
Alpha-Linolenic Acid Impedes Cadmium-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration in Mouse Brain

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Drug Treatments
- Control mice treated with saline as a vehicle for 2 weeks (IP).
- Mice treated with Cd chloride 5 mg/kg, an alternative day for 2 weeks (IP).
- Mice treated with Cd chloride 5 mg/kg/day and (ALA, 60 mg/kg/body weight/day/p.o) for 6 weeks.
2.2. Protein Extraction
2.3. Measurement of ROS Production
2.4. Western Blot Analysis
2.5. Antibodies
2.6. Brain Tissue Collection and Sample Preparation
2.7. Immunofluorescence Staining
2.8. Statistical Analysis
2.9. Cresyl Violet (Nissl) Staining
2.10. Molecular Docking
3. Results
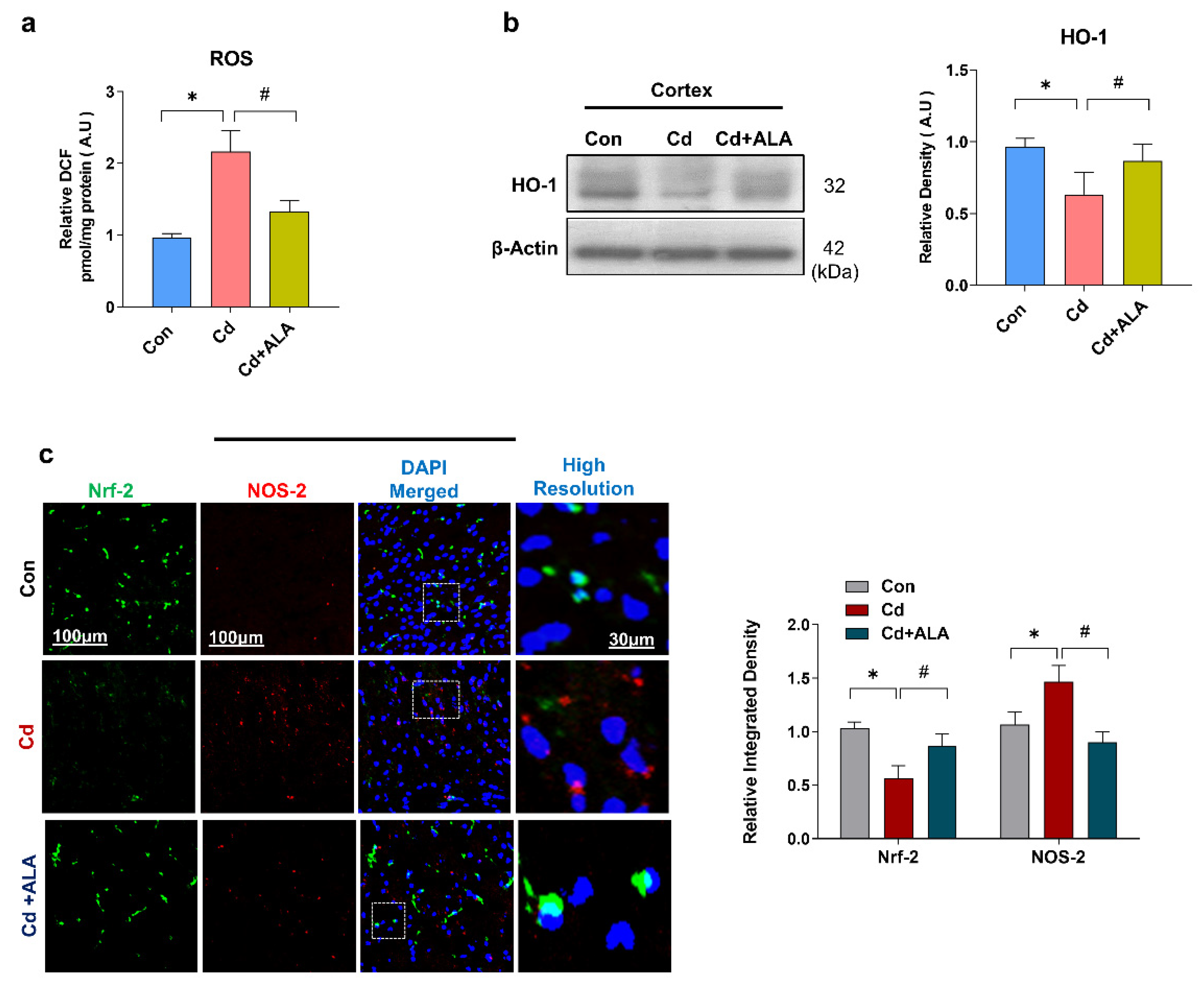
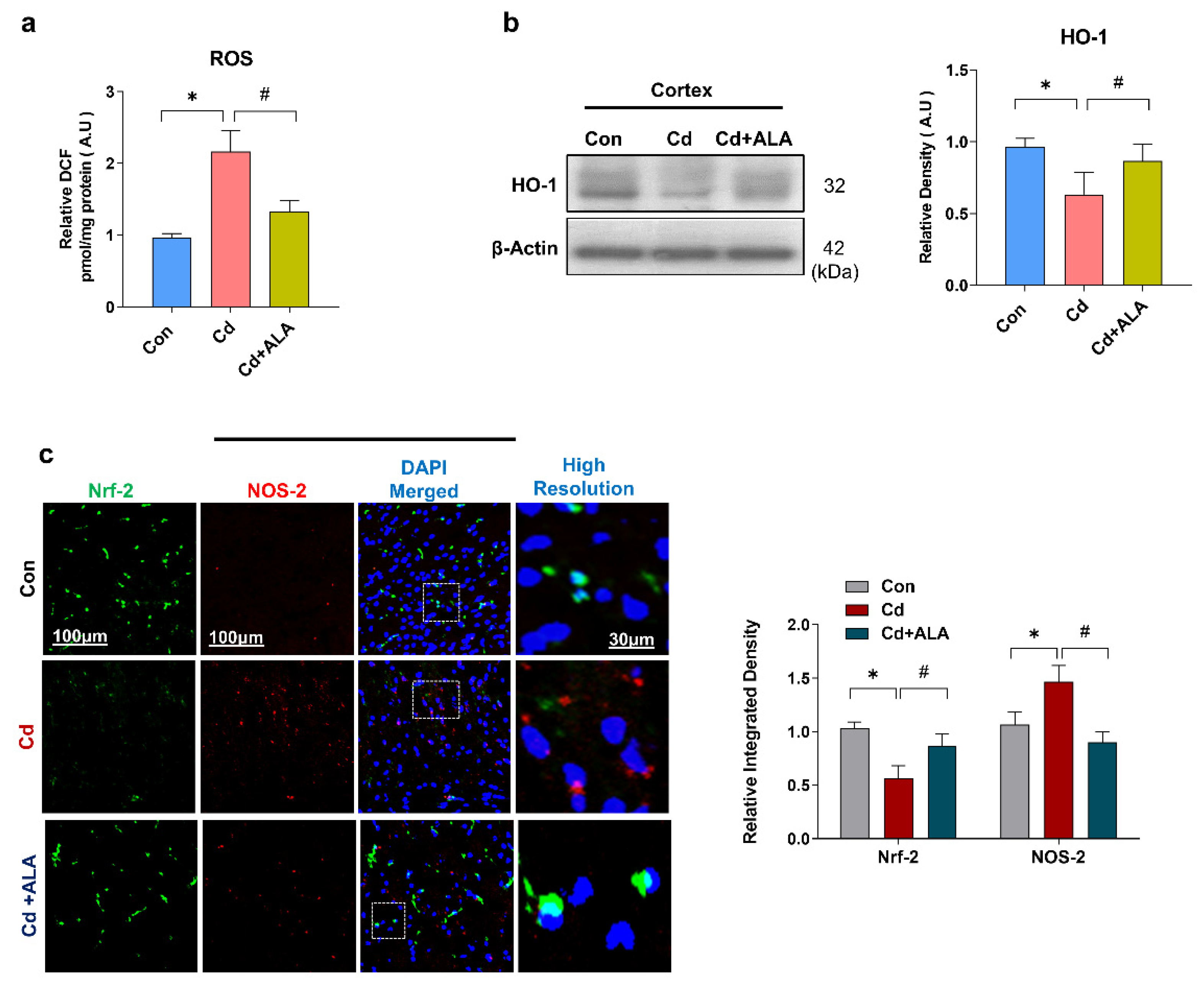
3.1. ALA Rescues Oxidative Stress by Improving Nrf2/HO-1 Signaling after Cd Administration in the Mouse Brain
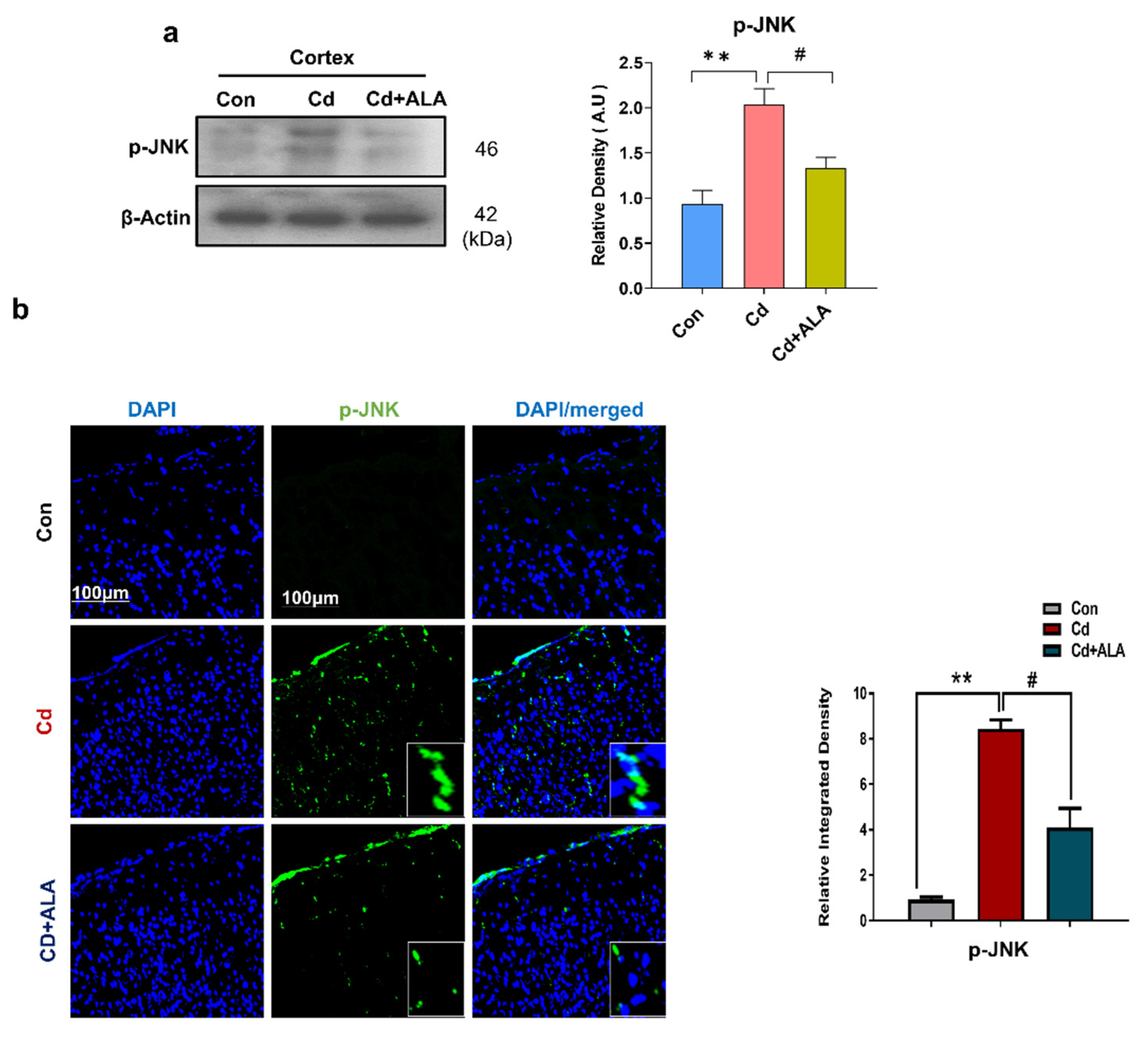
3.2. ALA Exerts Neuroprotection by Inhibiting JNK against Cd in Mouse Brain
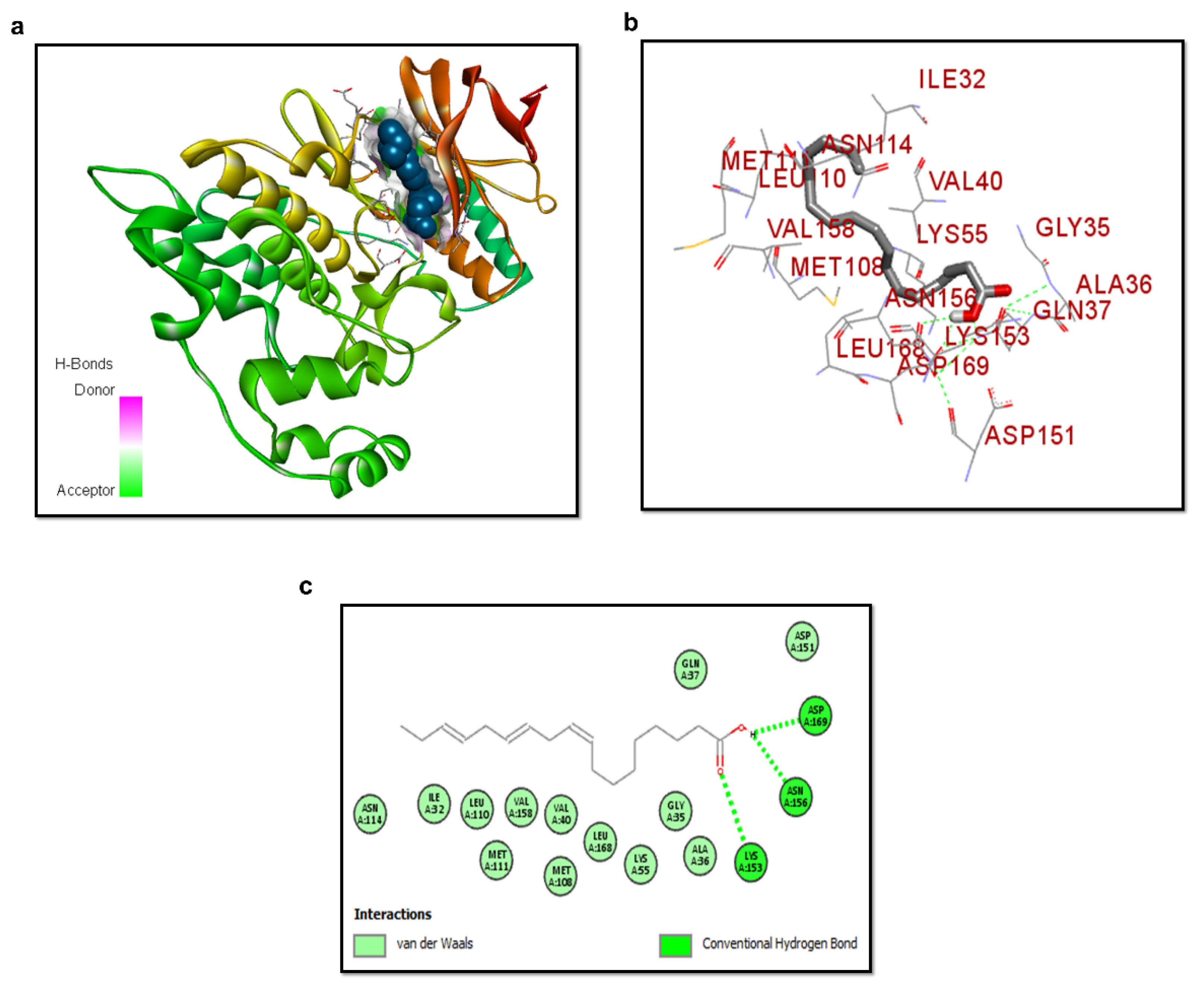
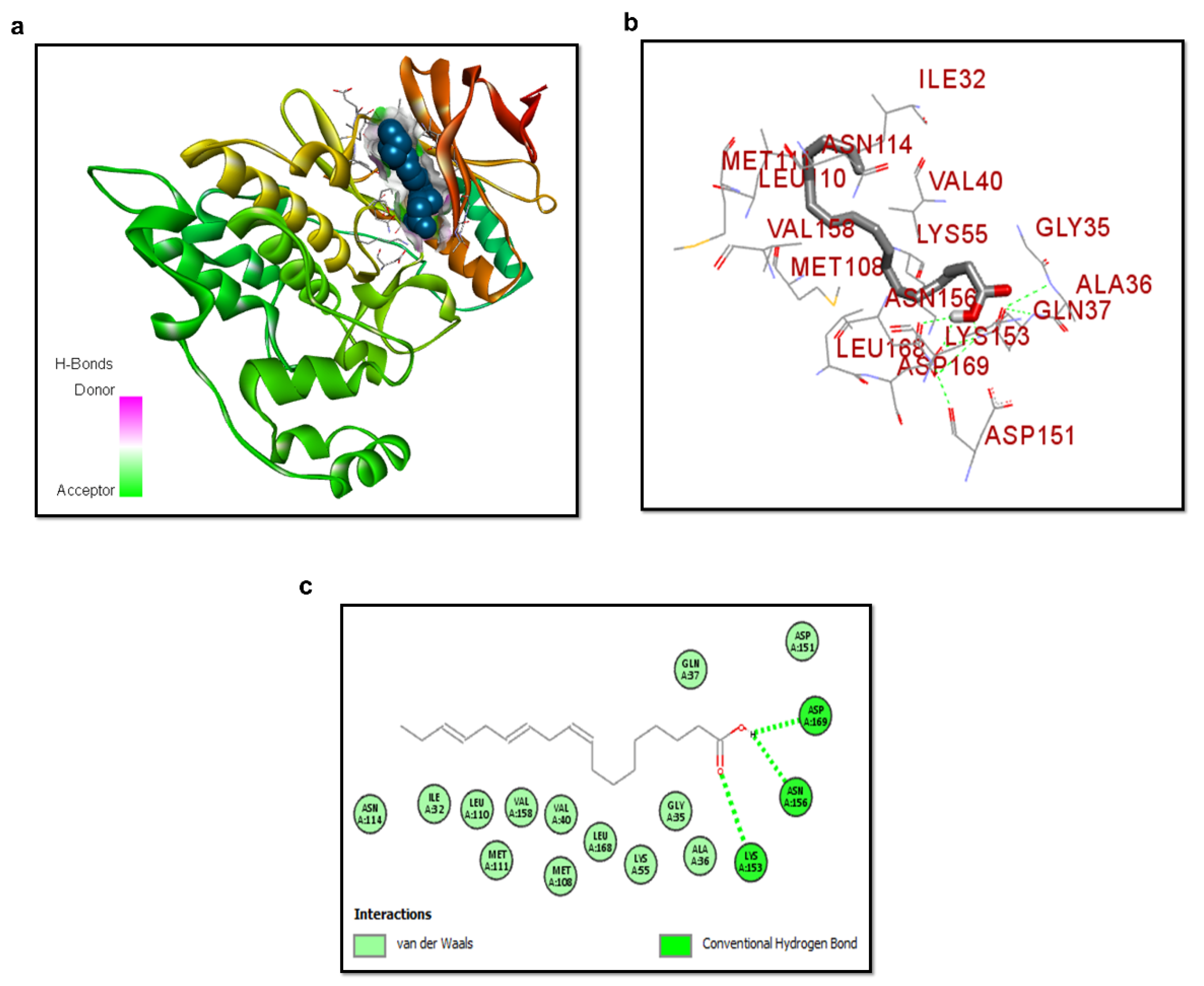
3.3. Molecular Docking of ALA with p-JNK
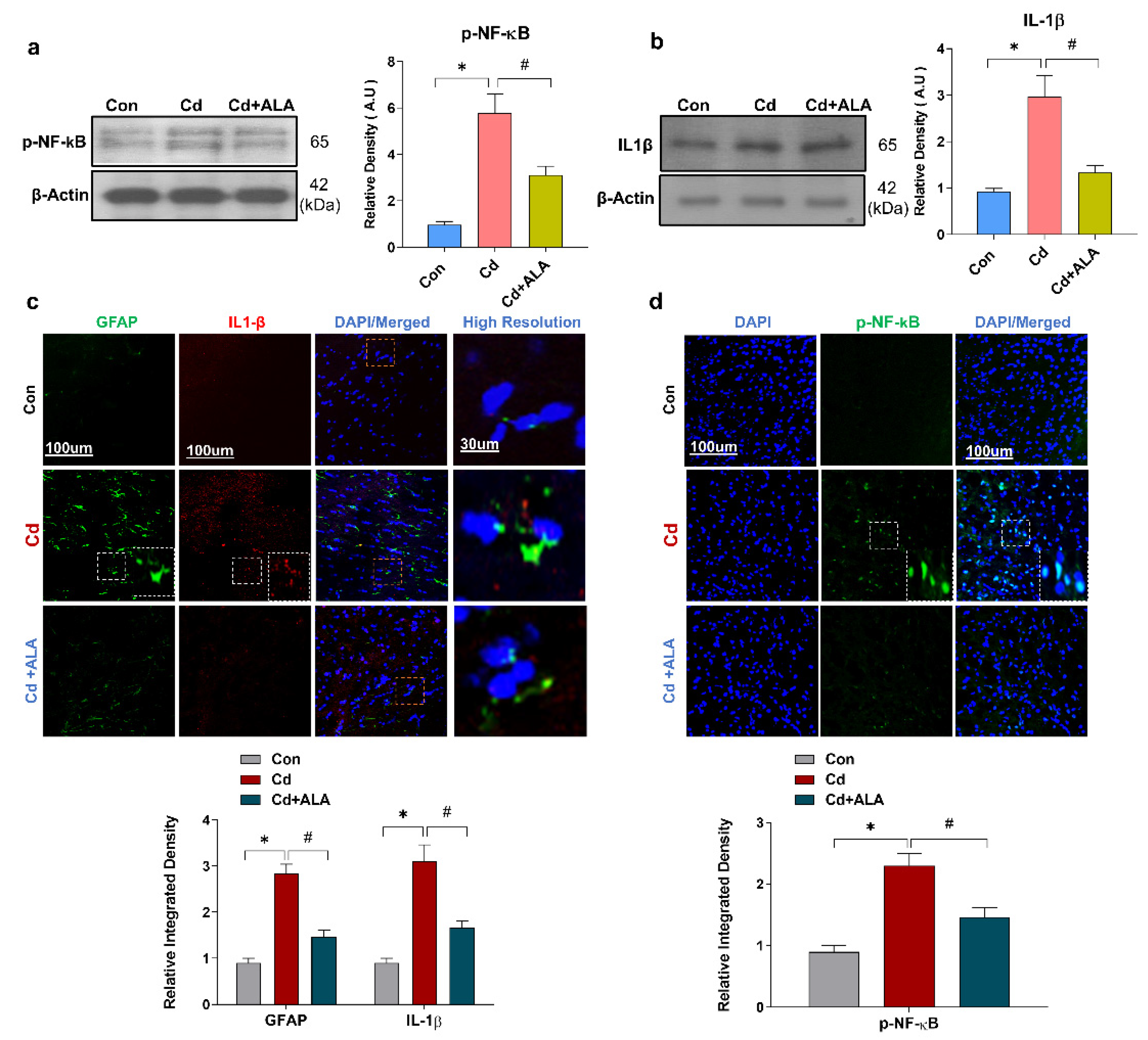
3.4. ALA Suppressed Cd-Induced Activated GFAP and Release of Inflammatory Cytokines in Mouse Brain
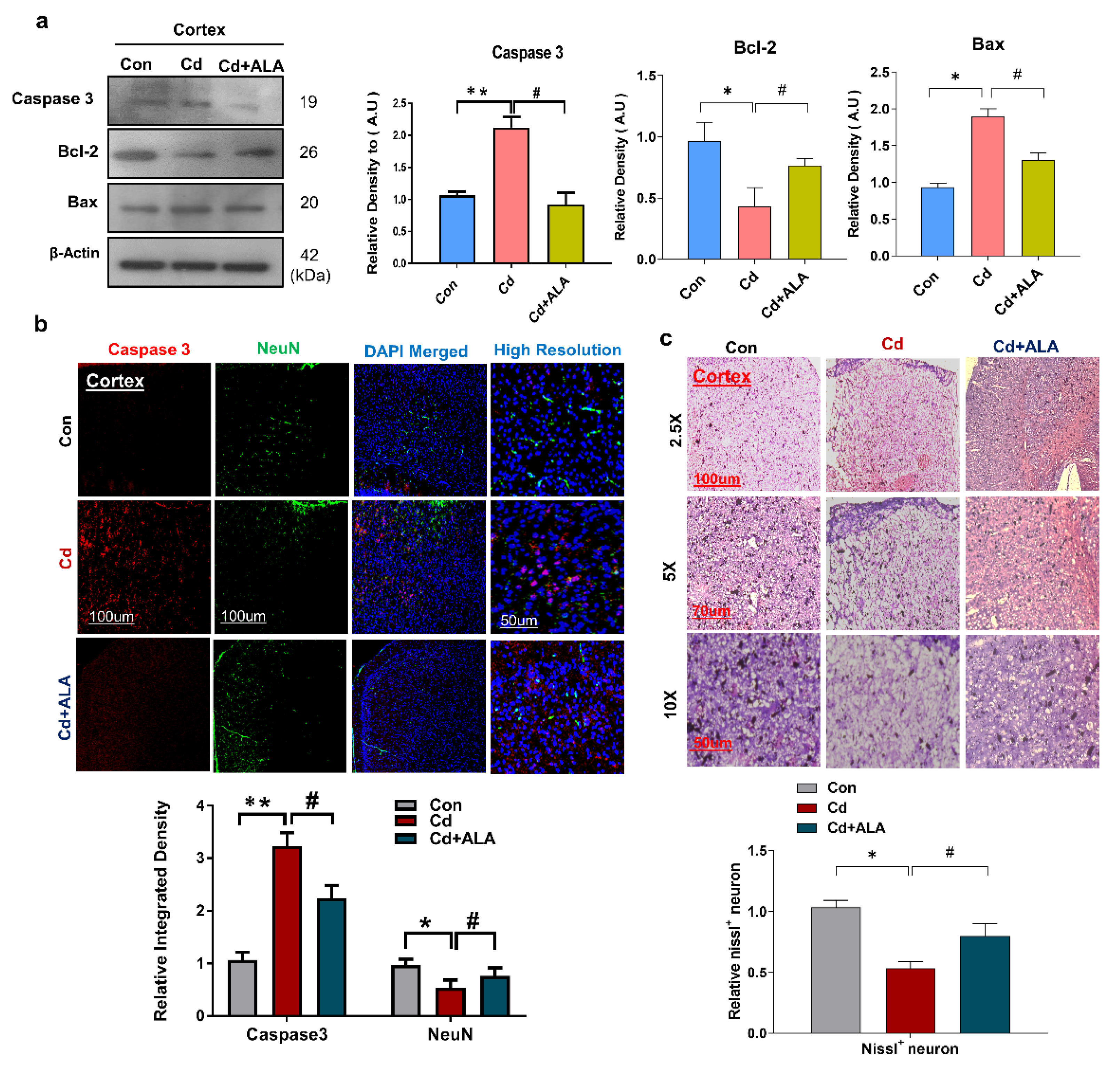
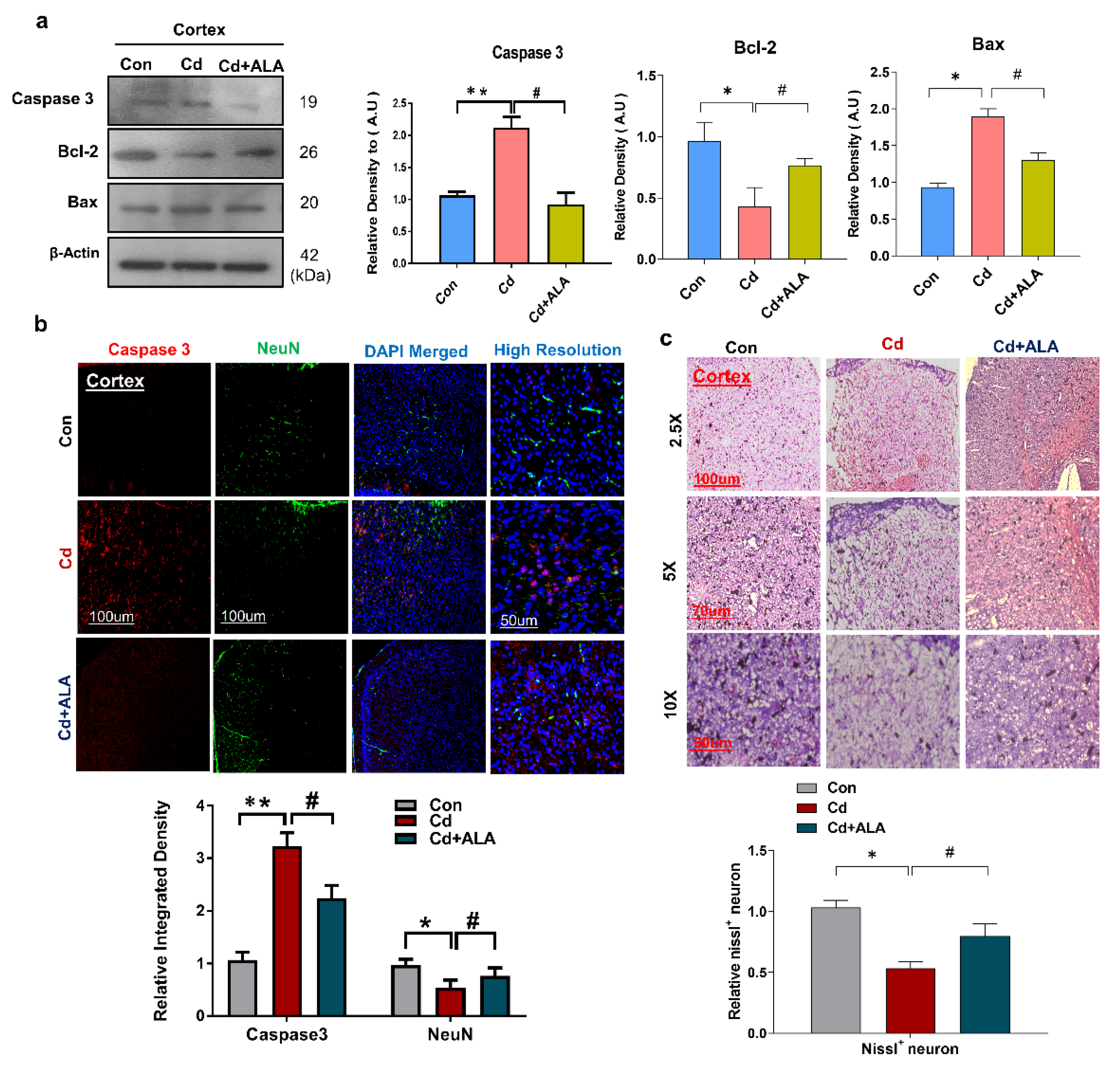
3.5. ALA Protects Cd-Induced Increase Neuronal Apoptosis and Neurodegeneration in Mouse Brain
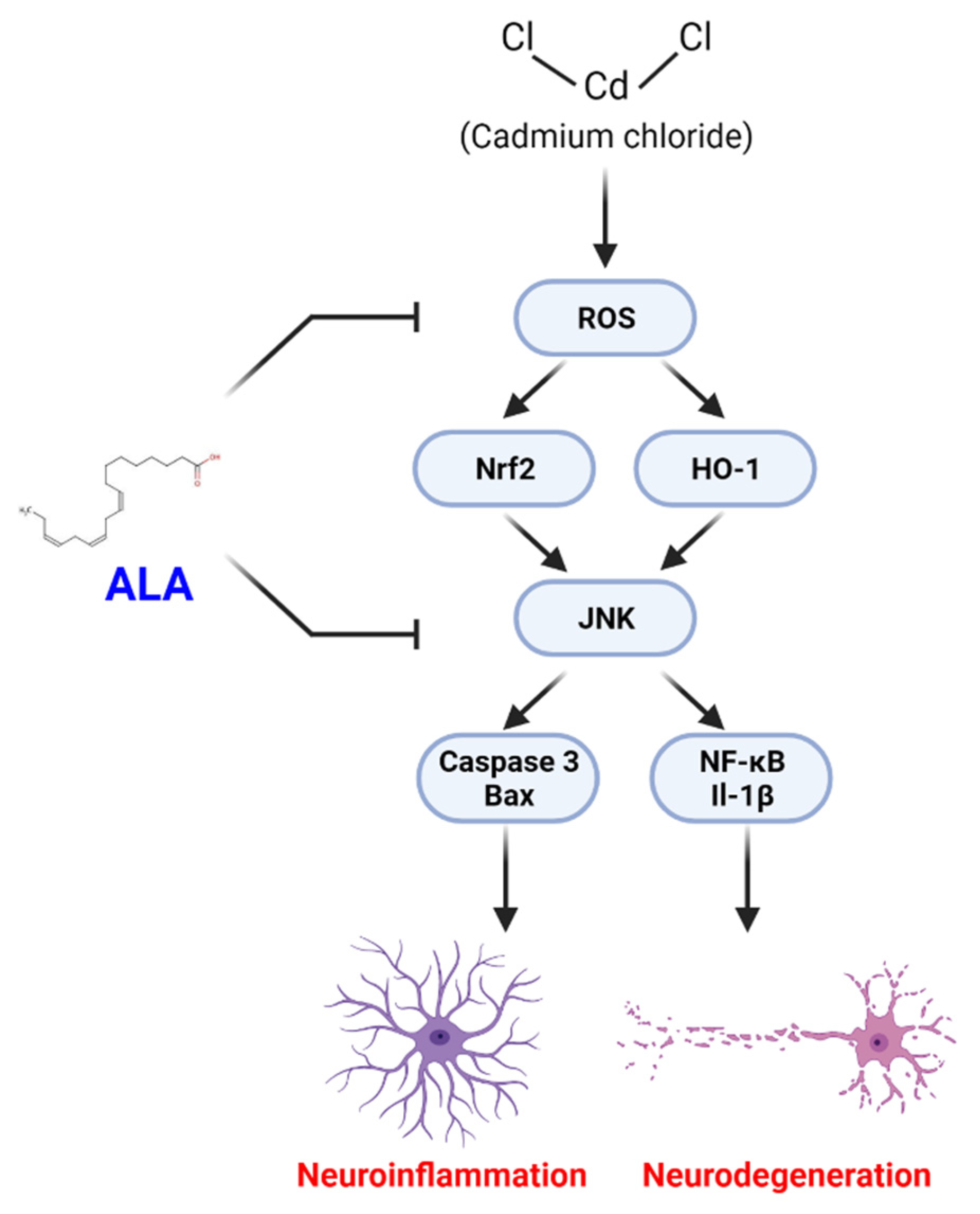
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peterson, T.C.; Hoane, M.R.; McConomy, K.S.; Farin, F.M.; Bammler, T.K.; MacDonald, J.W.; Kantor, E.D.; Anderson, G.D. A Combination Therapy of Nicotinamide and Progesterone Improves Functional Recovery following Traumatic Brain Injury. J. Neurotrauma 2015, 32, 765–779. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Ikram, M.; Muhammad, T.; Park, J.; Kim, M.O. Caffeine Modulates Cadmium-Induced Oxidative Stress, Neuroinflammation, and Cognitive Impairments by Regulating Nrf-2/HO-1 In Vivo and In Vitro. J. Clin. Med. 2019, 8, 680. [Google Scholar] [CrossRef] [Green Version]
- Godt, J.; Scheidig, F.; Grosse-Siestrup, C.; Esche, V.; Brandenburg, P.; Reich, A.; Groneberg, D.A. The toxicity of cadmium and resulting hazards for human health. J. Occup. Med. Toxicol. 2006, 1, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, P.; Faroon, O.; Pappas, R.S. Cadmium and Cadmium/Zinc Ratios and Tobacco-Related Morbidities. Int. J. Environ. Res. Public Health 2017, 14, 1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branca, J.J.V.; Morucci, G.; Pacini, A. Cadmium-induced neurotoxicity: Still much ado. Neural Regen. Res. 2018, 13, 1879–1882. [Google Scholar] [CrossRef]
- Del Pino, J.; Zeballos, G.; Anadon, M.J.; Moyano, P.; Diaz, M.J.; Garcia, J.M.; Frejo, M.T. Cadmium-induced cell death of basal forebrain cholinergic neurons mediated by muscarinic M1 receptor blockade, increase in GSK-3beta enzyme, beta-amyloid and tau protein levels. Arch. Toxicol. 2016, 90, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, Y.; Long, M.; Luo, T.; Bian, J.; Liu, X.; Gu, J.; Zou, H.; Song, R.; Wang, Y.; et al. Beclin-1-mediated Autophagy Protects Against Cadmium-activated Apoptosis via the Fas/FasL Pathway in Primary Rat Proximal Tubular Cell Culture. Sci. Rep. 2017, 7, 977. [Google Scholar] [CrossRef] [Green Version]
- Thevenod, F.; Lee, W.K. Toxicology of cadmium and its damage to mammalian organs. Met. Ions Life Sci. 2013, 11, 415–490. [Google Scholar] [CrossRef] [PubMed]
- Daniel, S.; Limson, J.L.; Dairam, A.; Watkins, G.M.; Daya, S. Through metal binding, curcumin protects against lead- and cadmium-induced lipid peroxidation in rat brain homogenates and against lead-induced tissue damage in rat brain. J. Inorg. Biochem. 2004, 98, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Pi, H.; Li, M.; Tian, L.; Yang, Z.; Yu, Z.; Zhou, Z. Enhancing lysosomal biogenesis and autophagic flux by activating the transcription factor EB protects against cadmium-induced neurotoxicity. Sci. Rep. 2017, 7, 43466. [Google Scholar] [CrossRef]
- Kwakye, G.F.; Jimenez, J.A.; Thomas, M.G.; Kingsley, B.A.; Mcilvin, M.; Saito, M.A.; Korley, E.M. Heterozygous huntingtin promotes cadmium neurotoxicity and neurodegeneration in striatal cells via altered metal transport and protein kinase C delta dependent oxidative stress and apoptosis signaling mechanisms. Neurotoxicology 2019, 70, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Abdel Moneim, A.E.; Bauomy, A.A.; Diab, M.M.; Shata, M.T.; Al-Olayan, E.M.; El-Khadragy, M.F. The protective effect of Physalis peruviana L. against cadmium-induced neurotoxicity in rats. Biol. Trace Elem. Res. 2014, 160, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Fiorillo, C.; Carrino, D.; Paternostro, F.; Taddei, N.; Gulisano, M.; Pacini, A.; Becatti, M. Cadmium-Induced Oxidative Stress: Focus on the Central Nervous System. Antioxidants 2020, 9, 492. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, P.; Shen, Q.; Wang, Q.; Liu, D.; Li, J.; Wang, L. The effects of cadmium exposure on the oxidative state and cell death in the gill of freshwater crab Sinopotamon henanense. PLoS ONE 2013, 8, e64020. [Google Scholar] [CrossRef] [Green Version]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, L.J.M.; Kuijper, I.A.; Schimming, J.P.; Wolters, L.; Braak, B.T.; Langenberg, J.P.; Noort, D.; Beltman, J.B.; van de Water, B. A systematic analysis of Nrf2 pathway activation dynamics during repeated xenobiotic exposure. Arch. Toxicol. 2019, 93, 435–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Armenta, M.; Villeda-Hernandez, J.; Barroso-Moguel, R.; Nava-Ruiz, C.; Jimenez-Capdeville, M.E.; Rios, C. Brain regional lipid peroxidation and metallothionein levels of developing rats exposed to cadmium and dexamethasone. Toxicol. Lett. 2003, 144, 151–157. [Google Scholar] [CrossRef]
- Geret, F.; Serafim, A.; Barreira, L.; Bebianno, M.J. Effect of cadmium on antioxidant enzyme activities and lipid peroxidation in the gills of the clam Ruditapes decussatus. Biomarkers Biochem. Indic. Expo. Response Susceptibility Chem. 2002, 7, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Whelan, J.; Fritsche, K. Linoleic acid. Adv. Nutr. 2013, 4, 311–312. [Google Scholar] [CrossRef]
- Castellano, J.M.; Garcia-Rodriguez, S.; Espinosa, J.M.; Millan-Linares, M.C.; Rada, M.; Perona, J.S. Oleanolic Acid Exerts a Neuroprotective Effect Against Microglial Cell Activation by Modulating Cytokine Release and Antioxidant Defense Systems. Biomolecules 2019, 9, 683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, U.N. COX-2 inhibitors and metabolism of essential fatty acids. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2005, 11, RA233–RA237. [Google Scholar]
- Ali, Y.M.; Kadir, A.A.; Ahmad, Z.; Yaakub, H.; Zakaria, Z.A.; Abdullah, M.N. Free radical scavenging activity of conjugated linoleic acid as single or mixed isomers. Pharm. Biol. 2012, 50, 712–719. [Google Scholar] [CrossRef] [Green Version]
- Simopoulos, A.P. Omega-3 fatty acids and antioxidants in edible wild plants. Biol. Res. 2004, 37, 263–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, S.I.; Chang, M.C.; Spector, A.A. Delivery and turnover of plasma-derived essential PUFAs in mammalian brain. J. Lipid Res. 2001, 42, 678–685. [Google Scholar] [CrossRef]
- Hegde, M.V.; Zanwar, A.A.; Adekar, S.P. Omega-3 Fatty Acids: Keys to Nutritional Health. In Omega-3 Fatty Acids; Springer International: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- Ali, W.; Ikram, M.; Park, H.Y.; Jo, M.G.; Ullah, R.; Ahmad, S.; Abid, N.B.; Kim, M.O. Oral Administration of Alpha Linoleic Acid Rescues Abeta-Induced Glia-Mediated Neuroinflammation and Cognitive Dysfunction in C57BL/6N Mice. Cells 2020, 9, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhammad, T.; Ali, T.; Ikram, M.; Khan, A.; Alam, S.I.; Kim, M.O. Melatonin Rescue Oxidative Stress-Mediated Neuroinflammation/ Neurodegeneration and Memory Impairment in Scopolamine-Induced Amnesia Mice Model. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2019, 14, 278–294. [Google Scholar] [CrossRef]
- Shah, S.A.; Yoon, G.H.; Chung, S.S.; Abid, M.N.; Kim, T.H.; Lee, H.Y.; Kim, M.O. Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuropathological deficits. Mol. Psychiatry 2017, 22, 407–416. [Google Scholar] [CrossRef]
- Ali, T.; Rehman, S.U.; Khan, A.; Badshah, H.; Abid, N.B.; Kim, M.W.; Jo, M.H.; Chung, S.S.; Lee, H.G.; Rutten, B.P.F.; et al. Adiponectin-mimetic novel nonapeptide rescues aberrant neuronal metabolic-associated memory deficits in Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 23. [Google Scholar] [CrossRef]
- Alam, S.I.; Jo, M.G.; Park, T.J.; Ullah, R.; Ahmad, S.; Rehman, S.U.; Kim, M.O. Quinpirole-Mediated Regulation of Dopamine D2 Receptors Inhibits Glial Cell-Induced Neuroinflammation in Cortex and Striatum after Brain Injury. Biomedicines 2021, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Ali, T.; Kim, M.W.; Jo, M.H.; Chung, J.I.; Kim, M.O. Anthocyanins Improve Hippocampus-Dependent Memory Function and Prevent Neurodegeneration via JNK/Akt/GSK3beta Signaling in LPS-Treated Adult Mice. Mol. Neurobiol. 2019, 56, 671–687. [Google Scholar] [CrossRef]
- Zulfiqar, Z.; Shah, F.A.; Shafique, S.; Alattar, A.; Ali, T.; Alvi, A.M.; Rashid, S.; Li, S. Repurposing FDA Approved Drugs as JNK3 Inhibitor for Prevention of Neuroinflammation Induced by MCAO in Rats. J. Inflamm. Res. 2020, 13, 1185–1205. [Google Scholar] [CrossRef]
- Al Kury, L.T.; Zeb, A.; Abidin, Z.U.; Irshad, N.; Malik, I.; Alvi, A.M.; Khalil, A.A.K.; Ahmad, S.; Faheem, M.; Khan, A.U.; et al. Neuroprotective effects of melatonin and celecoxib against ethanol-induced neurodegeneration: A computational and pharmacological approach. Drug Des. Dev. Ther. 2019, 13, 2715–2727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Kury, L.T.; Dayyan, F.; Ali Shah, F.; Malik, Z.; Khalil, A.A.K.; Alattar, A.; Alshaman, R.; Ali, A.; Khan, Z. Ginkgo biloba Extract Protects against Methotrexate-Induced Hepatotoxicity: A Computational and Pharmacological Approach. Molecules 2020, 25, 2540. [Google Scholar] [CrossRef] [PubMed]
- Campese, V.M.; Ye, S.; Zhong, H.; Yanamadala, V.; Ye, Z.; Chiu, J. Reactive oxygen species stimulate central and peripheral sympathetic nervous system activity. Am. J. Physiology. Heart Circ. Physiol. 2004, 287, H695–H703. [Google Scholar] [CrossRef] [PubMed]
- Badshah, H.; Ikram, M.; Ali, W.; Ahmad, S.; Hahm, J.R.; Kim, M.O. Caffeine May Abrogate LPS-Induced Oxidative Stress and Neuroinflammation by Regulating Nrf2/TLR4 in Adult Mouse Brains. Biomolecules 2019, 9, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.W.; Wang, Y.; Wang, T.; Zhang, K.B.; Yuan, Y.; Bian, J.C.; Liu, X.Z.; Gu, J.H.; Zhu, J.Q.; Liu, Z.P. Cadmium-induced autophagy is mediated by oxidative signaling in PC-12 cells and is associated with cytoprotection. Mol. Med. Rep. 2015, 12, 4448–4454. [Google Scholar] [CrossRef] [PubMed]
- Savage, M.J.; Lin, Y.G.; Ciallella, J.R.; Flood, D.G.; Scott, R.W. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer’s disease model is associated with amyloid deposition. J. Neurosci. 2002, 22, 3376–3385. [Google Scholar] [CrossRef] [Green Version]
- Hunot, S.; Vila, M.; Teismann, P.; Davis, R.J.; Hirsch, E.C.; Przedborski, S.; Rakic, P.; Flavell, R.A. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 665–670. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xu, Y.; Cheng, Z.; Su, H.; Liu, X.; Xu, D.; Kapron, C.; Liu, J. Low-dose cadmium activates the JNK signaling pathway in human renal podocytes. Int. J. Mol. Med. 2018, 41, 2359–2365. [Google Scholar] [CrossRef] [Green Version]
- Joardar, S.; Dewanjee, S.; Bhowmick, S.; Dua, T.K.; Das, S.; Saha, A.; De Feo, V. Rosmarinic Acid Attenuates Cadmium-Induced Nephrotoxicity via Inhibition of Oxidative Stress, Apoptosis, Inflammation and Fibrosis. Int. J. Mol. Sci. 2019, 20, 2027. [Google Scholar] [CrossRef] [Green Version]
- Unsal, C.; Kanter, M.; Aktas, C.; Erboga, M. Role of quercetin in cadmium-induced oxidative stress, neuronal damage, and apoptosis in rats. Toxicol. Ind. Health 2015, 31, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Thevenod, F. Cadmium and cellular signaling cascades: To be or not to be? Toxicol. Appl. Pharmacol. 2009, 238, 221–239. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Du, Y. Cadmium and its neurotoxic effects. Oxidative Med. Cell. Longev. 2013, 2013, 898034. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.R.; Degheselle, O.; Smeets, K.; Van Kerkhove, E.; Cuypers, A. Cadmium-Induced Pathologies: Where Is the Oxidative Balance Lost (or Not)? Int. J. Mol. Sci. 2013, 14, 6116–6143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petri, S.; Korner, S.; Kiaei, M. Nrf2/ARE Signaling Pathway: Key Mediator in Oxidative Stress and Potential Therapeutic Target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemmiche, S.; Chabane-Sari, D.; Kadri, M.; Guiraud, P. Cadmium-induced apoptosis in the BJAB human B cell line: Involvement of PKC/ERK1/2/JNK signaling pathways in HO-1 expression. Toxicology 2012, 300, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, X.; Gu, C.; Zhang, H.; Zhang, R.; Dong, X.; Liu, C.; Hu, X.; Ji, X.; Huang, S.; et al. Celastrol ameliorates Cd-induced neuronal apoptosis by targeting NOX2-derived ROS-dependent PP5-JNK signaling pathway. J. Neurochem. 2017, 141, 48–62. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, S.-I.; Kim, M.-W.; Shah, F.A.; Saeed, K.; Ullah, R.; Kim, M.-O. Alpha-Linolenic Acid Impedes Cadmium-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration in Mouse Brain. Cells 2021, 10, 2274. https://doi.org/10.3390/cells10092274
Alam S-I, Kim M-W, Shah FA, Saeed K, Ullah R, Kim M-O. Alpha-Linolenic Acid Impedes Cadmium-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration in Mouse Brain. Cells. 2021; 10(9):2274. https://doi.org/10.3390/cells10092274
Chicago/Turabian StyleAlam, Sayed-Ibrar, Min-Woo Kim, Fawad Ali Shah, Kamran Saeed, Rahat Ullah, and Myeong-Ok Kim. 2021. "Alpha-Linolenic Acid Impedes Cadmium-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration in Mouse Brain" Cells 10, no. 9: 2274. https://doi.org/10.3390/cells10092274