTFA), we decided to investigate the behavior and the synthesis of some new thallium(III) salts. Thus, inspired by the article by Arseniadys and co-workers concerning the reaction of lead(IV) acetate in
(S)-2-acetoxypropionic acid [
16], thallium tris-[
(S)-2-acetoxypropionate] (TTAP) was chosen as a candidate to further explore the oxidation of the substrate
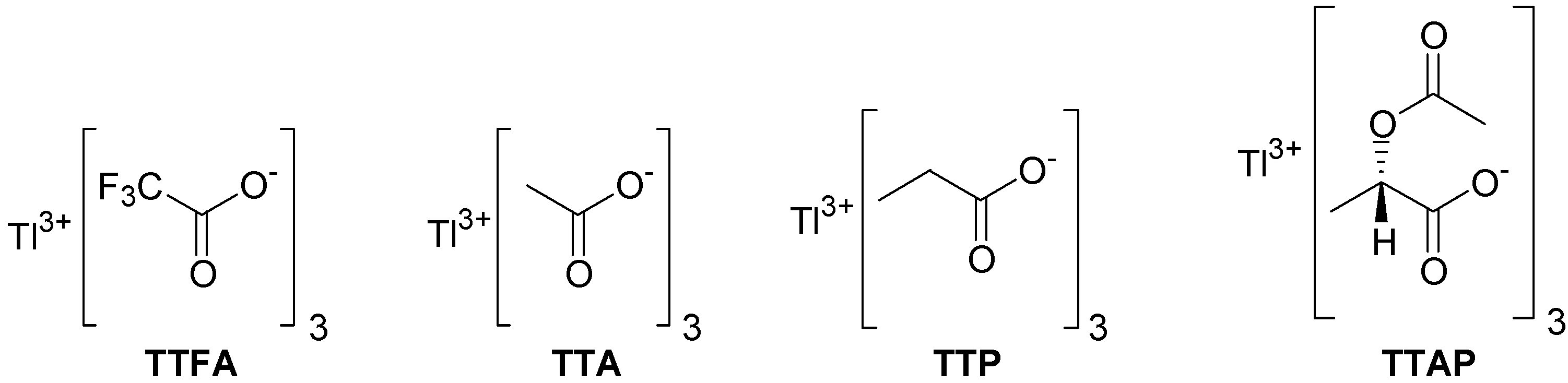
1. We expect that eventually the chiral non-racemic TTAP will find useful applications in asymmetric oxidations. For comparison with either TTA or TTAP, the preparation of thallium tripropionate (TTP) was also conceived (
Figure 1).
Figure 1.
Structures of thallium(III) carboxylates.
Figure 1.
Structures of thallium(III) carboxylates.
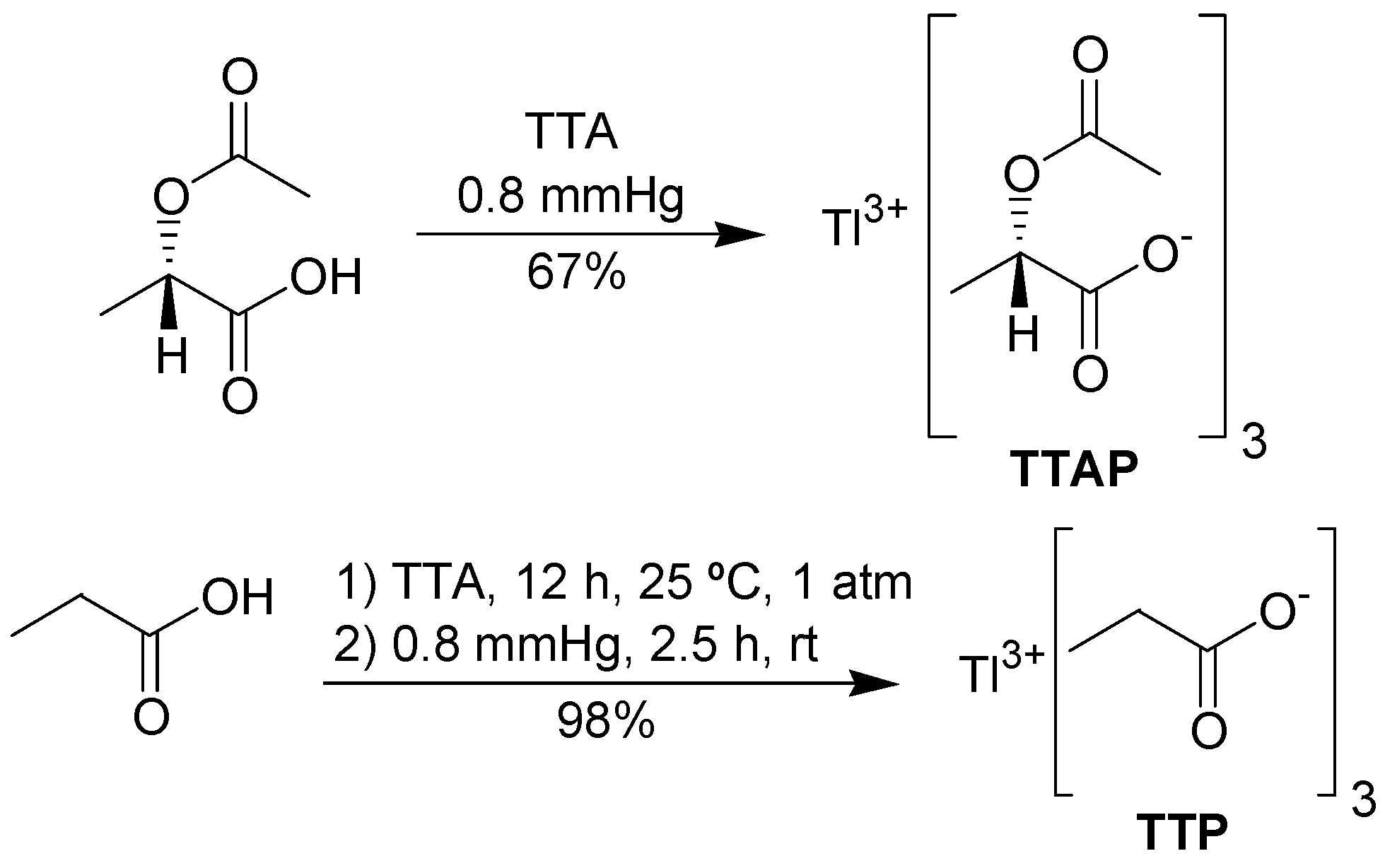
To prepare TTAP, it was necessary to obtain
(S)-(-)-2-acetoxypropionic acid in an efficient manner. In our hands, this was best achieved by acetylation of
(S)-(+)-lactic acid with acetyl chloride in THF, avoiding an aqueous work-up. The synthesis of TTAP was performed according to the procedure of Mosher and Kher [
17], who prepared several lead carboxylates. The required metathesis reaction occurred by mixing TTA and
(S)-(-)-2-acetoxypropionic acid under reduced pressure, leading to TTAP in good yield. TTAP was obtained in a pure form as a white solid, showing a single set of signals in the NMR spectrum. However, this salt is unstable, becoming a highly viscous yellow oil when exposed to the air. Thus, thallium(I) content varied from 14 to 22%. TTP was prepared in a similar fashion. However, the mixture of TTA and propionic acid must be stirred for 12 h at room pressure and for 2.5 h at 0.8 mmHg (
Scheme 2), because under the same conditions used for TTAP, the metathesis occurs only partially. TTP is a stable white solid and its thallium(I) content was zero.
The thallium(III) mediated cyclization of the monoterpene isopulegol has been carefully studied by Ferraz and co-workers [
18]. This oxidation occurs either with TTN (
Table 1, entry 1) or with TTA (entry 2) under a variety of different conditions [
19], leading always to the corresponding cyclic ether in a very pure form [
20]. For these reasons, this reaction was selected to test the reactivity of TTAP and of TTP. When isopulegol was treated with TTP (entry 3) or with TTAP (entry 4) in similar conditions to that used with TTA or with TTN, the corresponding cyclic hydroxyether was obtained in good isolated yield. The reaction time clearly shows a difference of reactivity among the thallium salts, where the more reactive is the TTN and the less is the TTAP.
Table 1.
Cyclization of Isopulegol with Thallium(III) Salts
![Molecules 10 01419 i001]()
Table 1.
Cyclization of Isopulegol with Thallium(III) Salts ![Molecules 10 01419 i001]()
| Entry | Conditions | Yield |
|---|
| 1 | TTN, AcOH/H2O (1:1), 5 min, rt | 86% [18] |
| 2 | TTA, AcOH/H2O (1:1), 40 min, rt | 92% [20] |
| 3 | TTP, AcOH/H2O (2:1), 2 h, rt | 71% |
| 4 | TTAP, AcOH/H2O (2:1), 4 h, rt | 69% |
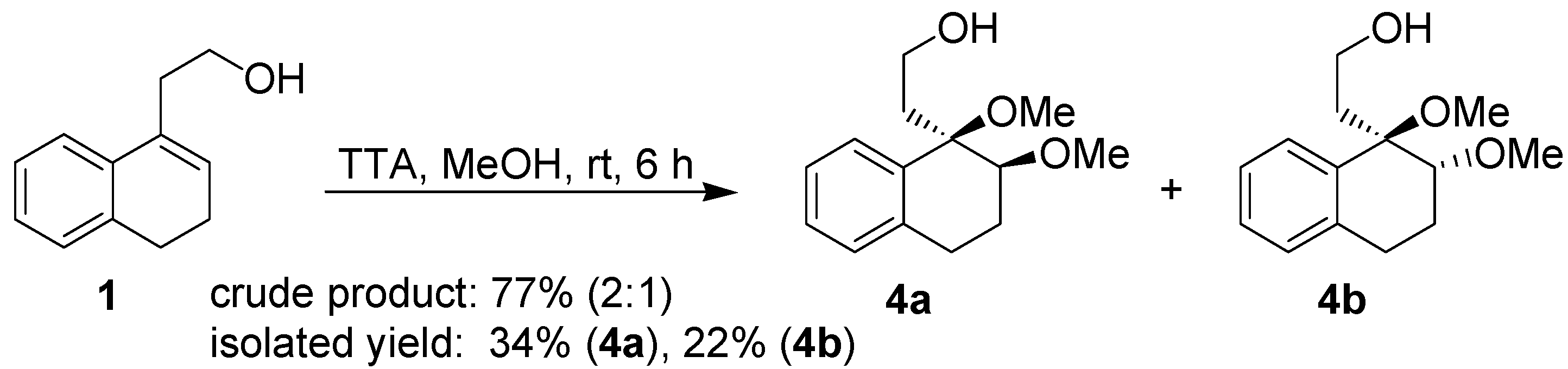
After the positive results obtained in the cyclization of isopulegol, the behavior of the substrate
1 towards thallium(III) was carefully investigated. This study was initiated by performing the reaction of
1 with TTA in two different solvents, namely methanol and aqueous acetic acid. Treatment of the homoallylic alcohol
1 with TTA in methanol gavea 2:1 mixture of the addition products
4a and
4b, respectively, as determined by
1H-NMR analysis of the crude product. However, a different ratio was obtained for the pure isolated products (
Scheme 3).
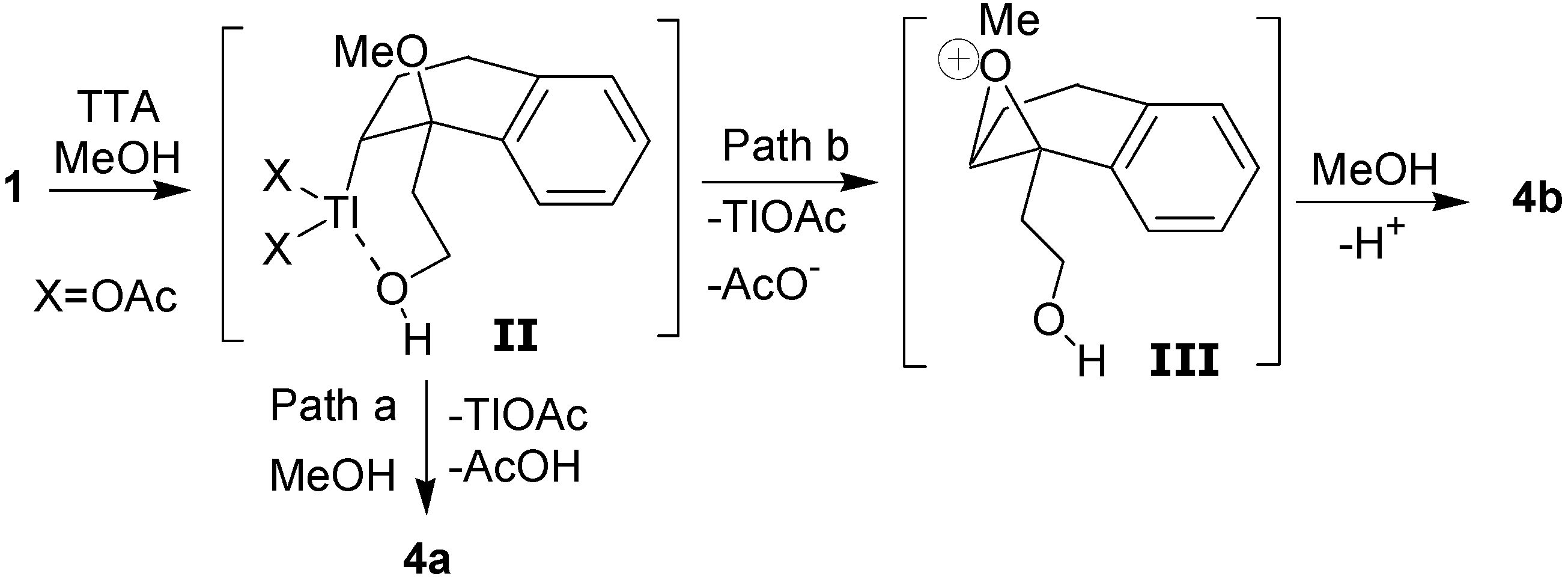
The formation of the
cis- and
trans-dimethoxylated compounds
4a and
4b can be explained by the mechanism shown in
Scheme 4. The first step would be the electrophilic addition of the thallium(III), which is assisted by the hydroxyl group, giving the intermediate
II. The
cis diastereomer
4a would result from a reductive displacement of thallium(III) by the methanol in the oxythallated adduct
II (Path a). The intermediate
II could also lead to the trans compound
4b through the oxonium ion
III, which is formed by an intramolecular displacement of the thallium(III) (Path b).
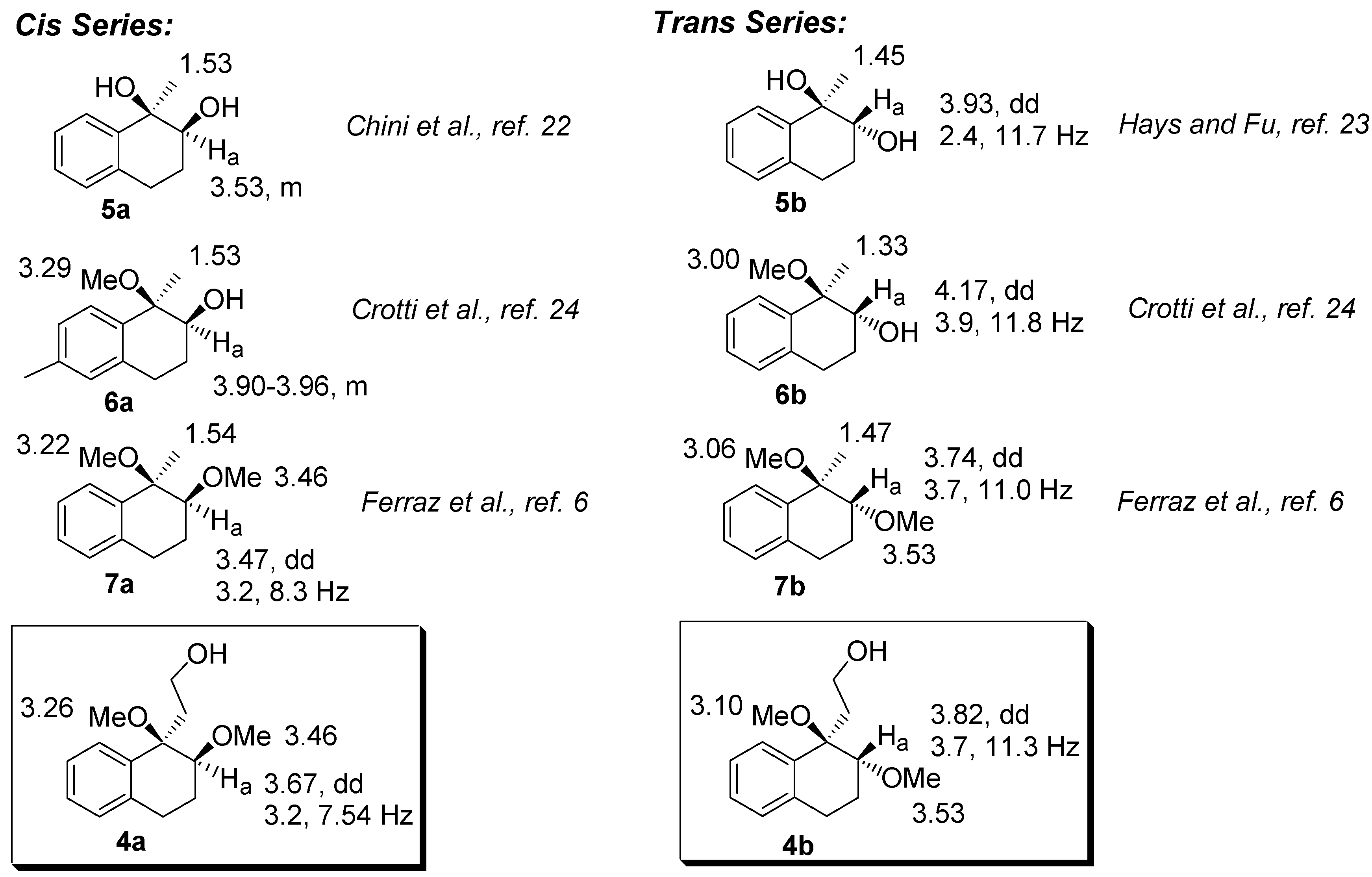
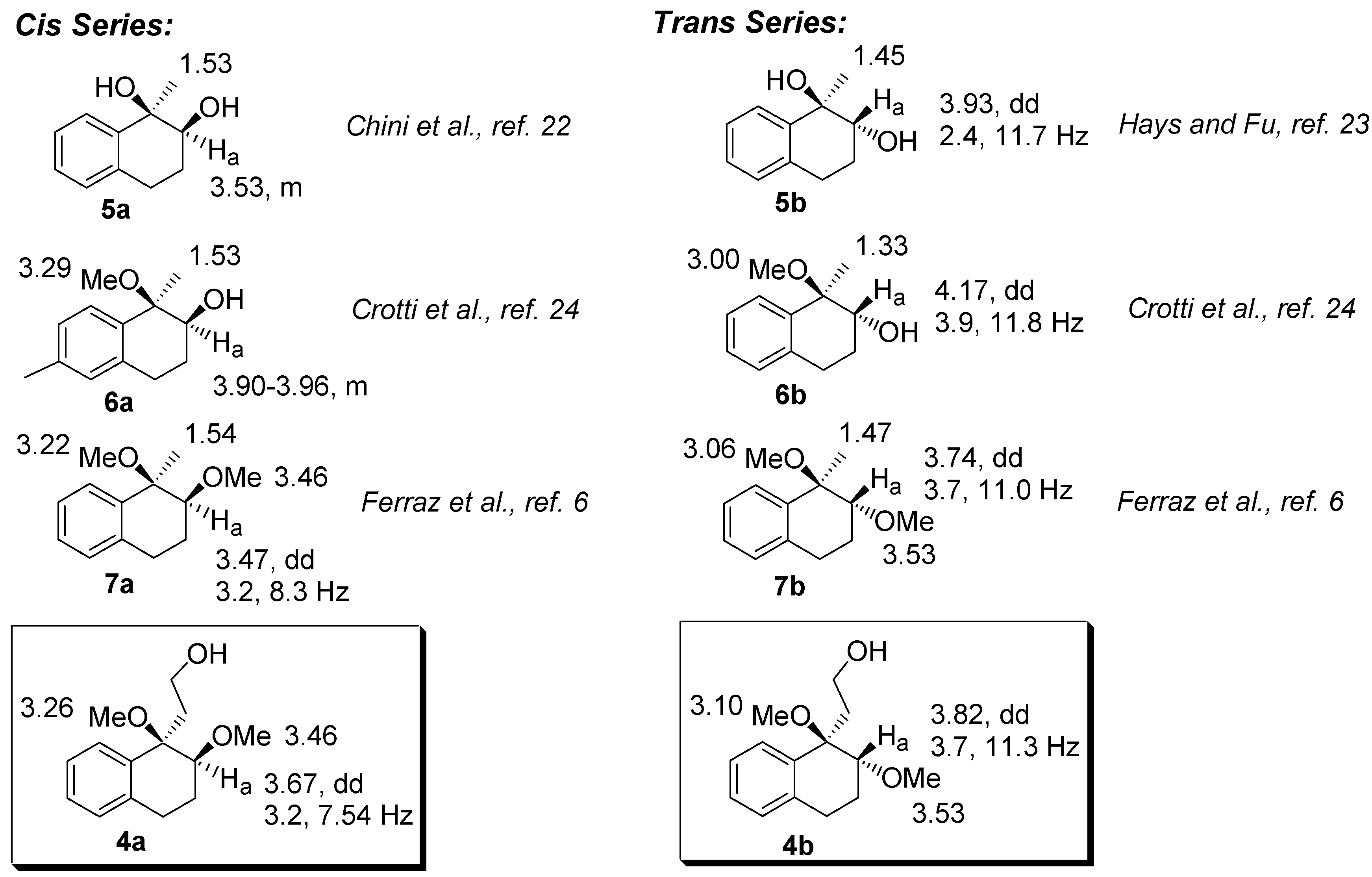
The relative configurations of
4a-b were assigned by comparison with the NMR data of analogous dihydroxy-, dimethoxy, and hydroxy-methoxy-tetrahydronaphthalenes derivatives [
6,
21,
22,
23]. Tabulating these data, we observed that the hydrogen bonded to the C2 carbon (H
a) in the
trans series is deshielded when compared to the corresponding
cis isomer. In addition, in the
trans compounds, H
a appears as a dd with coupling constants ranging from 2.4-3.9 and 11.0-11.8. Furthermore, the hydrogens of the methyl group bounded to the C1 carbon are deshielded in the
cis compounds. A mnemonic device to differentiate the
cis from the
trans diastereomer in the dimethoxylated compounds (
4 and
7, for example) could be established by noting the distance of the singlets corresponding to the hydrogens of the methoxyl group. In the
cis diastereomers, the singlets are closer than in the
trans.
Figure 2.
Selected chemical shifts of cis- and trans-dimethoxylated compounds
Figure 2.
Selected chemical shifts of cis- and trans-dimethoxylated compounds
Contrasting with the reaction in methanol, the indan
2 was obtained in 57% yield when the substrate
1 was treated with TTA in aqueous acetic acid (
Table 2, Entry 1). This value is lower than that obtained in the reaction with TTN [
5]. After the optimization of the reaction conditions for TTA, the ring contraction of
1 with TTAP or with TTP appeared to be straightforward, considering the experiments performed with isopulegol. However, when
1 was treated with TTAP or with TTP in aqueous acetic acid, a complex mixture of compounds was obtained, in which the ring contraction product was not detected by TLC and GC analysis. So far, we cannot explain why the ring contraction of
1 with TTAP or with TTP did not take place in AcOH similarly to the reaction using TTA, whereas the cyclization of isopulegol occurs with all thallium salts in AcOH. The disappointing result obtained in the reactions with TTAP and with TTP showed that further information concerning the ring contraction of
1 with thallium(III) was required. Thus, we decided to investigate the oxidation of
1 with another commercially available thallium(III) carboxylate: thallium tris-trifluoroacetate (TTFA). The two different solvents – TFA/H
2O and CH
2Cl
2 [
11] – normally used in the reactions with TTFA were tested in the oxidation of
1. Under both conditions, the desired ring contraction product could be obtained in good yield (Entries 2 and 3). At this stage, we realized an important feature for the reaction of
1 with thallium(III):
the best solvent is an aqueous solution of the carboxylic acid corresponding to the carboxylate anion of the thallium salt (compare entries 1 and 2). Thus, the reaction of
1 was performed with TTP in a mixture of propionic acid and H
2O. Indeed, under such a condition, the indan
2 was obtained (entry 4). Similarly, treatment of
1 with TTAP in aqueous
(S)-(-)-2-acetoxypropionic acid gave the ring contraction product
2 (entry 5). CG analysis of this product using chiral column was then performed. Unfortunately, no chiral induction was observed in the formation of
2 promoted by the chiral non-racemic TTAP. Finally, two other reaction conditions were tested to promote the rearrangement of
1 into
2 using TTAP. In CH
2Cl
2 the substrate was recovered without the formation of any
2, whereas in
(S)-(-)-2-acetoxypropionic acid a complex mixture was obtained.
Table 2.
Ring Contraction of
1 promoted by Thallium(III)
![Molecules 10 01419 i002]()
Table 2.
Ring Contraction of 1 promoted by Thallium(III) ![Molecules 10 01419 i002]()
| Entry | Conditions | Yield |
|---|
| 1 | TTA, AcOH/H2O (2:1), 4 h, rt | 57% |
| 2 | TTFA, TFA/H2O (2:1), 2 h, rt | 67% |
| 3 | TTFA, CH2Cl2, 2 h, rt | 63% |
| 4 | TTP, H3CCH2COOH/H2O (2:1), 4 h, rt | 59% |
| 5 | TTAP, (S)-(-)-2-acetoxypropionic acid/H2O (2:1), 18 h, rt | 32% |
Analysis of the respective yields and reaction times of the reactions of the alkenol
1 with TTN (
Scheme 1), TTA (entry 1), TTFA (entry 2), TTP (entry 4), and TTAP (entry 5) allows one to conclude that the order of reactivity is as follows: TTAP < TTP ≈ TTA < TTFA < TTN, which agrees with that observed in the reaction of isopulegol (cf.
Table 1).
Experimental
General
Information concerning general experimental methods was recently published [
5]. Flash chromatography was performed using silica gel 200-400 Mesh and the indicated eluents. Technical grade isopulegol was purified by this technique (10% AcOEt in hexanes). The [α]
D measurements were performed in a JASCO Digital Polarimeter Model DIP-370. The amount of thallium(I) salt in TTAP and TTP was determined by titration [
24].
1H- and
13C-NMR spectra were recorded on Bruker spectrometers. IR spectra were measured on a Perkin-Elmer 1750-FT. GG analyses were performed on a HP-6890 series II. GC-MS analyses were performed using Finnigan-MAT INCOS 50B and GC Varian 2400. Elemental analyses were performed using Perkin-Elmer 2400 apparatus.
(S)-(-)-2-Acetoxypropionic acid
To stirred solution of (+)-lactic acid (99%, 1.27, 14.2 mmol) in anhydrous THF (2 mL), was added acetyl chloride [
25] (2.25 g, 28.2 mmol). The mixture was stirred at the room temperature for 2 h. The THF was removed under reduced pressure and the resulting oil was distilled (100-103 °C/0.7 mmHg) giving
(S)-(-)-2-acetoxypropionic acid [
25] (1.81 g, 13.7 mmol, 98%), as a colorless oil.
Thallium tris-[(S)-(-)-2-acetoxypropionic acid] (TTAP)
To a three-neck flask were added TTA (0.787 g, 1.92 mmol) and (S)-(-)-2-acetoxypropionic acid (3.0 eq., 0.77 g, 5.8 mmol) under an inert atmosphere. After the addition, the mixture was stirred under reduced pressure (0.8 mmHg). A few minutes later, the temperature was increased to 60 ºC for 3 h. A pale yellow high viscous mixture was obtained which became a white solid after trituration with Et2O. The solid was filtered giving TTAP (0.784 g, 1.31 mmol, 67%) as a white flocculent solid. mp 60.6-61.9 ºC; [α]D25 -70.6 º (c 3.5, CHCl3). TTAP is unstable, which precluded obtaining a satisfactory elemental analysis. 1H-NMR (200 MHz, CDCl3) δ: 1.52 (d, J = 7.0 Hz, 3H), 2.11 (s, 3H), 5.11 (q, J = 7.0 Hz, 1H); 13C-NMR (50 MHz, CDCl3) δ: 17.6, 20.9, 70.5, 171.3, 177.1; IR (KBr) cm-1: 2990, 1722, 1594, 1408, 1374, 1260, 1094.
Thallium tripropionate (TTP)
To a three-neck flask were added TTA (3.67 g, 9.01 mmol) and propionic acid (3 eq., 2.01 g, 27.0 mmol) under inert atmosphere. The mixture was stirred for 12 h at room pressure and temperature. Then, the mixture was stirred under reduced pressure (0.8 mmHg) for 10 minutes at room temperature and the temperature was then increased to 60 ºC and stirring continued for an additional 3 h under reduced pressure (0.8 mmHg). A pale yellow high viscous liquid with a suspended white solid was obtained which became a white solid after trituration with Et2O. The ether was evaporated giving TTP (3.78 g, 26.4 mmol, 98%), as a white solid. mp 111.5-112.1 ºC. 1H-NMR (200 MHz, CDCl3) δ: 1.18 (d, J = 7.4 Hz, 3H), 2.43 (q, J = 7.4 Hz, 2H); 13C-NMR (50 MHz, CDCl3) δ: 10.1, 26.9, 182.6; IR (KBr) cm-1: 2981, 2942, 2884, 1564, 1464, 1410, 1293; Anal. Calc. for C9H15O6Tl: C, 25.33; H, 3.54. Found: C, 25.23; H, 3.49.
Reaction of isopulegol with TTAP in AcOH/H2O
The reaction was performed following the general procedure, but using isopulegol (0.108 g, 0.718 mmol), acetic acid/H
2O (2:1, 3 mL), TTA (1.1 eq., 0.44 g, 0.72 mmol) and reaction time of 4 h. The crude product was purified by flash chromatography (10-50% gradient of AcOEt in hexanes) giving the cyclic ether
3 [
18] (0.0776 g, 0.502 mmol, 69%).
Reaction of 2-(3,4-dihydronaphthalen-1-yl)-ethanol (1) with TTA in AcOH/H2O
The reaction was performed following the general procedure, but using 2-(3,4-dihydronaphthalen-1-yl)-ethanol (0.117 g, 0.718 mmol), AcOH/H
2O (2:1, 3.5 mL), TTA (1.1 eq., 0.35 g, 0.78 mmol) and a reaction time of 4 h. The crude product was purified by flash chromatography (10-50% gradient of AcOEt in hexanes) giving the indan
2 [
5] (0.0774 g, 0.410 mmol, 57%), as a colorless oil.
Reaction of 2-(3,4-dihydronaphthalen-1-yl)-ethanol (1) with TTAP in (S)-2-acetoxypropionic acid/H2O
The reaction was performed following the general procedure, but using 2-(3,4-dihydro-naphthalen-1-yl)-ethanol (0.102 g, 0.581 mmol),
(S)-2-acetoxypropionic acid/H
2O (2:1, 3 mL), TTAP (2.3 eq., 0.81 g, 1.4 mmol), and reaction time of 18 h. The crude product was purified by flash chromatography (10-50% gradient of AcOEt in hexanes) giving the indan
2 [
5] (0.0351 g, 0.180 mmol, 32%), as a colorless oil.
Reaction of 2-(3,4-dihydronaphthalen-1-yl)-ethanol (1) with TTP in propionic acid/H2O
The reaction was performed following the general procedure, but using the 2-(3,4-dihydronaphthalen-1-yl)-ethanol (0.142 g, 0.791 mmol), propionic acid/H
2O (2:1, 4 mL), TTP (1.1 eq., 0.37 g, 0.87 mmol) and reaction time of 6 h. The crude product was purified by flash chromatography (15-50% gradient of AcOEt in hexanes) giving the indan
2 [
5] (0.0976 g, 0.460 mmol, 59%), as a colorless oil.
Reaction of 2-(3,4-dihydronaphthalen-1-yl)-ethanol (1) with TTFA in trifluoroacetic acid/H2O
The reaction was performed following the general procedure, but using the 2-(3,4-dihydronaphthalen-1-yl)-ethanol (0.152 g, 0.861 mmol), TFA/H
2O (2:1, 5 mL), TTFA (1.1 eq., 0.51 g, 0.94 mmol) and reaction time of 2 h. The crude product was purified by flash chromatography (15-50% gradient AcOEt in hexanes) giving the indan
2 [
5] (0.123 g, 0.631 mmol, 67%), as a colorless oil.
Reaction of 2-(3,4-dihydronaphthalen-1-yl)-ethanol (1) with TTFA in CH2Cl2
The reaction was performed following the general procedure, but using 2-(3,4-dihydro-naphthalen-1-yl)-ethanol (0.156 g, 0.910 mmol), CH
2Cl
2 (5 mL), TTFA (1.1 eq., 0.53 g, 0.97 mmol) and a reaction time of 2 h. Instead of neutralization with NaHCO
3, the mixture was filtered through a silica gel pad (10 cm, 70-230 Mesh) using CH
2Cl
2 as eluent (100 mL). The mixture was concentrated under reduced pressure. The crude product was purified by flash chromatography (15-50% gradient of AcOEt in hexanes) giving the indan
2 [
5] (0.120 g, 0.610 mmol, 63%), as a colorless oil.
Preparation of cis- and trans-2-(1,2,3,4-tetrahydro-1,2-dimethoxynaphthalen-1-yl)-ethanol (4a-b)
The reaction was performed following the general procedure, using the 2-(3,4-dihydronaphthalen-1-yl)-ethanol (0.132 g, 0.760 mmol), MeOH (3 mL), TTA (1.2 eq., 0.372g, 0.908 mmol) and reaction time of 6 h. Instead of neutralization with NaHCO3, the mixture was filtered through a silica gel pad (10 cm, 70-230 Mesh) using CH2Cl2 as eluent (100 mL). The mixture was concentrated under reduced pressure. The crude product was purified by flash chromatography (15-50% gradient of AcOEt in hexane) affording cis-2-(1,2,3,4-tetrahydro-1,2-dimethoxynaphthalen-1-yl)ethanol (4a, 0.0626 g, 0.265 mmol, 35%), as a colorless oil: 1H-NMR (300 MHz, CDCl3) δ: 2.00-2.18 (m, 4H), 2.64-3.03 (m, 3H), 3.26 (s, 3H), 3.46 (s, 3H), 3.66-3.74 (m, 3H), 7.10-7.26 (m, 3H), 7.44-7.48 (m, 1H); 13C-NMR (75 MHz, CDCl3) δ: 22.6, 25.8, 41.0, 52.7, 56.4, 59.2, 79.3, 81.2, 125.7, 127.3, 127.4, 128.8, 136.6, 137.0; IR (Film) cm-1: 3520, 3070, 2940, 1463, 1427, 1104, 1073 and trans-2-(1,2,3,4-tetrahydro-1,2-dimethoxynaphthalen-1-yl)ethanol (4b, 0.0400 g, 0.170 mmol, 22%), also as a colorless oil: 1H-NMR (500 MHz, CDCl3) δ: 1.88-1.97 (m, 2H), 2.22-2.37 (m, 2H), 2.85-2.97 (m, 2H), 3.10 (s, 3H), 3.53 (s, 4H), 3.67 (m, 1H), 3.80-3.90 (m, 2H), 7.09-7.43 (m, 4H); 13C-NMR (75 MHz, CDCl3) δ: 22.9, 26.1, 41.3, 53.0, 56.8, 59.5, 79.7, 81.6, 126.1, 127.6, 127.7, 129.1, 136.9, 137.3; GC-MS (m/z): 236 (M+ , 1), 119 (100); Anal. Calc. for C9H15O6: C, 71.16; H, 8.53. Found: C, 71.40; H, 8.45.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}