
Hypoxia and the Receptor for Advanced Glycation End Products (RAGE) Signaling in Cancer

Department of Pharmaceutical Sciences, North Dakota State University, Fargo, ND 58105, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(15), 8153; https://doi.org/10.3390/ijms22158153
Submission received: 21 May 2021
/
Revised: 25 July 2021
/
Accepted: 26 July 2021
/
Published: 29 July 2021
(This article belongs to the Special Issue Hypoxia Signaling in Human Diseases)
Abstract
:Hypoxia is characterized by an inadequate supply of oxygen to tissues, and hypoxic regions are commonly found in solid tumors. The cellular response to hypoxic conditions is mediated through the activation of hypoxia-inducible factors (HIFs) that control the expression of a large number of target genes. Recent studies have shown that the receptor for advanced glycation end products (RAGE) participates in hypoxia-dependent cellular adaptation. We review recent evidence on the role of RAGE signaling in tumor biology under hypoxic conditions.
1. Physiological and Pathological Hypoxia
Hypoxia is a condition characterized by low levels of oxygen in tissues and organs. Two main types of hypoxia have been described: physiological and pathological hypoxia [1]. Physiological hypoxia is observed when oxygen levels are slightly lower than typical under healthy conditions and when these levels can be easily reversed by homeostatic mechanisms [2]. For instance, in peripheral tissues, where oxygen levels vary between about 3% to 7%, physiological hypoxia is observed at oxygen levels between 2% and 6% [1]. On the other hand, pathological hypoxia is characterized by oxygen levels significantly lower than those in healthy tissues and can reach levels of less than 1% oxygen saturation [1]. Pathological hypoxia develops because homeostatic mechanisms are not able to restore the supply of the tissue with oxygen to its physiological level [3]. Pathological hypoxia is observed in many solid tumors and has been shown to contribute to tumor growth, metastasis, and chemoresistance [1,4]. On the cellular level, hypoxia is sensed by transcription factors named hypoxia-inducible factors (HIFs). HIFs regulate the transcription of hundreds of genes, some of them critically involved in cancer processes contributing to tumorigenesis, chemoresistance, and metastasis [5,6].
2. Hypoxia-Inducible Factors: Structure and Mechanisms
The first HIF was discovered in 1991 by Semenza et al. as a protein interacting with a DNA response element present on the erythropoietin gene [7]. Since its initial discovery, two additional hypoxia-responsive members of the HIF family have been identified. Collectively, the HIFs regulate the transcription of a large number of genes involved in many aspects of cancer, such as metabolic reprogramming, cell proliferation and invasion, metastasis, and chemoresistance [5,8,9]. The functions of the three HIFs in the cellular response to hypoxia are complementary yet distinct [10,11].
2.1. HIF Subunits
HIFs function as heterodimers consisting of two subunits: an oxygen-sensitive α-subunit and an oxygen-independent β-subunit. The genes encoding the HIF-1α, HIF-2α (approved gene symbol EPAS1, endothelial PAS domain protein 1), and HIF-3α are located on chromosomes 14, 2, and 19, respectively. Two HIF β-subunits have been described and are known as the aryl hydrocarbon nuclear translocator ARNT1 (HIF-1β) and ARNT2 (HIF-2β) [12].
All three HIF-α subunits can form heterodimers with HIF-1β, bind to hypoxia response elements (HREs) of HIF-1α target genes, and modulate their expressions, resulting in cellular adaptation to hypoxia [5,7,13,14]. HIF-α forms are differentially expressed in tissues. At the RNA level, HIF-1α is found expressed in most tissues, whereas the expression of HIF-2α is limited mostly to vascular tissues (lung, heart, placenta, and kidney) [15,16,17]. HIF-3a mRNAs have been detected in adult thymus, lung, heart, and kidney [18,19,20]. Splice variants have been reported for HIF-1α and, in particular, for HIF-3α [21]. The expression of HIF-3a splice variants appears to be tissue-specific, with different variants being expressed at different levels in distinct tissues [22]. Interestingly, some splice variants of HIF-3α appear capable of binding HIF-1α and of suppressing HIF-1α activity [23].
2.2. Domain Organization
The HIF-1α and HIF-2α paralogs share an identical domain architecture and 48% sequence similarity [5,15,18]. HIF-3α differs from the other two HIF-αs in the C-terminal domain organization and is about 200 amino acids shorter in length [16,24] (Figure 1).
Structurally, all three HIF-α and the HIF-1β subunits contain a basic helix-loop-helix (bHLH) and a twin Per-ARNT-Sim (PAS) domain in the N-terminal part of the protein. These domains are responsible for heterodimerization between the α and β subunits (Figure 1) [25].
All three HIF-α forms are oxygen-sensitive due to the presence of an oxygen-dependent degradation (ODD) domain [26]. The ODD domains are defined through functional studies rather than by a conserved three-dimensional protein fold. In addition, HIF-1α and HIF-2α subunits contain two functional transactivation domains: the N-terminal transactivation domain (N-TAD) and the C-terminal transactivation domain (C-TAD). Functional studies have identified some sequence overlap between the ODD and N-TAD regions [27].
In association with transcriptional coactivators such as the coactivator binding protein (CBP) and the p300 protein, C-TAD controls the transcription of HIF-1α and HIF-2α target genes while N-TAD protects these α subunits against oxygen-dependent degradation [24,28]. In contrast, HIF-3α contains a leucine zipper (LZIP) domain instead of the C-TAD domain [12,29]. The LZIP domain participates in protein-protein interactions [30]. HIF-1β lacks both the ODD and N-TAD domains but possesses a C-TAD domain (Figure 1) [30].
2.3. Regulation Mechanisms
The HIFs themselves cannot sense oxygen directly and depend on cellular enzymes to introduce posttranslational modifications into HIF-α in an oxygen-dependent manner. Two posttranslational mechanisms of HIF-α modifications regulate the proteolytic degradation of HIF-α. The HIF-β subunits are not responsive to cellular oxygen concentrations.
The first mechanism is the oxygen-dependent hydroxylation of specific proline residues in the ODD region and adjacent to the N-TAD domain of HIF-α subunits. In HIF-1α and HIF-2a, two conserved proline residues (P402 and P564, and P405 and P531, respectively) are hydroxylated by proline-hydroxylases (PHDs) under physiological oxygen levels. These hydroxylated proline residues are recognized by the von Hippel-Lindau protein (pVHL), which then forms a larger complex with additional proteins such as elongin C, elongin B, cullin-2, and Rbx1, resulting in ubiquitination and subsequent proteosomal degradation of HIF-α [31,32,33]. In HIF-3a, only hydroxylation of P492 is necessary for recognition by pVHL [31]. Under hypoxic conditions, PHDs lose their activity and cannot hydroxylate proline residues, resulting in the stabilization of HIF-α subunits [12,33]. Subsequently, the HIF-α subunits translocate to the nucleus and heterodimerize with HIF-1β. The heterodimers then bind to HREs of target genes, resulting in the activation of a variety of signaling pathways [34]. Because the HIF-1β subunit does not contain an ODD domain, HIF-1β expression levels are not dependent on the presence of oxygen [5]. The interaction between HIF-1α and pVHL has also been shown to be enhanced following acetylation of Lysine residue 532 present in the ODD domain of HIF-1α through the activation of an oxygen-independent acetyltransferase (arrest-defective-1 (ARD1)) [35]. Besides being degraded through the pVHL-dependent pathway, HIF-1α can be ubiquitinated and later degraded through the activation of other pathways, including the oncogenic E3 ubiquitin ligase murine double minute 2 (MDM2) and the p53 protein [36].
The second main mechanism of oxygen-dependent regulation of HIF-1α activity involves the hydroxylation of a specific asparagine residue in the C-terminal TAD domain. Under normoxic conditions, hydroxylation of asparagine 803 in the C-TAD domain of HIF-1α by an asparaginyl hydroxylase enzyme named factor-inhibiting HIF1 (FIH) prevents the interaction between HIF-1α with transcriptional coactivators [37]. In order to be fully active, HIF-1α needs to form complexes through the N-TAD and C-TAD domains with transcriptional coactivators such as CREB binding proteins (CBP) and p300, SRC-1, and TIF2 [38,39,40,41]. At the physiological oxygen level, hydroxylation of Asp 803 prevents assembly with coactivators. Under hypoxic conditions, hydroxylation of asparagine 803 does not occur, thus resulting in the formation of active transcriptional complexes between the C-TAD of HIF-1α and the transcriptional coactivators [42,43].
HIF-1α can also be regulated through oxygen-independent mechanisms. One mechanism involves the interaction between HIF-1α and the hypoxia-associated factor (HAF), a specific E3 ubiquitin ligase, resulting in oxygen-independent proteosomal degradation of HIF-1α [44]. Interestingly, the interaction of HAF with HIF-2α does not result in the same outcome: HIF-2α does not become ubiquitinylated and is not degraded. Instead, the interaction between HAF and HIF-2α results in increased activity of HIF-2α [45].
In addition, HIFs have been found to be regulated through direct phosphorylation by the MAP kinases p38 and ERK1/2 or through the phosphorylation of coactivators of HIFs, in a cell type-dependent manner [46,47,48,49]. HIF-1α expression has also been shown to be regulated through the activation of growth factor receptors by their ligands (such as insulin or insulin-like growth factor 1) or other external stimuli through the activation of the PI3K pathway [50,51,52,53]. HIF-α can also be regulated directly through the activation of NF-kB, and binding sites for NF-kB have been identified in the promoter region of HIF-1α [54].
3. Effect of Hypoxia in Tumors
Many tumor tissues show higher expression levels of HIF-1α than normal tissues. For instance, immunohistochemical studies of tissues from bladder, breast, colon, glial, hepatocellular, ovarian, pancreatic, prostate, and renal tumors showed positive staining for either HIF-1α or HIF-2α or both, as compared to negatively stained control adjacent tissues, with the exception of bone marrow macrophages [49,50]. In these tissues, staining was found to be mostly nuclear [55,56]. The presence of hypoxic regions in tumors is explained in part by the fast-growing rate of tumors, where the demand for oxygen exceeds its supply, and also because of defective vascularization [57]. Indeed, the tumor vasculature is leaky and more fragile than the vasculature found in healthy tissues [58]. In response to hypoxia, a large cellular reprograming is initiated and coordinated by HIFs. Activation of HIFs has been shown to contribute to many cellular processes described as cancer “hallmarks”, including metabolic reprogramming, angiogenesis, apoptosis, and invasion and metastasis [1,4,59,60].
3.1. Metabolic Reprogramming
Under hypoxic conditions, cancer cells reprogram their metabolism by switching from oxygen-dependent mitochondrial oxidative phosphorylation to oxygen-independent glycolysis in order to meet their energy requirements; this metabolic switch is referred to as the Warburg effect [61]. This switch is controlled by the HIF-dependent upregulation of a large number of enzymes of the glycolytic pathway as well as glucose transporters (reviewed in the work of [5]).
3.2. Angiogenesis
Another critical response to hypoxia by cancer cells is angiogenesis. Angiogenesis is triggered by the upregulation of proangiogenic factors, including vascular endothelial growth factor (VEGF) (reviewed in the work of [62]). The formation of new blood vessels promotes tumor cell survival by increasing the delivery of nutrients and oxygen to tumor cells [63,64].
3.3. Apoptosis
The effect of hypoxia on apoptosis is complex and conflicting results have been observed where activation of HIF-1α resulted in either increased or decreased apoptosis [65,66,67]. The p53 protein is a transcription factor with critical roles in apoptosis [68]. Hypoxia has been shown to increase apoptosis in tumors through the accumulation of p53 [69]. HIF-1α has also been suggested to stabilize p53 through its direct interaction with the Mdm2 protein and by inhibiting Mdm2-mediated p53 degradation [70,71]. However, although hypoxia can lead to apoptosis and reduced cell proliferation, hypoxia correlates with tumor aggressiveness and is associated with poor patient prognosis in many cancer types [72]. To explain this paradoxical effect of hypoxia on apoptosis, it has been suggested that hypoxic conditions can serve as a selective pressure in growing tumors, favoring the elimination of wild-type p53 expressing cells and the growth of cells with inactive p53 [73]. Another study also suggested that rather than enhancing the transactivation properties of p53, hypoxia could lead to increases in p53 transrepression activities, resulting in a significant decrease in p53 dependent apoptosis [74]. The role of p53 in apoptosis is still open for investigation [68], and future studies will certainly enhance our understanding of its role in hypoxic conditions.
3.4. Cell Invasion and Metastasis
The roles of hypoxia and HIF-1α on tumor cell invasion and metastasis are well established [75]. It has been shown that intratumoral hypoxia is associated with metastasis (reviewed in the work of [60]). Under hypoxic conditions, cancer cells can undergo epithelial to mesenchymal transition (EMT) that is associated with higher cell motility and reduced cell-cell adhesion [76]. Hypoxia also increases cell invasion through the activation of matrix metalloproteases (MMPs) [77]. In addition, hypoxic conditions have been shown to support cancer stem-cell self-renewal through the activation of HIF-dependent activation of several genes, including OCT4 [78]. Under hypoxic conditions, the colonization of tumor cells at distant sites is enhanced through the upregulation of angiopoietin-like 4 (ANGPTL4) that disrupts contacts between endothelial cells and favors the passage of tumors cells through the blood vessels [76].
3.5. Tumor Microenvironment and Inflammation
Hypoxia has been linked to inflammation in the tumor microenvironment through the activity of HIFs as well through the activation of the master regulator of inflammation, NF-κB [79,80,81]. In the tumor, under hypoxic conditions, HIF-1α expression and activity are regulated by NF-κB [82,83]. The tumor microenvironment contains many types of non-cancer cells, including immune cells, and hypoxia has been shown to modulate the function of immune cells and to result in inflammation [84,85,86,87,88,89]. Importantly, the tumor microenvironment is rich in damaged-associated molecular patterns (DAMPs) or alarmins that further contribute to an inflamed tumor microenvironment [90,91]. Alarmins or DAMPs influence tumor growth through their activation with their receptors; one of these receptors is the receptor for advanced glycation end products (RAGE) [92,93,94].
4. The Receptor for Advanced Glycation End Products
The receptor for advanced glycation end products (RAGE) was first identified in endothelial cells of bovine lungs as a binding partner for advanced glycation end products (AGEs) [95]. RAGE belongs to the large immunoglobulin superfamily, and its gene is located in the class III major histocompatibility complex (MHC) region on chromosome 6 [96]. The presence of the RAGE gene within this complex suggests a critical role of RAGE in inflammation [97].
4.1. RAGE: Domain Structure and Isoforms
The full-length form of RAGE consists of three main regions: an extracellular part (amino acids 23–342) itself divided into a variable-like (V) and two constant-like (C1 and C2) immunoglobulin (Ig) domains, a transmembrane domain (amino acids 343–363), and a cytoplasmic domain (amino acids 364–404) (Figure 2) [95]. Two other isoforms of RAGE are the N-truncated RAGE (amino acids 124–404), where the N-terminal V-like domain is deleted [98], and the dominant negative isoform (amino acids 23–363), where the cytoplasmic domain is missing. In addition, two different types of soluble isoforms of RAGE (sRAGE) (amino acids 23–342) have been described [99]. sRAGE contains the extracellular part but lacks the transmembrane and cytoplasmic domains. sRAGE can be generated as a result of a splicing event or by proteolytic cleavage of full-length RAGE [100].
4.2. RAGE Signaling Pathways in Cancer
RAGE signaling contributes to many physiological and pathophysiological processes and diseases such as innate immune responses, tissue repair, bone homeostasis, complications of diabetes, Alzheimer’s disease, as well as cancers (brain, breast, colon, colorectal, prostate, pancreatic oral squamous cell, ovarian cancer, and melanoma) [101,102,103,104,105,106,107,108,109]. In general, RAGE signaling is initiated by the binding of its ligands to the extracellular part of the receptor. The outcome of this interaction depends on the type of ligand, its concentration, as well as the cell type (reviewed in the work of [110]). Upon RAGE engagement by its ligand, the cytoplasmic domain of RAGE transmits signals with the help of adaptor proteins [111,112,113]. Three adaptor proteins have been identified: diaphanous-1 (Dia-1), TIRAP, and MyD88. Dia-1 has been shown to play an important role in RAGE-dependent cell migration through the activation of Rac1 and Cdc42 [114]. TIRAP and MyD88 also function as adaptors to toll-like receptors (TLR) 2 and 4 [115].
The three main signaling pathways activated in response to RAGE/ligand interactions are the MAPK/ERK, PI3K/Akt, and Jak/STAT pathways (Figure 3). Activation of the MAPK/ERK and PI3K/AKT pathways both lead to activation of NF-κB, resulting in the transcription of genes involved in cellular proliferation, apoptosis, and tumor growth [116,117,118,119,120,121,122,123]. In PC3 prostate cancer cells, activation of the PI3K/Akt pathway has been shown to regulate cell cycle progression through the phosphorylation of the retinoblastoma (Rb) protein, resulting in EF2-dependent gene transcription [124]. Similarly, in SiHa cervical squamous cancer cells, RAGE-dependent activation of the PI3K/Akt signaling pathway has been shown to promote cell cycle progression through the upregulation of the proliferating cell nuclear antigen (PCNA) [125].
In the JAK/STAT pathway, JAK phosphorylates STAT3 in response to IL-6, resulting in the translocation of STAT3 to the nucleus and enhanced cell proliferation [126,127,128,129]. Kang et al. reported that in pancreatic tumors, after its phosphorylation with JAK, STAT3 translocated to the mitochondria and increased ATP production, favoring tumor cell proliferation as a result of RAGE activation [128].
4.3. Hypoxia and RAGE Signaling in Cancer Tumors
The promoter region of RAGE contains at least one functional HRE (sequence: 5’-RCGTG-3’, where R is a purine base), and increased expression levels of RAGE have been observed during hypoxia in murine and rat models of brain and liver ischemic hypoxia [130,131,132,133,134,135]. These data suggest that RAGE is a potential target gene for both HIF-1a and HIF-2a since the HREs for both transcription factors (HIF-1a and HIF-2a) are identical [136]. RAGE signaling has also been associated with increased inflammation, as well as increased expression levels of RAGE ligands [131,132,137,138,139].
Few studies so far have investigated the role of RAGE in cancer tumors under hypoxic conditions (Figure 3) [140,141]. Two independent studies, the first one in breast and head and neck tumors, and the second one in pancreatic tumors, have shown that RAGE expression level was increased in these tumors under hypoxic conditions [140,141]. Tafani et al. reported that in breast and cervical cancer cell lines, the increase in RAGE expression levels was HIF-1α-dependent and was associated with the activation of ERK1/2, PI3K, as well as NF-κB, resulting in increased cell migration and invasion through the activation of MMPs [140]. These authors also showed that increases in HIF-1α resulted in direct activation of NF-κB as well as an indirect activation of NF-κB through the activation of RAGE [140].
Hypoxia has also been shown to modulate RAGE-dependent signaling pathways in pancreatic tumors [141]. Kang et al. showed that RAGE expression levels increased in hypoxic conditions, resulting in the activation of two different pathways: the KRAS dependent RAF/MEK/ERK pathway as well as the PI3K/Akt pathway [141]. Activation of both pathways was shown to lead to NF-κB activation as well as HIF-1α activation [141]. In this study, hypoxic conditions resulted in a RAGE-dependent activation of HIF-1α, but not in an HIF-1α dependent RAGE upregulation, as reported by Tafani et al. in breast and head and neck cancer tumors, suggesting distinct mechanisms of RAGE activity in hypoxia in different types of cancers [140]. Kang et al. further reported that under hypoxic conditions, RAGE activation was positively associated with tumor cell survival through increased autophagy and decreased apoptosis [141]
4.4. RAGE Ligands in Hypoxic Tumors
RAGE ligands consist of a large number of molecules structurally very diverse. RAGE ligands include advanced glycation end products (AGE) that provided the name to the receptor, S100 proteins, β-amyloid peptides, high mobility group box 1 (HMGB1) protein, transthyretin, the Mac-1 α2 integrin, and the C3a and C1q complement proteins [108,142,143,144]. We will only discuss here selected RAGE ligands that have been reported to be involved in the hypoxic response in tumors.
4.4.1. HMGB1
HMGB1 has been found overexpressed in many types of tumors, including colon, colorectal, prostate, gastric, pancreatic, glioma, neuroblastoma, and melanoma tumors (reviewed in the work of [145]) [93,116,146,147,148,149,150,151]. HMGB1 has multiple functions depending on its cellular localization. In the nucleus, HMGB1 is a DNA binding protein and modulates DNA transcription and recombination [152,153,154,155]. HMGB1 interacts with DNA via two DNA binding regions called box A and box B [156,157]. Nuclear HMGB1 plays the role of DNA chaperone, regulates DNA damage repair, and contributes to the maintenance of genome stability [158]. Besides its function as a nuclear protein, HMGB1 has important cytoplasmic and extracellular functions [156,159,160,161]. Cytoplasmic HMGB1 plays a role in tumor progression by promoting autophagy and activating the inflammasome [162,163,164]. HMGB1 can also be secreted from cells either via an active process involving hyperacetylation and packaging of HMGB1 into secretory vesicles or via a passive process during cell necrosis [162]. Extracellular HMGB1 interacts with RAGE as well as with other cell surface receptors such as TLRs [160,165,166,167,168,169]. Activation of the HMGB1/RAGE axis has been shown to result in many cellular processes, including proliferation and migration, inflammation, and chemotaxis [170,171,172,173,174].
In hypoxia, melanoma cells have been shown to release higher levels of HMGB1 than in normoxia [150]. Huber et al. showed that HMGB1 released under hypoxia facilitated melanoma growth and metastasis, as well as tumor infiltration of tumor-promoting M2-like macrophages [150,175]. In addition, these macrophages were shown to synthesize IL-10 in a RAGE-dependent manner [150]. IL-10 is an inflammatory cytokine and has been shown to correlate with melanoma tumor progression and metastases formation [176]. In hepatocellular carcinoma (HCC) tumors, HMGB1 was shown to mediate tumor growth, under hypoxic conditions, through the interaction with intracellular TLR-9 [177] and to attract macrophages to the tumor site, resulting in increased metastasis [178]. In this study, however, the authors did not investigate if HMGB1 influenced RAGE signaling [177]. In a different study, Yan et al. showed that in HCC cells exposed to hypoxia, HMGB1 secreted extracellularly interacted with both TLR-4 and RAGE to promote cellular invasion through the activation of caspase 1, resulting in the cleavage and subsequent activation of the pro-inflammatory cytokines IL-1β and IL-18 [179]. HMGB1 is also a predictor of poor outcomes in glioblastoma patients [180]. Under hypoxic conditions, U118 glioblastoma cells were shown to release up to 50-fold higher HMGB1 levels than under normoxic conditions. HMGB1 containing condition media was shown to stimulate cell proliferation and invasion of these cells [180]. The mechanisms by which nuclear HMGB1 translocates to the extracellular milieu in hypoxic conditions are currently unknown.
4.4.2. S100 Proteins
S100 proteins are small calcium-binding proteins that transmit calcium signals through their interaction with target proteins. S100 proteins exert both intracellular and extracellular functions [181,182]. Many S100 proteins interact with RAGE extracellularly and contribute to cancer cell proliferation, invasion, and metastasis [183,184]. Calcium has been shown to be released under hypoxic conditions and to stimulate HIF-1α expression and stabilization [185,186]. Changes in intracellular calcium levels in hypoxic cells could result in the activation of S100 dependent signaling pathways. S100 proteins could thus play critical functions during hypoxia. In this review, we will focus on six S100 members (S100B, S100P, S100A4, S100A7, S100A8, and S100A9) that have been investigated in the context of hypoxia in cancer.
4.4.3. S100B
S100B is one of the best-characterized members of the S100 protein family [181,182]. S100B expression is highest in the brain, and high levels of S100B are associated with neurodegeneration and other neurologic disorders [187,188]. Interestingly, increased levels of S100B have been reported in the brain during ischemia [188], and S100B levels in umbilical cord blood have been suggested to be a potential biomarker of hypoxic-ischemic encephalopathy in asphyxiated newborns [189]. In human colon adenocarcinoma Caco-2 cells, activation of RAGE by extracellular S100B has been shown to upregulate HIF-1α through the activation of the p38/MAPK and Akt/mTOR signaling pathways, resulting in increased cell proliferation and angiogenesis [190]. This study was performed in normoxic conditions and showed that activation of the RAGE signaling pathways can regulate HIF-1α and the transcription of HIF-1α dependent genes.
4.4.4. S100P
S100P was first isolated from the placenta and herein the letter P [191]. In tumors, S100P was found to promote tumor cell proliferation and survival, as well as metastasis through its interaction with RAGE [192,193,194,195]. A recent study showed that S100P played an important function during hypoxia by promoting cell migration and tumor metastasis in hepatocarcinoma cells and tumors [196]. In this study, the authors showed that under hypoxia, the levels of both caveolin 1 and S100P were increased, resulting in increased cell motility and metastasis, and that these effects were dependent on NF-κB [196]. Although the authors did not interrogate RAGE signaling pathways in their study, it is probable that the effect of S100P on cell migration, tumor growth, and metastasis was partially mediated by RAGE.
4.4.5. S100A4
S100A4 is another ligand of RAGE that has been shown to stimulate tumor proliferation and metastasis in various cancer types through its interaction with RAGE [197,198,199,200,201]. In cancer tumors, S100A4 has been shown to increase cell motility, modulate cell proliferation and apoptosis, stimulate angiogenesis and trigger extracellular matrix protein remodeling (reviewed in the work of [199,200]).
In colorectal cancer (CRC), higher levels of HIF-1α were observed upon RAGE stimulation with human recombinant S100A4 [197]. Because HIF-1α activity in these tumors has been shown to enhance cellular motility and increased resistance to apoptosis and chemotherapy, it is reasonable to speculate that activation of the RAGE/S100A4 axis in CRC tumors favors tumor progression and chemoresistance through HIF-mediated mechanisms [202,203,204]. S100A4 expression levels are also regulated by hypoxia due to the presence of HRE in the gene coding S100A4 [205]. Hypoxic regulation of S100A4 was demonstrated in BGC823 gastric cancer cells where exposure to hypoxic conditions resulted in higher transcript levels of S100A4 as compared to normoxic conditions [205]. In a different study, Horiuchi et al. reported that in ovarian cancer cells, hypoxic-dependent expression of S100A4 was regulated by epigenetic mechanisms involving methylation of the HRE of S100A4 [206]. Increased levels of S100A4 transcripts and proteins were also observed in esophageal squamous cancer cells (ESCC) exposed to cobalt chloride, a hypoxia-mimetic agent that upregulates HIF-α [207,208].
4.4.6. S100A7
S100A7 was initially named psoriasin because of its high expression level in psoriatic lesions [209]. Since its discovery, S100A7 has been shown to contribute to the progression of many cancers, including bladder, prostate, and skin cancers [210,211,212,213], and has been most studied in the context of breast cancer [12]. In breast cancer cells, intracellular S100A7 was shown to form a complex with the cofactor Jab1, resulting in the translocation of Jab1 to the nucleus and its interaction with HIF-1a, resulting in the transcription of HIF-1a dependent genes [214]. Interestingly, extracellular S100A7 has also been shown to stimulate hypoxic-dependent processes, such as angiogenesis, through its interaction with RAGE, in an insulin-like growth factor (IGF) dependent manner [215]. However, in this study, the authors did not investigate if the changes in angiogenesis w HIF dependent [215].
4.4.7. S100A8/9
S100A8 and S100A9 are most commonly found as heterodimers [181,182,216]. Extracellular S100A8/A9 can bind to RAGE and other receptors described as S100 soil sensor receptors (SSSRs) [217], that include extracellular matrix metalloproteinase inducer (EMMPRIN), activated leukocyte cell adhesion molecule (ALCAM), TLR-4, neuroplastin β, and melanoma cells adhesion molecule (MCAM) [218,219]. In cancer, S100A8/A9 has been shown to have paradoxical functions by either promoting apoptosis or favoring tumor development, depending on the cellular context [220]. In tumors, activation of the S100A8/A9/RAGE pathway has been shown to promote tumor progression in several types of cancer, including breast, prostate, and colorectal cancer, through the activation of the MAPK pathway and NF-kB [178,221,222].
5. Conclusions
Hypoxia in tumors results in large cellular reprogramming. Recent studies have shown that activation of the RAGE signaling pathway could be a key player in cellular adaption to hypoxia. In this review, we discussed recent evidence on the role of RAGE and its ligands in stimulating tumor cell survival and favoring metastasis in hypoxic conditions. This emerging aspect of RAGE signaling in hypoxia deserves further attention, and inhibition of the RAGE signaling pathway could become a promising approach to reduce tumor growth in hypoxic regions of tumors.
Author Contributions
Conceptualization: E.L., writing—original draft preparation, S.T.; writing—review and editing, E.L. and S.W.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by the College of Health Professions at NDSU and by the NIH grant U54GM128729.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AGEs | Advanced glycation endproducts |
| ALCAM | Activated leukocyte cell adhesion molecule |
| ANGPTL4 | Angiopoietin-like 4 |
| ARNT1 | Aryl hydrocarbon receptor nuclear translocator 1 |
| bHLH | Basic-helix-loop-helix |
| CBP/p300 | CREB binding protein/p300 coactivator |
| Cdc42 | Cell division control protein 42 |
| C-TAD | C-terminal transactivation domain |
| DAMP | Damage-associated molecular pattern |
| Dia-1 | Diaphanous 1 |
| EMMPRIN | Extracellular matrix metalloproteinase inducer |
| EMT | Epithelial mesenchymal transition |
| ERK | Extracellular-regulated kinase |
| HAF | Hypoxia-associated factor |
| HIF | Hypoxia-inducible factor |
| HIF-1α | Hypoxia-inducible Factor 1α |
| HMGB1 | High mobility group box 1 |
| HRE | Hypoxia-responsive elements |
| Ig | Immunoglobulin |
| Jak | Janus kinase |
| LZIP | Leucine zipper domain |
| MAP | Mitogen-activated protein |
| MCAM | Melanoma cells adhesion molecule |
| MDM2 | Mouse double minute 2 homolog |
| MHC | Major histocompatibility complex |
| MMP | Matrix metalloproteinases |
| mTOR | Mechanistic target of rapamycin kinase |
| MyD88 | Myeloid differentiation primary response 88 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| N-TAD | N-terminal transactivation domain |
| NPTN | Neuroplastin β |
| OCT4 | Octamer binding transcription factor 4 |
| ODD | Oxygen-dependent degradation |
| PAS | Per-ARNT-Sim domains |
| PCNA | Proliferating cell nuclear antigen |
| PHD | Proline-hydroxylases |
| PI3K | Phosphoinositide 3-kinase |
| Rac 1 | Ras-related C3 botulinum toxin substrate 1 |
| RAGE | Receptor of advanced glycation end products |
| SRC-1 | Nuclear receptor coactivator 1 |
| SSSR | S100 soil sensor receptors |
| STAT | Signal transducer and activator of transcription |
| TIF2 | Transcriptional mediator/intermediary factor 2 |
| TIRAP | TIR domain-containing adaptor protein |
| TLR | Toll-like receptor |
| VHL | von Hippel-Lindau protein |
| VEGF | Vascular endothelial growth factor |
References
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours—implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [Green Version]
- Michiels, C. Physiological and Pathological Responses to Hypoxia. Am. J. Pathol. 2004, 164, 1875–1882. [Google Scholar] [CrossRef] [Green Version]
- Hockel, M.; Vaupel, P. Tumor Hypoxia: Definitions and Current Clinical, Biologic, and Molecular Aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.R.; Hay, M. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-Inducible Factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Shen, S.-M.; Zhao, X.-Y.; Chen, G.-Q. Targeted genes and interacting proteins of hypoxia inducible factor. Int. J. Biochem. Mol. Boil. 2012, 3, 165–178. [Google Scholar]
- Rankin, E.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downes, N.; Laham-Karam, N.; Kaikkonen, M.U.; Ylä-Herttuala, S. Differential but Complementary HIF1α and HIF2α Transcriptional Regulation. Mol. Ther. 2018, 26, 1735–1745. [Google Scholar] [CrossRef] [Green Version]
- Ratcliffe, P.J. HIF-1 and HIF-2: Working alone or together in hypoxia? J. Clin. Investig. 2007, 117, 862–865. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.; Keogh, C.E.; Crean, D.; Taylor, C.T. The role of HIF in immunity and inflammation. Mol. Asp. Med. 2016, 47–48, 24–34. [Google Scholar] [CrossRef]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively ex-pressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [Green Version]
- Wiener, C.M.; Bootha, G.; Semenza, G.L. In Vivo Expression of mRNAs Encoding Hypoxia-Inducible Factor. Biochem. Biophys. Res. Commun. 1996, 225, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.-Z.; Moran, S.M.; HogenEsch, J.B.; Wartman, L.; Bradfield, C.A. Molecular Characterization and Chromosomal Localization of a Third α-Class Hypoxia Inducible Factor Subunit, HIF3α. Gene Expr. 2018, 7, 205–213. [Google Scholar]
- Hara, S.; Hamada, J.; Kobayashi, C.; Kondo, Y.; Imura, N. Expression and Characterization of Hypoxia-Inducible Factor (HIF)-3α in Human Kidney: Suppression of HIF-Mediated Gene Expression by HIF-3α. Biochem. Biophys. Res. Commun. 2001, 287, 808–813. [Google Scholar] [CrossRef]
- Li, Q.F.; Wang, X.R.; Yang, Y.W.; Lin, H. Hypoxia upregulates hypoxia inducible factor (HIF)-3α expression in lung epithelial cells: Characterization and comparison with HIF-1α. Cell Res. 2006, 16, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Maynard, M.A.; Qi, H.; Chung, J.; Lee, E.H.L.; Kondo, Y.; Hara, S.; Conaway, R.C.; Conaway, J.; Ohh, M. Multiple Splice Variants of the Human HIF-3α Locus Are Targets of the von Hippel-Lindau E3 Ubiquitin Ligase Complex. J. Biol. Chem. 2003, 278, 11032–11040. [Google Scholar] [CrossRef] [Green Version]
- Pasanen, A.; Heikkila, M.; Rautavuoma, K.; Hirsilä, M.; Kivirikko, K.I.; Myllyharju, J. Hypoxia-inducible factor (HIF)-3α is subject to extensive alternative splicing in human tissues and cancer cells and is regulated by HIF-1 but not HIF-2. Int. J. Biochem. Cell Biol. 2010, 42, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Maynard, M.A.; Evans, J.A.; Hosomi, T.; Hara, S.; Jewett, A.S.M.; Ohh, M. Human HIF-3alpha4 is a dominant-negative regulator of HIF-1 and is down-regulated in renal cell carcinoma. FASEB J. 2005, 19, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ko, H.P.; Whitlock, J.P. Induction of Phosphoglycerate Kinase 1 Gene Expression by Hypoxia. J. Biol. Chem. 1996, 271, 21262–21267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Potluri, N.; Lu, J.; Kim, Y.; Rastinejad, F. Structural integration in hypoxia-inducible factors. Nat. Cell Biol. 2015, 524, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [Green Version]
- Klinger, P.; Schietke, E.R.; Warnecke, C.; Swoboda, B.; Wiesener, M.; Hennig, F.F.; Gelse, K. Deletion of the oxygen-dependent degradation domain results in impaired transcriptional activity of hy-poxia-inducible factors. Transcription 2011, 2, 269–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, K.; Discher, D.J.; Hu, J.; Bishopric, N.H.; Webster, K.A. Molecular Regulation of the Endothelin-1 Gene by Hypoxia. J. Biol. Chem. 2001, 276, 12645–12653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Nock, S.; Nesper, M.; Sprinzl, A.M.; Sigler, P.B. Structure and Importance of the Dimerization Domain in Elongation Factor Ts from Thermus thermophilus. Biochemistry 1996, 35, 10269–10278. [Google Scholar] [CrossRef]
- Ravenna, L.; Salvatori, L.; Russo, M.A. HIF3alpha: The little we know. FEBS J. 2016, 283, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.D.; Coleman, M.; Pugh, C.W. Hypoxia, hypoxia-inducible factors (HIF), HIF hydroxylases and oxygen sensing. Cell. Mol. Life Sci. 2009, 66, 3539–3554. [Google Scholar] [CrossRef]
- Koh, M.Y.; Spivak-Kroizman, T.R.; Powis, G. HIF-1 regulation: Not so easy come, easy go. Trends Biochem. Sci. 2008, 33, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hip-pel-Lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Activation of the HIF pathway in cancer. Curr. Opin. Genet. Dev. 2001, 11, 293–299. [Google Scholar] [CrossRef]
- Jeong, J.-W.; Bae, M.-K.; Ahn, M.-Y.; Kim, S.-H.; Sohn, T.-K.; Bae, M.-H.; Yoo, M.-A.; Song, E.J.; Lee, K.-J.; Kim, K.-W. Regulation and Destabilization of HIF-1α by ARD1-Mediated Acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genome Res. 2000, 14, 34–44. [Google Scholar]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arany, Z.; Huang, E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef] [Green Version]
- Ebert, B.L.; Bunn, H.F. Regulation of transcription by hypoxia requires a multiprotein complex that includes hypox-ia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol. Cell Biol. 1998, 18, 4089–4096. [Google Scholar] [CrossRef] [Green Version]
- Kallio, P.J.; Okamoto, K.; O’Brien, S.; Carrero, P.; Makino, Y.; Tanaka, H.; Poellinger, L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 co-activator by the hypoxia-inducible factor-1alpha. EMBO J. 1998, 17, 6573–6586. [Google Scholar] [CrossRef]
- Ema, M.; Hirota, K.; Mimura, J.; Abe, H.; Yodoi, J.; Sogawa, K.; Poellinger, L.; Fujii-Kuriyama, Y. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: Their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999, 18, 1905–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lando, D.; Peet, D.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine Hydroxylation of the HIF Transactivation Domain: A Hypoxic Switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypox-ia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, M.Y.; Darnay, B.G.; Powis, G. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hy-poxia-inducible factor 1alpha, leading to its oxygen-independent degradation. Mol. Cell Biol. 2008, 28, 7081–7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, M.Y.; Lemos, R.; Liu, X.; Powis, G. The hypoxia-associated factor switches cells from HIF-1alpha- to HIF-2alpha-dependent signaling pro-moting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrows, N.; Babur, M.; Resch, J.; Williams, K.J.; Brabant, G. Hypoxia-Inducible Factor in Thyroid Carcinoma. J. Thyroid. Res. 2011, 2011, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, D.E.; Berra, E.; Gothié, E.; Roux, D.; Pouysségur, J. p42/p44 Mitogen-activated Protein Kinases Phosphorylate Hypoxia-inducible Factor 1α (HIF-1α) and Enhance the Transcriptional Activity of HIF-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef] [Green Version]
- Gradin, K.; Takasaki, C.; Fujii-Kuriyama, Y.; Sogawa, K. The Transcriptional Activation Function of the HIF-like Factor Requires Phosphorylation at a Conserved Threonine. J. Biol. Chem. 2002, 277, 23508–23514. [Google Scholar] [CrossRef] [Green Version]
- Kietzmann, T.; Mennerich, D.; Dimova, E.Y. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef]
- Stiehl, D.P.; Jelkmann, W.; Wenger, R.H.; Hellwig-Bürgel, T. Normoxic induction of the hypoxia-inducible factor 1alpha by insulin and interleukin-1beta involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002, 512, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Davidson, G.; Huang, Y.; Jiang, B.-H.; Shi, X.; Costa, M.; Huang, C. Nickel compounds act through phosphatidylinositol-3-kinase/Akt-dependent, p70(S6k)-independent pathway to induce hypoxia inducible factor transactivation and Cap43 expression in mouse epidermal Cl41 cells. Cancer Res. 2004, 64, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Beppu, K.; Nakamura, K.; Linehan, W.M.; Rapisarda, A.; Thiele, C.J. Topotecan blocks hypoxia-inducible factor-1alpha and vascular endothelial growth factor expression in-duced by insulin-like growth factor-I in neuroblastoma cells. Cancer Res. 2005, 65, 4775–4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohlin, S.; Hamidian, A.; von Stedingk, K.; Bridges, E.; Wigerup, C.; Bexell, D.; Påhlman, S. PI3K-mTORC2 but not PI3K-mTORC1 regulates transcription of HIF2A/EPAS1 and vascularization in neuroblastoma. Cancer Res. 2015, 75, 4617–4628. [Google Scholar] [CrossRef] [Green Version]
- Van Uden, P.; Kenneth, N.S.; Webster, R.; Müller, H.A.; Mudie, S.; Rocha, S. Evolutionary conserved regulation of HIF-1beta by NF-kappaB. PLoS Genet. 2011, 7, e1001285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.; Pugh, C.; Ratcliffe, P.; Harris, A.L. The Expression and Distribution of the Hypoxia-Inducible Factors HIF-1α and HIF-2α in Normal Human Tissues, Cancers, and Tumor-Associated Macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussenbaum, F.; Herman, I.M. Tumor Angiogenesis: Insights and Innovations. J. Oncol. 2010, 2010, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Antiangiogenesis in cancer therapy—endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar] [CrossRef]
- Folkman, J.; Ingber, D. Inhibition of angiogenesis. Semin. Cancer Biol. 1992, 3, 89–96. [Google Scholar]
- Carmeliet, P.; Dor, Y.; Herbert, J.-M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nat. Cell Biol. 1998, 394, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Ryan, H.E.; Lo, J.; Johnson, R.S. HIF-1alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998, 17, 3005–3015. [Google Scholar] [CrossRef] [Green Version]
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef]
- Aubrey, B.; Kelly, G.L.; Janic, A.; Herold, M.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Graeber, T.G.; Peterson, J.F.; Tsai, M.; Monica, K.; Fornace, A.J.; Giaccia, A.J. Hypoxia induces accumulation of p53 protein, but activation of a G1-phase checkpoint by low-oxygen conditions is independent of p53 status. Mol. Cell. Biol. 1994, 14, 6264–6277. [Google Scholar] [CrossRef] [PubMed]
- An, B.; Johnson, D.E.; Jin, J.R.; Antoku, K.; Dou, Q.P. Bcl-2- and CrmA-inhibitable dephosphorylation and cleavage of retinoblastoma protein during etopo-side-induced apoptosis. Int. J. Mol. Med. 1998, 1, 131–136. [Google Scholar]
- Chen, D.; Li, M.; Luo, J.; Gu, W. Direct Interactions between HIF-1α and Mdm2 Modulate p53 Function. J. Biol. Chem. 2003, 278, 13595–13598. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef]
- Graeber, T.; Osmanian, C.; Jacks, T.; Housman, D.E.; Koch, C.J.; Lowe, S.W.; Giaccia, A.J. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nat. Cell Biol. 1996, 379, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Koumenis, C.; Alarcon, R.; Hammond, E.; Sutphin, P.; Hoffman, W.; Murphy, M.; Derr, J.; Taya, Y.; Lowe, S.W.; Kastan, M.; et al. Regulation of p53 by Hypoxia: Dissociation of Transcriptional Repression and Apoptosis from p53-Dependent Transactivation. Mol. Cell. Biol. 2001, 21, 1297–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, E.; Nam, J.-M.; Giaccia, A.J. Hypoxia: Signaling the Metastatic Cascade. Trends Cancer 2016, 2, 295–304. [Google Scholar] [CrossRef] [Green Version]
- De Bock, K.; Mazzone, M.; Carmeliet, P. Antiangiogenic therapy, hypoxia, and metastasis: Risky liaisons, or not? Nat. Rev. Clin. Oncol. 2011, 8, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Biophys. Acta 2016, 1863, 382–391. [Google Scholar] [CrossRef]
- Keith, B.; Simon, M.C. Hypoxia-Inducible Factors, Stem Cells, and Cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Botta, C.; Misso, G.; Martino, E.C.; Pirtoli, L.; Cusi, M.G.; Tassone, P.; Tagliaferri, P.; Caraglia, M.; Correale, P. The route to solve the interplay between inflammation, angiogenesis and anti-cancer immune response. Cell Death Dis. 2016, 7, e2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sitkovsky, M.; Lukashev, D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat. Rev. Immunol. 2005, 5, 712–721. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Greten, F.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Delovitch, T.L. Different subsets of natural killer T cells may vary in their roles in health and disease. Immunity 2014, 142, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Y.; Yang, D.; Oppenheim, J.J. Alarmins and Antitumor Immunity. Clin. Ther. 2016, 38, 1042–1053. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Xie, J.; Sun, X.; Zeh, H.J.; Kang, R.; Lotze, M.T.; Tang, D. DAMPs, ageing, and cancer: The ’DAMP Hypothesis’. Ageing Res. Rev. 2015, 24 Pt A, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D.; Schapiro, N.E.; Lotze, M.T.; Zeh, H.J. Abstract B96: The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Tumor. Biol. 2012, 33, 567–577. [Google Scholar] [CrossRef]
- Papale, M.; Buccarelli, M.; Mollinari, C.; Russo, M.A.; Pallini, R.; Ricci-Vitiani, L.; Tafani, M. Hypoxia, Inflammation and Necrosis as Determinants of Glioblastoma Cancer Stem Cells Progression. Int. J. Mol. Sci. 2020, 21, 2660. [Google Scholar] [CrossRef] [Green Version]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Sugaya, K.; Fukagawa, T.; Matsumoto, K.-I.; Mita, K.; Takahashi, E.-I.; Ando, A.; Inoko, H.; Ikemura, T. Three Genes in the Human MHC Class III Region near the Junction with the Class II: Gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int-3. Genomics 1994, 23, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Verweij, C.L. How RAGE turns in rage. Genes Immun. 2002, 3, 117–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370 Pt 3, 1097–1109. [Google Scholar] [CrossRef]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2007, 22, 1572–1580. [Google Scholar] [CrossRef]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef]
- Sorci, G.; Riuzzi, F.; Giambanco, I.; Donato, R. RAGE in tissue homeostasis, repair and regeneration. Biochim. Biophys. Acta 2013, 1833, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, L.I.; Essex, A.L.; Davis, H.M. RAGE Signaling in Skeletal Biology. Curr. Osteoporos. Rep. 2019, 17, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Liliensiek, B.; Weigand, M.A.; Bierhaus, A.; Nicklas, W.; Kasper, M.; Hofer, S.; Plachky, J.; Gröne, H.-J.; Kurschus, F.C.; Schmidt, A.M.; et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Investig. 2004, 113, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhu, L.; Zhang, Z.; Li, H.; Li, P.; Wang, Y.; Leng, M. HMGB1-RAGE signaling pathway in pPROM. Taiwan. J. Obstet. Gynecol. 2018, 57, 211–216. [Google Scholar] [CrossRef]
- Kwak, T.; Drews-Elger, K.; Ergonul, A.; Miller, P.C.; Braley, A.; Hwang, G.H.; Zhao, D.; Besser, A.; Yamamoto, Y.; El-Ashry, D.; et al. Targeting of RAGE-ligand signaling impairs breast cancer cell invasion and metastasis. Oncogene 2017, 36, 1559–1572. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. Tempering the wrath of RAGE: An emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Ann. Med. 2009, 41, 408–422. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Münch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Logsdon, C.D.; Fuentes, M.K.; Huang, E.H.; Arumugam, T. RAGE and RAGE ligands in cancer. Curr. Mol. Med. 2007, 7, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Morales, M.; Gonzalez, I.; Araya, P. Inhibition of RAGE Axis Signaling: A Pharmacological Challenge. Curr. Drug Targets 2019, 20, 340–346. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for advanced glycation end products (RAGE)-mediated neurite out-growth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef] [Green Version]
- Kislinger, T.; Tanji, N.; Wendt, T.; Qu, W.; Lu, Y.; Ferran, L.J., Jr.; Taguchi, A.; Olson, K.; Bucciarelli, L.; Goova, M.; et al. Receptor for Advanced Glycation End Products Mediates Inflammation and Enhanced Expression of Tissue Factor in Vasculature of Diabetic Apolipoprotein E–Null Mice. Arter. Thromb. Vasc. Biol. 2001, 21, 905–910. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, T.; Yan, S.F.; Belov, D.; Rong, L.L.; Sousa, M.; Andrassy, M.; Marso, S.P.; Duda, S.; Arnold, B.; Liliensiek, B.; et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J. Clin. Investig. 2003, 111, 959–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, B.I.; Kalea, A.Z.; del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, M.; Murata, H.; Yamamoto, K.-I.; Ono, T.; Sakaguchi, Y.; Motoyama, A.; Hibino, T.; Kataoka, K.; Huh, N.-H. TIRAP, an Adaptor Protein for TLR2/4, Transduces a Signal from RAGE Phosphorylated upon Ligand Binding. PLoS ONE 2011, 6, e23132. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, A.; Blood, D.C.; Del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE–amphoterin signalling suppresses tumour growth and metastases. Nat. Cell Biol. 2000, 405, 354–360. [Google Scholar] [CrossRef]
- Hermani, A.; De Servi, B.; Medunjanin, S.; Tessier, P.A.; Mayer, D. S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp. Cell Res. 2006, 312, 184–197. [Google Scholar] [CrossRef]
- Ghavami, S.; Kerkhoff, C.; Chazin, W.J.; Kadkhoda, K.; Xiao, W.; Zuse, A.; Hashemi, M.; Eshraghi, M.; Schulze-Osthoff, K.; Klonisch, T.; et al. S100A8/9 induces cell death via a novel, RAGE-independent pathway that involves selective release of Smac/DIABLO and Omi/HtrA2. Biochim. Biophys. Acta. 2008, 1783, 297–311. [Google Scholar] [CrossRef] [Green Version]
- Rouhiainen, A.; Kuja-Panula, J.; Tumova, S.; Rauvala, H. RAGE-Mediated Cell Signaling. Methods Mol. Biol. 2013, 963, 239–263. [Google Scholar]
- Sharaf, H.; Matou-Nasri, S.; Wang, Q.; Rabhan, Z.; Al-Eidi, H.; Al Abdulrahman, A.; Ahmed, N. Advanced glycation endproducts increase proliferation, migration and invasion of the breast cancer cell line MDA-MB-231. Biochim. Biophys. Acta 2015, 1852, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Azizan, N.; Suter, M.A.; Liu, Y.; Logsdon, C.D. RAGE maintains high levels of NFkappaB and oncogenic Kras activity in pancreatic cancer. Biochem. Biophys. Res. Commun. 2017, 493, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.-N.; Seo, J.-H.; Lee, M.-H.; Yoon, G.; Cho, S.-S.; Liu, K.; Choi, H.; Oh, K.B.; Cho, Y.-S.; Kim, H.; et al. Oridonin induces apoptosis in oral squamous cell carcinoma probably through the generation of reactive oxygen species and the p38/JNK MAPK pathway. Int. J. Oncol. 2018, 52, 1749–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dukic-Stefanovic, S.; Gasic-Milenkovic, J.; Deuther-Conrad, W.; Münch, G. Signal transduction pathways in mouse microglia N-11 cells activated by advanced glycation endproducts (AGEs). J. Neurochem. 2003, 87, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.-M.; He, M.-Y.; Liu, Y.-W.; Lu, Y.-J.; Hong, Y.-Q.; Luo, H.-H.; Ren, Z.-L.; Zhao, S.-C.; Jiang, Y. AGE/RAGE/Akt pathway contributes to prostate cancer cell proliferation by promoting Rb phosphorylation and degradation. Am. J. Cancer Res. 2015, 5, 1741–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Song, Y.; Zhou, L.; Li, W.; Zhu, X. Downregulation of RAGE Inhibits Cell Proliferation and Induces Apoptosis via Regulation of PI3K/AKT Path-way in Cervical Squamous Cell Carcinoma. Onco. Targets Ther. 2020, 13, 2385–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 Activation by IL-6 Transsignaling Promotes Progression of Pancreatic Intraepithelial Neoplasia and Development of Pancreatic Cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, A.; Wang, S.; Morris, J.P.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 Contribute to Pancreatic Ductal Adenocarcinoma Initiation and Progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh 3rd, H.J. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.; Danson, S. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Pichiule, P.; Chavez, J.C.; Schmidt, A.M.; Vannucci, S.J. Hypoxia-inducible Factor-1 Mediates Neuronal Expression of the Receptor for Advanced Glycation End Products following Hypoxia/Ischemia. J. Biol. Chem. 2007, 282, 36330–36340. [Google Scholar] [CrossRef] [Green Version]
- Kamide, T.; Kitao, Y.; Takeichi, T.; Okada, A.; Mohri, H.; Schmidt, A.M.; Kawano, T.; Munesue, S.; Yamamoto, Y.; Yamamoto, H.; et al. RAGE mediates vascular injury and inflammation after global cerebral ischemia. Neurochem. Int. 2012, 60, 220–228. [Google Scholar] [CrossRef]
- Hassid, B.G.; Nair, M.N.; Ducruet, A.F.; Otten, M.L.; Komotar, R.J.; Pinsky, D.J.; Schmidt, A.M.; Yan, S.F.; Connolly, E.S. Neuronal RAGE expression modulates severity of injury following transient focal cerebral ischemia. J. Clin. Neurosci. 2009, 16, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Zhai, D.-X.; Kong, Q.-F.; Xu, W.-S.; Bai, S.-S.; Peng, H.-S.; Zhao, K.; Li, G.-Z.; Wang, D.-D.; Sun, B.; Wang, J.-H.; et al. RAGE expression is up-regulated in human cerebral ischemia and pMCAO rats. Neurosci. Lett. 2008, 445, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Feirt, N.; Goldstein, M.; Guarrera, J.; Ippagunta, N.; Ekong, U.; Dun, H.; Lu, Y.; Qu, W.; Schmidt, A.M.; et al. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology 2004, 39, 422–432. [Google Scholar] [CrossRef]
- Zeng, S.; Dun, H.; Ippagunta, N.; Rosario, R.; Zhang, Q.Y.; Lefkowitch, J.; Yan, S.F.; Schmidt, A.M.; Emond, J.C. Receptor for advanced glycation end product (RAGE)-dependent modulation of early growth response-1 in hepatic ischemia/reperfusion injury. J. Hepatol. 2009, 50, 929–936. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors—Similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef]
- Kiewitz, R.; Acklin, C.; Minder, E.; Huber, P.R.; Schafer, B.; Heizmann, C.W. S100A1, a New Marker for Acute Myocardial Ischemia. Biochem. Biophys. Res. Commun. 2000, 274, 865–871. [Google Scholar] [CrossRef]
- Mori, T.; Tan, J.; Arendash, G.W.; Koyama, N.; Nojima, Y.; Town, T. Overexpression of human S100B exacerbates brain damage and periinfarct gliosis after permanent focal is chemia. Stroke 2008, 39, 2114–2121. [Google Scholar] [CrossRef] [Green Version]
- Pelinka, L.E.; Harada, N.; Szalay, L.; Jafarmadar, M.; Redl, H.; Bahrami, S. Release of S100B Differs During Ischemia and Reperfusion of the Liver, the Gut, and the Kidney in Rats. Shock 2004, 21, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Tafani, M.; Schito, L.; Pellegrini, L.; Villanova, L.; Marfe, G.; Anwar, T.; Rosa, R.; Indelicato, M.; Fini, M.; Pucci, B.; et al. Hypoxia-increased RAGE and P2X7R expression regulates tumor cell invasion through phosphorylation of Erk1/2 and Akt and nuclear translocation of NF-[14]B. Carcinogenesis 2011, 32, 1167–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Hou, W.; Zhang, Q.; Chen, R.; Lee, Y.J.; Bartlett, D.L.; Lotze, M.T.; Tang, D.; Zeh, H.J. RAGE is essential for oncogenic KRAS-mediated hypoxic signaling in pancreatic cancer. Cell Death Dis. 2014, 5, e1480. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Rai, V.; Hudson, B.I.; Song, F.; Schmidt, A.M.; Barile, G.R. RAGE binds C1q and enhances C1q-mediated phagocytosis. Cell. Immunol. 2012, 274, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Ruan, B.H.; Li, X.; Winkler, A.R.; Cunningham, K.M.; Kuai, J.; Greco, R.M.; Nocka, K.H.; Fitz, L.J.; Wright, J.F.; Pittman, D.D.; et al. Complement C3a, CpG Oligos, and DNA/C3a Complex Stimulate IFN-α Production in a Receptor for Advanced Glycation End Product-Dependent Manner. J. Immunol. 2010, 185, 4213–4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.M.; Hofmann, M.; Taguchi, A.; Yan, S.D.; Stern, D.M. RAGE: A multiligand receptor contributing to the cellular response in diabetic vasculopathy and in-flammation. Semin. Thromb Hemost. 2000, 26, 485–493. [Google Scholar] [CrossRef]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in Inflammation and Cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Volp, K. Increased expression of high mobility group box 1 (HMGB1) is associated with an elevated level of the antiapoptotic c-IAP2 protein in human colon carcinomas. Gut 2006, 55, 234–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiguro, H.; Nakaigawa, N.; Miyoshi, Y.; Fujinami, K.; Kubota, Y.; Uemura, H. Receptor for advanced glycation end products (RAGE) and its ligand, amphoterin are overexpressed and associated with prostate cancer development. Prostate 2005, 64, 92–100. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Chihara, Y.; Kondo, H. Differential effects between amphoterin and advanced glycation end products on colon cancer cells. Int. J. Cancer 2003, 104, 722–727. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Oue, N.; Wakikawa, A.; Shigeishi, H.; Matsutani, N.; Kuraoka, K.; Ito, R.; Yokozaki, H.; Yasui, W. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the in-vasive and metastatic activity of gastric cancer. J. Pathol. 2002, 196, 163–170. [Google Scholar] [CrossRef]
- Huber, R.; Meier-Schiesser, B.; Otsuka, A.; Fenini, G.; Satoh, T.; Gehrke, S.; Widmer, D.; Levesque, M.P.; Mangana, J.; Kerl, K.; et al. Tumour hypoxia promotes melanoma growth and metastasis via High Mobility Group Box-1 and M2-like macrophages. Sci. Rep. 2016, 6, 29914. [Google Scholar] [CrossRef] [Green Version]
- Pezzolo, A.; Marimpietri, D.; Raffaghello, L.; Cocco, C.; Pistorio, A.; Gambini, C.; Cilli, M.; Horenstein, A.; Malavasi, F.; Pistoia, V. Failure of antitumor-derived endothelial cell immunotherapy depends on augmentation of tumor hypoxia. Oncotarget 2014, 5, 10368–10381. [Google Scholar] [CrossRef] [Green Version]
- Vicentino, A.R.R.; Carneiro, V.; Allonso, D.; Guilherme, R.D.F.; Benjamim, C.; Dos Santos, H.A.M.; Xavier, F.; Pyrrho, A.; Gomes, J.; Fonseca, M.; et al. Emerging Role of HMGB1 in the Pathogenesis of Schistosomiasis Liver Fibrosis. Front. Immunol. 2018, 9, 1979. [Google Scholar] [CrossRef] [Green Version]
- Gerlitz, G.; Hock, R.; Ueda, T.; Bustin, M. The dynamics of HMG protein-chromatin interactions in living cells. Biochem. Cell Biol. 2009, 87, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Travers, A.A. Priming the nucleosome: A role for HMGB proteins? EMBO Rep. 2003, 4, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, G.H.; Sanders, C.; Johns, E.W. A New Group of Chromatin-Associated Proteins with a High Content of Acidic and Basic Amino Acids. JBIC J. Biol. Inorg. Chem. 1973, 38, 14–19. [Google Scholar] [CrossRef]
- Andersson, U.; Erlandsson-Harris, H.; Yang, H.; Tracey, K.J. HMGB1 as a DNA-binding cytokine. J. Leukoc. Biol. 2002, 72, 1084–1091. [Google Scholar]
- Thomas, J.O.; Travers, A.A. HMG1 and 2, and related ’architectural’ DNA-binding proteins. Trends Biochem. Sci. 2001, 26, 167–174. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Y. HMGB1 in inflammation and cancer. J. Hematol. Oncol. 2020, 13, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Iii, H.J.Z.; Lotze, M.T. High-mobility group box 1 and cancer. Biochim. Biophys. Acta 2010, 1799, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, S.; Dragoi, A.M.; Wang, X.; Dallacosta, C.; Louten, J.; Musco, G.; Sitia, G.; Yap, G.S.; Wan, Y.; Biron, C.A.; et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 2007, 110, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Parkkinen, J.; Raulo, E.; Merenmies, J.; Nolo, R.; Kajander, E.O.; Baumann, M.; Rauvala, H. Amphoterin, the 30-kDa protein in a family of HMG1-type polypeptides. Enhanced expression in transformed cells, leading edge localization, and interactions with plasminogen activation. J. Biol. Chem. 1993, 268, 19726–19738. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Chavan, S.S.; Andersson, U. High Mobility Group Box Protein 1 (HMGB1): The Prototypical Endogenous Danger Molecule. Mol. Med. 2015, 21, S6–S12. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.-G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasahira, T.; Kirita, T.; Oue, N.; Bhawal, U.K.; Yamamoto, K.; Fujii, K.; Ohmori, H.; Luo, Y.; Yasui, W.; Bosserhoff, A.K.; et al. High mobility group box-1-inducible melanoma inhibitory activity is associated with nodal metastasis and lymphangiogenesis in oral squamous cell carcinoma. Cancer Sci. 2008, 99, 1806–1812. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, H.; Rauvala, H. Amphoterin as an extracellular regulator of cell motility: From discovery to disease. J. Intern. Med. 2004, 255, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Riuzzi, F.; Sorci, G.; Donato, R. The amphoterin (HMGB1)/receptor for advanced glycation end products (RAGE) pair modulates myoblast proliferation, apoptosis, adhesiveness, migration, and invasiveness. Functional inactivation of RAGE in L6 myoblasts results in tumor formation in vivo. J. Biol. Chem. 2006, 281, 8242–8253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todorova, J.; Pasheva, E. High mobility group B1 protein interacts with its receptor RAGE in tumor cells but not in normal tissues. Oncol. Lett. 2011, 3, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Media-tion of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef] [Green Version]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Criollo, A.; Ortiz, C.; Lidereau, R.; Mariette, C.; Chaput, N.; Mira, J.P.; Delaloge, S.; et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radio-therapy. Immunol. Rev. 2007, 220, 47–59. [Google Scholar] [CrossRef]
- Li, W.; Xu, Q.; Deng, Y.; Yang, Z.; Xing, S.; Zhao, X.; Zhu, P.; Wang, X.; He, Z.; Gao, Y. High-mobility group box 1 accelerates lipopolysaccharide-induced lung fibroblast proliferation in vitro: Involvment of the NF-κB signaling pathway. Lab. Investig. 2015, 95, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.-C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High Mobility Group 1 Protein (Hmg-1) Stimulates Proinflammatory Cytokine Synthesis in Human Monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef]
- El Gazzar, M. HMGB1 modulates inflammatory responses in LPS-activated macrophages. Inflamm. Res. 2007, 56, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Fages, C.; Nolo, R.; Huttunen, H.J.; Eskelinen, E.; Rauvala, H. Regulation of cell migration by amphoterin. J. Cell Sci. 2000, 113, 611–620. [Google Scholar] [CrossRef]
- Degryse, B.; Bonaldi, T.; Scaffidi, P.; Müller-Knapp, S.; Resnati, M.; Sanvito, F.; Arrigoni, G.; Bianchi, M.E. The High Mobility Group (Hmg) Boxes of the Nuclear Protein Hmg1 Induce Chemotaxis and Cytoskeleton Reorganization in Rat Smooth Muscle Cells. J. Cell Biol. 2001, 152, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Po-tential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Huang, R.-R.; Wen, D.-R.; Paul, E.; Wünsch, P.H.; Cochran, A.J. IL-10 expression by primary tumor cells correlates with melanoma progression from radial to vertical growth phase and development of metastatic competence. Mod. Pathol. 2011, 24, 801–809. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, W.; Tohme, S.; Chen, M.; Fu, Y.; Tian, D.; Lotze, M.; Tang, D.; Tsung, A. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular car-cinoma through Toll-like receptor. J. Hepatol. 2015, 63, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Wang, G.Z.; Wang, Y.; Huang, H.-Z.; Li, W.-T.; Qu, X.-D. Hypoxia-induced HMGB1 expression of HCC promotes tumor invasiveness and metastasis via regulating macrophage-derived IL-6. Exp. Cell Res. 2018, 367, 81–88. [Google Scholar] [CrossRef]
- Yan, W.; Chang, Y.; Liang, X.; Cardinal, J.S.; Huang, H.; Thorne, S.H.; Monga, S.P.S.; Geller, D.A.; Lotze, M.T.; Tsung, A. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology 2012, 55, 1863–1875. [Google Scholar] [CrossRef]
- Cheng, P.; Ma, Y.; Gao, Z.; Duan, L. High Mobility Group Box 1 (HMGB1) Predicts Invasion and Poor Prognosis of Glioblastoma Multiforme via Activating AKT Signaling in an Autocrine Pathway. Med Sci. Monit. 2018, 24, 8916–8924. [Google Scholar] [CrossRef]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclerc, E.; Fritz, G.; Vetter, S.W.; Heizmann, C.W. Binding of S100 proteins to RAGE: An update. Biochim. Biophys. Acta 2009, 1793, 993–1007. [Google Scholar] [CrossRef] [Green Version]
- Donato, R. Intracellular and extracellular roles of S100 proteins. Microsc. Res. Tech. 2003, 60, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Seta, K.A.; Yuan, Y.; Spicer, Z.; Lu, G.; Bedard, J.; Ferguson, T.K.; Pathrose, P.; Cole-Strauss, A.; Kaufhold, A.; Millhorn, D.E. The role of calcium in hypoxia-induced signal transduction and gene expression. Cell Calcium 2004, 36, 331–340. [Google Scholar] [CrossRef]
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Han, H.J. Role of HIF1α Regulatory Factors in Stem Cells. Int. J. Stem Cells 2019, 12, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, R.; Sorci, G.; Riuzzi, F.; Arcuri, C.; Bianchi, R.; Brozzi, F.; Tubaro, C.; Giambanco, I. S100B’s double life: Intracellular regulator and extracellular signal. Biochim. Biophys. Acta 2009, 1793, 1008–1022. [Google Scholar] [CrossRef] [Green Version]
- Rothermundt, M.; Peters, M.; Prehn, J.; Arolt, V. S100B in brain damage and neurodegeneration. Microsc. Res. Tech. 2003, 60, 614–632. [Google Scholar] [CrossRef]
- Zaigham, M.; Lundberg, F.; Olofsson, P. Protein S100B in umbilical cord blood as a potential biomarker of hypox-ic-ischemic encephalopathy in asphyxiated newborns. Early Hum. Dev. 2017, 112, 48–53. [Google Scholar] [CrossRef]
- Seguella, L.; Capuano, R.; Pesce, M.; Annunziata, G.; Pesce, M.; De Conno, B.; Sarnelli, G.; Aurino, L.; Esposito, G. S100B Protein Stimulates Proliferation and Angiogenic Mediators Release through RAGE/pAkt/mTOR Pathway in Human Colon Adenocarcinoma Caco-2 Cells. Int. J. Mol. Sci. 2019, 20, 3240. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Hu, H.; Tong, X.; Jiang, Q.; Zhu, H.; Zhang, S. Calcium-binding protein S100P and cancer: Mechanisms and clinical relevance. J. Cancer Res. Clin. Oncol. 2011, 138, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Simeone, D.M.; Schmidt, A.M.; Logsdon, C.D. S100P Stimulates Cell Proliferation and Survival via Receptor for Activated Glycation End Products (RAGE). J. Biol. Chem. 2004, 279, 5059–5065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penumutchu, S.R.; Chou, R.H.; Yu, C. Structural insights into calcium-bound S100P and the V domain of the RAGE complex. PLoS ONE 2014, 9, e103947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, M.; Wang, G.; Barsukov, I.L.; Gross, S.; Smith, R.; Rudland, P.S. Direct interaction of metastasis-inducing S100P protein with tubulin causes enhanced cell migration without changes in cell adhesion. Biochem. J. 2020, 477, 1159–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Ito, T.; Nakahara, T.; Nagae, K.; Fuyuno, Y.; Nakao, M.; Akahoshi, M.; Nakagawa, R.; Tu, Y.; Uchi, H.; et al. Upregulation of S100P, receptor for advanced glycation end products and ezrin in malignant melanoma. J. Dermatol. 2013, 40, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Sze Wong, S.Y.; Ting Tse, E.Y.; Fat Ko, F.C.; Tey, S.K.; Yeung, Y.S.; Man, K.; Lam Lo, R.C.; Lin Ng, I.O.; Ping Yam, J.W. Mechanisms through Which Hypoxia-Induced Caveolin-1 Drives Tumorigenesis and Metastasis in Hepato-cellular Carcinoma. Cancer Res. 2016, 76, 7242–7253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlmann, M.; Okhrimenko, A.; Marcinkowski, P.; Osterland, M.; Herrmann, M.; Smith, J.; Heizmann, C.W.; Schlag, P.M.; Stein, U. RAGE mediates S100A4-induced cell motility via MAPK/ERK and hypoxia signaling and is a prognos-tic biomarker for human colorectal cancer metastasis. Oncotarget 2014, 5, 3220–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herwig, N.; Belter, B.; Wolf, S.; Haase-Kohn, C.; Pietzsch, J. Interaction of extracellular S100A4 with RAGE prompts prometastatic activation of A375 melanoma cells. J. Cell. Mol. Med. 2016, 20, 825–835. [Google Scholar] [CrossRef]
- Fei, F.; Qu, J.; Zhang, M.; Li, Y.; Zhang, S. S100A4 in cancer progression and metastasis: A systematic review. Oncotarget 2017, 8, 73219–73239. [Google Scholar] [CrossRef] [Green Version]
- Boye, K.; Maelandsmo, G.M. S100A4 and metastasis: A small actor playing many roles. Am. J. Pathol. 2010, 176, 528–535. [Google Scholar] [CrossRef]
- Garrett, S.C.; Varney, K.M.; Weber, D.J.; Bresnick, A.R. S100A4, a Mediator of Metastasis. J. Biol. Chem. 2006, 281, 677–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.-X.; Wu, X.-Y.; Wang, X.-H.; Fu, H.C. Effect of hypoxia-inducible factor 1-α on Survivin in colorectal cancer. Mol. Med. Rep. 2010, 3, 409–415. [Google Scholar] [CrossRef] [Green Version]
- Mao, Q.; Zhang, Y.; Fu, X.; Xue, J.; Guo, W.; Meng, M.; Zhou, Z.; Mo, X.; Lu, Y. A tumor hypoxic niche protects human colon cancer stem cells from chemotherapy. J. Cancer Res. Clin. Oncol. 2012, 139, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Hongo, K.; Tsuno, N.H.; Kawai, K.; Sasaki, K.; Kaneko, M.; Hiyoshi, M.; Murono, K.; Tada, N.; Nirei, T.; Sunami, E.; et al. Hypoxia enhances colon cancer migration and invasion through promotion of epithelial-mesenchymal transition. J. Surg. Res. 2013, 182, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Fu, H.; Chen, D.; Hua, J.; Hu, Y.; Sun, K.; Sun, X. Subcellular distribution of S100A4 and its transcriptional regulation under hypoxic conditions in gastric cancer cell line BGC823. Cancer Sci. 2010, 101, 1141–1146. [Google Scholar] [CrossRef]
- Horiuchi, A.; Hayashi, T.; Kikuchi, N.; Hayashi, A.; Fuseya, C.; Shiozawa, T.; Konishi, I. Hypoxia upregulates ovarian cancer invasiveness via the binding of HIF-1α to a hypoxia-induced, methylation-free hypoxia response element of S100A4 gene. Int. J. Cancer 2012, 131, 1755–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Yang, G.; Shahzidi, S.; Tkacz-Stachowska, K.; Suo, Z.; Nesland, J.M.; Peng, Q. Induction of hypoxia-inducible factor-1α overexpression by cobalt chloride enhances cellular resistance to photodynamic therapy. Cancer Lett. 2006, 244, 182–189. [Google Scholar] [CrossRef]
- Xuan, X.; Li, Q.; Zhang, Z.; Du, Y.; Liu, P. Increased expression levels of S100A4 associated with hypoxia-induced invasion and metastasis in esopha-geal squamous cell cancer. Tumour. Biol. 2014, 35, 12535–12543. [Google Scholar] [CrossRef]
- Madsen, P.S.; Rasmussen, H.H.; Leffers, H.; Honoré, B.; Dejgaard, K.; Olsen, E.; Kiil, J.; Walbum, E.; Andersen, A.H.; Basse, B.; et al. Molecular Cloning, Occurrence, and Expression of a Novel Partially Secreted Protein “Psoriasin” That Is Highly Up-Regulated in Psoriatic Skin. J. Investig. Dermatol. 1991, 97, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Sun, P.-H.; Martin, T.A.; Sanders, A.J.; Mason, M.D.; Jiang, W. Psoriasin (S100A7) is a positive regulator of survival and invasion of prostate cancer cells. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 1576–1583. [Google Scholar] [CrossRef]
- Yao, R.; Lopez-Beltran, A.; MacLennan, G.T.; Montironi, R.; Eble, J.N.; Cheng, L. Expression of S100 protein family members in the pathogenesis of bladder tumors. Anticancer. Res. 2007, 27, 3051–3058. [Google Scholar]
- Bhalla, S.; Kaur, H.; Dhall, A.; Raghava, G.P.S. Prediction and Analysis of Skin Cancer Progression using Genomics Profiles of Patients. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkie, T.; Verma, A.K.; Zhao, H.; Charan, M.; Ahirwar, D.K.; Kant, S.; Pancholi, V.; Mishra, S.; Ganju, R.K. Lipopolysaccharide from the commensal microbiota of the breast enhances cancer growth: Role of S100A7 and TLR4. Mol. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Emberley, E.D.; Murphy, L.C.; Watson, P. S100 proteins and their influence on pro-survival pathways in cancer. Biochem. Cell Biol. 2004, 82, 508–515. [Google Scholar] [CrossRef]
- Muoio, M.; Talia, M.; Lappano, R.; Sims, A.; Vella, V.; Cirillo, F.; Manzella, L.; Giuliano, M.; Maggiolini, M.; Belfiore, A.; et al. Activation of the S100A7/RAGE Pathway by IGF-1 Contributes to Angiogenesis in Breast Cancer. Cancers 2021, 13, 621. [Google Scholar] [CrossRef] [PubMed]
- Kerkhoff, C.; Klempt, M.; Sorg, C. Novel insights into structure and function of MRP8 (S100A8) and MRP14 (S100A9). Biochim. Biophys. Acta 1998, 1448, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Tomonobu, N.; Kinoshita, R.; Sakaguchi, M. S100 Soil Sensor Receptors and Molecular Targeting Therapy against Them in Cancer Metastasis. Transl. Oncol. 2020, 13, 100753. [Google Scholar] [CrossRef]
- Hibino, T.; Sakaguchi, M.; Miyamoto, S.; Yamamoto, M.; Motoyama, A.; Hosoi, J.; Shimokata, T.; Ito, T.; Tsuboi, R.; Huh, N.-H. S100A9 Is a Novel Ligand of EMMPRIN That Promotes Melanoma Metastasis. Cancer Res. 2013, 73, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Ruma, I.M.; Putranto, E.W.; Kondo, E.; Murata, H.; Watanabe, M.; Huang, P.; Kinoshita, R.; Futami, J.; Inoue, Y.; Yamauchi, A.; et al. MCAM, as a novel receptor for S100A8/A9, mediates progression of malignant melanoma through prominent activation of NF-κB and ROS formation upon ligand binding. Clin. Exp. Metastasis 2016, 33, 609–627. [Google Scholar] [CrossRef]
- Ghavami, S.; Chitayat, S.; Hashemi, M.; Eshraghi, M.; Chazin, W.J.; Halayko, A.J.; Kerkhoff, C. S100A8/A9: A Janus-faced molecule in cancer therapy and tumorgenesis. Eur. J. Pharmacol. 2009, 625, 73–83. [Google Scholar] [CrossRef]
- Ghavami, S.; Rashedi, I.; Dattilo, B.M.; Eshraghi, M.; Chazin, W.J.; Hashemi, M.; Wesselborg, S.; Kerkhoff, C.; Los, M. S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J. Leukoc. Biol. 2008, 83, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Turovskaya, O.; Foell, D.; Sinha, P.; Vogl, T.; Newlin, R.; Nayak, J.; Nguyen, M.; Olsson, A.; Nawroth, P.P.; Bierhaus, A.; et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis 2008, 29, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Grebhardt, S.; Veltkamp, C.; Ströbel, P.; Mayer, D. Hypoxia and HIF-1 increase S100A8 and S100A9 expression in prostate cancer. Int. J. Cancer 2012, 131, 2785–2794. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Domain architecture of HIF-α and HIF-1β subunits. All subunits contain bHLH and twin PAS domains. These domains contain the DNA binding and heterodimerization sites. All three HIF-α forms possess an oxygen-dependent degradation (ODD) domain. HIF-1α and HIF-2α possess two transactivation domains (N-TAD and C-TAD). N-TAD is responsible for stabilizing all three forms of HIF-α under hypoxic conditions, while C-TAD controls the transcription of HIF-α target genes. The N-TAD domains are part of the ODD domains. HIF-3α possesses a leucine zipper (LZIP) domain instead of a C-TAD domain. HIF-1β lacks both the ODD and N-TAD domains.
Figure 1.
Domain architecture of HIF-α and HIF-1β subunits. All subunits contain bHLH and twin PAS domains. These domains contain the DNA binding and heterodimerization sites. All three HIF-α forms possess an oxygen-dependent degradation (ODD) domain. HIF-1α and HIF-2α possess two transactivation domains (N-TAD and C-TAD). N-TAD is responsible for stabilizing all three forms of HIF-α under hypoxic conditions, while C-TAD controls the transcription of HIF-α target genes. The N-TAD domains are part of the ODD domains. HIF-3α possesses a leucine zipper (LZIP) domain instead of a C-TAD domain. HIF-1β lacks both the ODD and N-TAD domains.
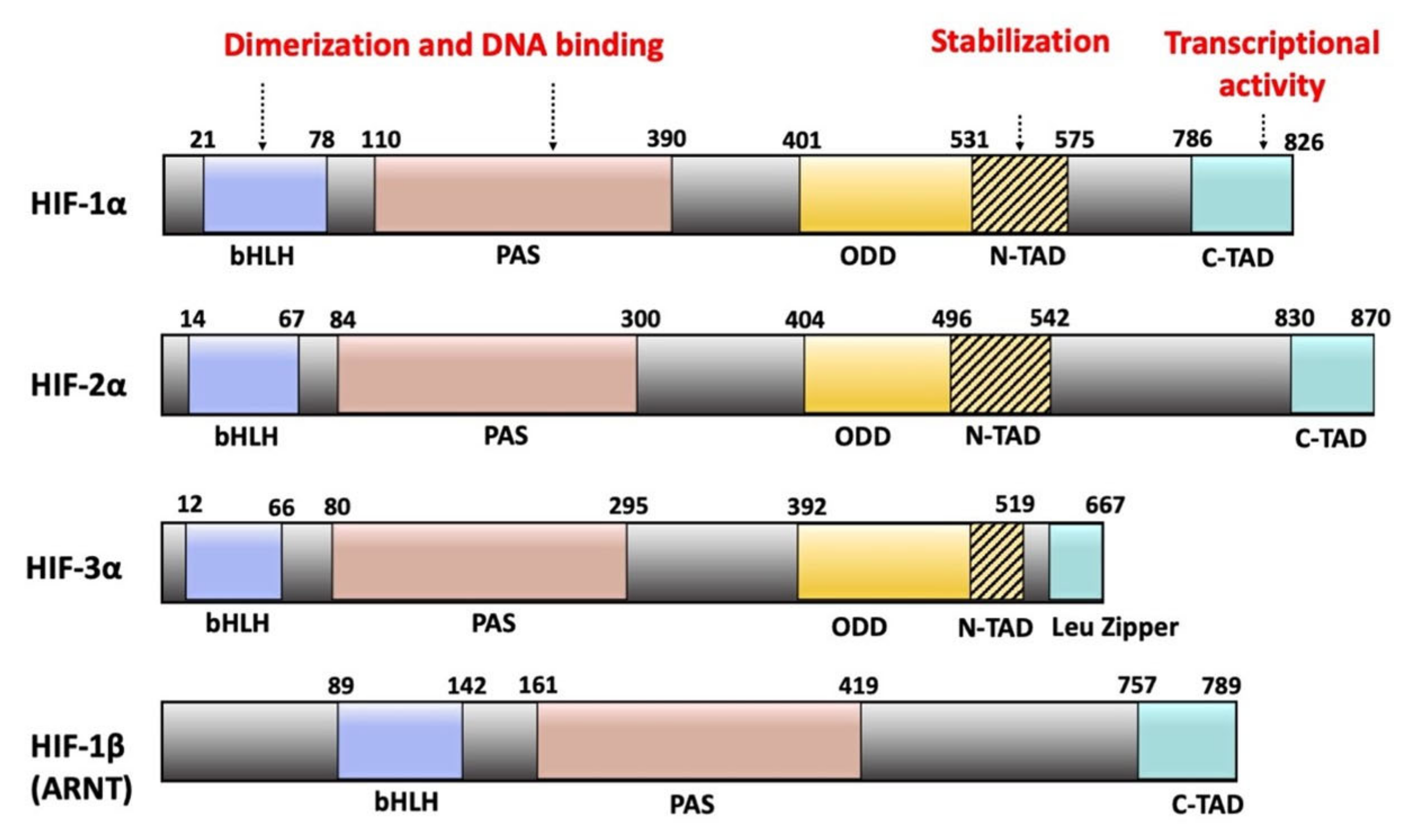
Figure 2.
RAGE structure and isoforms. Full-length RAGE consists of three regions: an extracellular part itself divided into a variable-like (V), two constant-like Ig (C1 and C2) domains, a transmembrane domain, and a cytoplasmic domain. The soluble form of RAGE (sRAGE) contains the extracellular region but lacks both the transmembrane and cytoplasmic domains. The dominant negative isoform lacks the cytoplasmic domain, while N-truncated RAGE is deficient in the N-terminal V-like domain.
Figure 2.
RAGE structure and isoforms. Full-length RAGE consists of three regions: an extracellular part itself divided into a variable-like (V), two constant-like Ig (C1 and C2) domains, a transmembrane domain, and a cytoplasmic domain. The soluble form of RAGE (sRAGE) contains the extracellular region but lacks both the transmembrane and cytoplasmic domains. The dominant negative isoform lacks the cytoplasmic domain, while N-truncated RAGE is deficient in the N-terminal V-like domain.
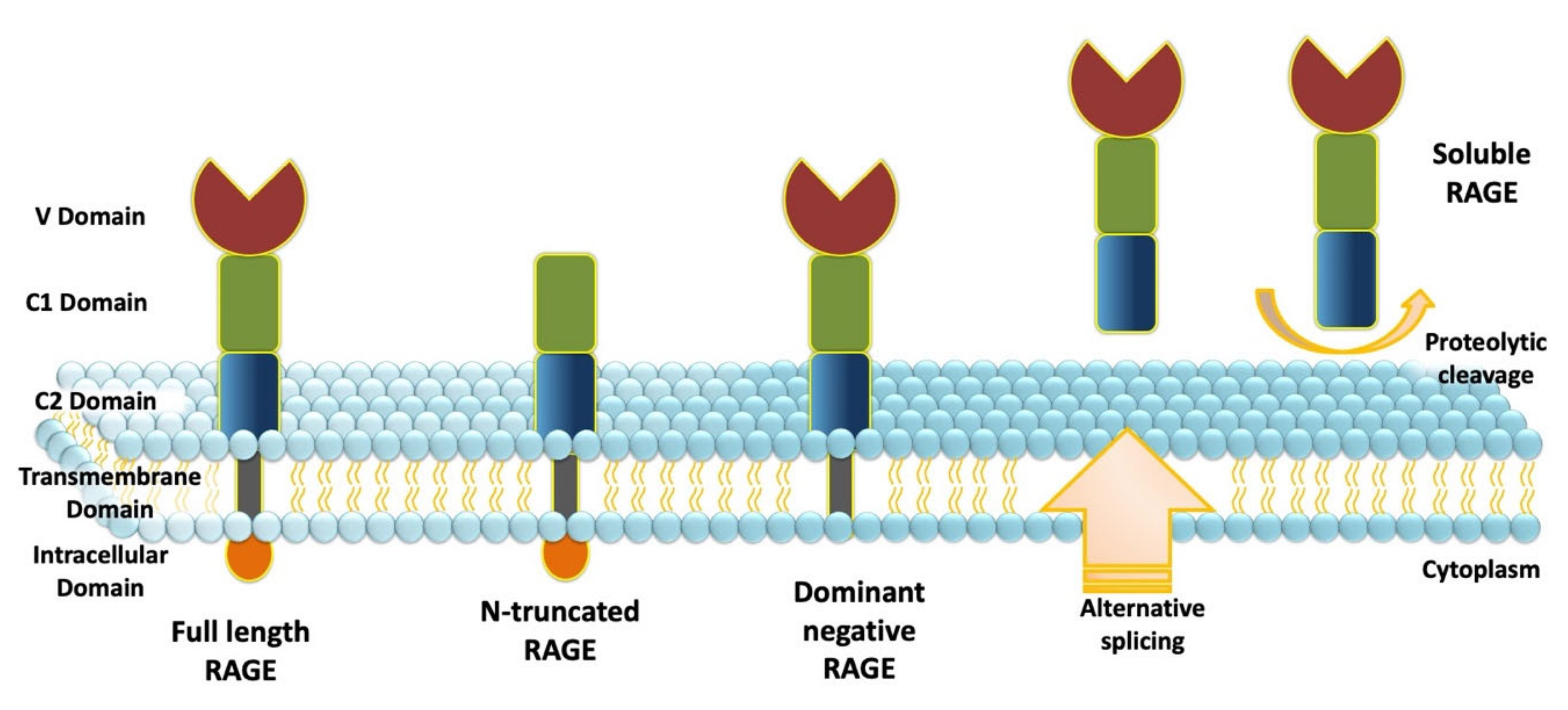
Figure 3.
RAGE signaling pathways in normoxia and hypoxia. (A) In normoxia, RAGE activation by its ligands results in the activation of the MEK/ERK, PI3K/Akt, and JAK/STAT pathways, resulting in gene expression through the activation of NF-kB and STAT3. Because the expression of RAGE is under the control of NF-kB, RAGE activation results in RAGE upregulation through positive feedback. (B) In hypoxia, RAGE activation has been shown to stimulate HIF-1α through the activation of the PI3K/Akt and the RAS/RAF/MEK/ERK pathways. In hypoxia, nuclear HMGB1 translocates to the extracellular space through an unknown mechanism and interacts with RAGE. S100A4 and S100A8/A9 are upregulated and secreted in the tumor microenvironment and interact with RAGE. RAGE interaction with HMGB1, S100A4, and S100A8/A9 results in the activation of the MEK/ERK and PI3K/Akt pathways, resulting in changes in cancer-related gene expressions through the activation of NF-kB. NF-kB can further induce HIF-1a expression.
Figure 3.
RAGE signaling pathways in normoxia and hypoxia. (A) In normoxia, RAGE activation by its ligands results in the activation of the MEK/ERK, PI3K/Akt, and JAK/STAT pathways, resulting in gene expression through the activation of NF-kB and STAT3. Because the expression of RAGE is under the control of NF-kB, RAGE activation results in RAGE upregulation through positive feedback. (B) In hypoxia, RAGE activation has been shown to stimulate HIF-1α through the activation of the PI3K/Akt and the RAS/RAF/MEK/ERK pathways. In hypoxia, nuclear HMGB1 translocates to the extracellular space through an unknown mechanism and interacts with RAGE. S100A4 and S100A8/A9 are upregulated and secreted in the tumor microenvironment and interact with RAGE. RAGE interaction with HMGB1, S100A4, and S100A8/A9 results in the activation of the MEK/ERK and PI3K/Akt pathways, resulting in changes in cancer-related gene expressions through the activation of NF-kB. NF-kB can further induce HIF-1a expression.
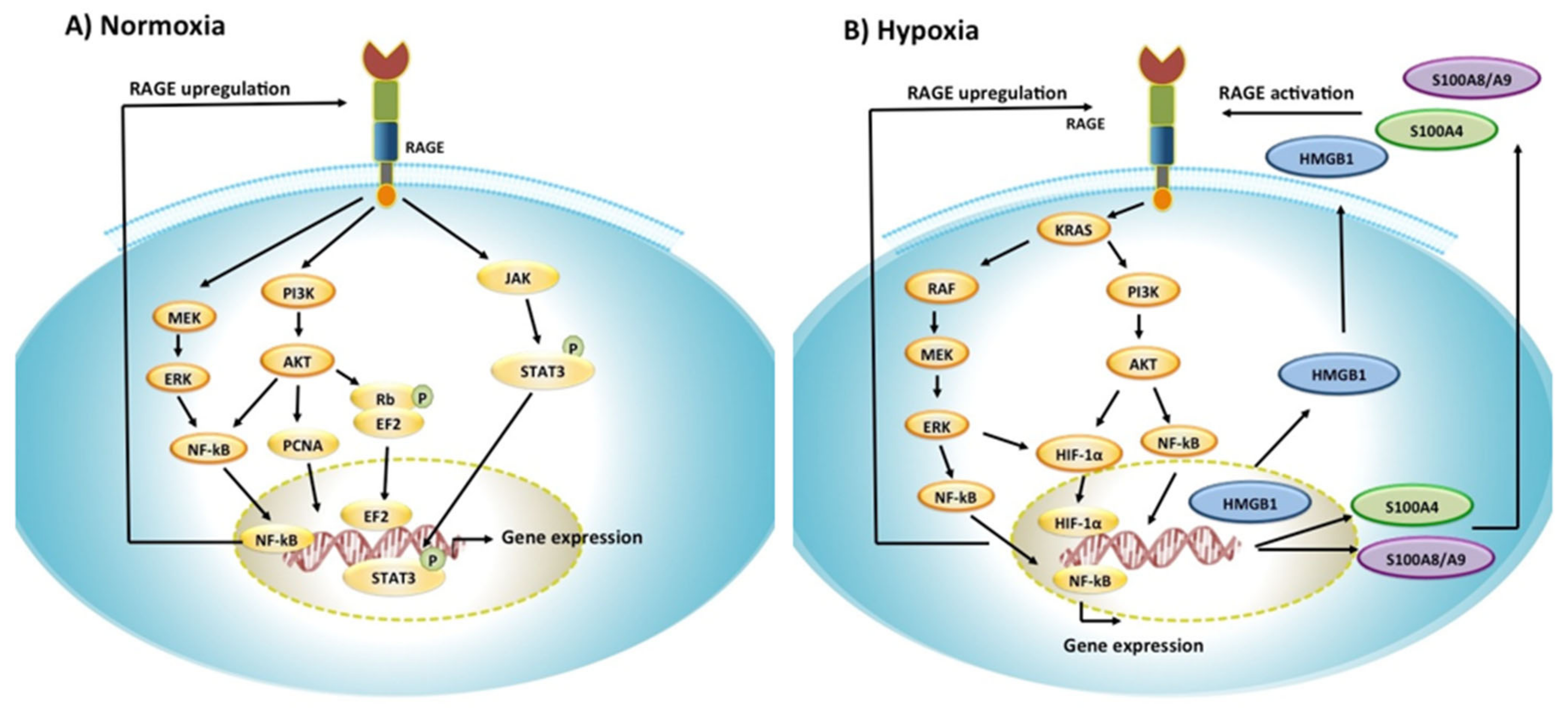

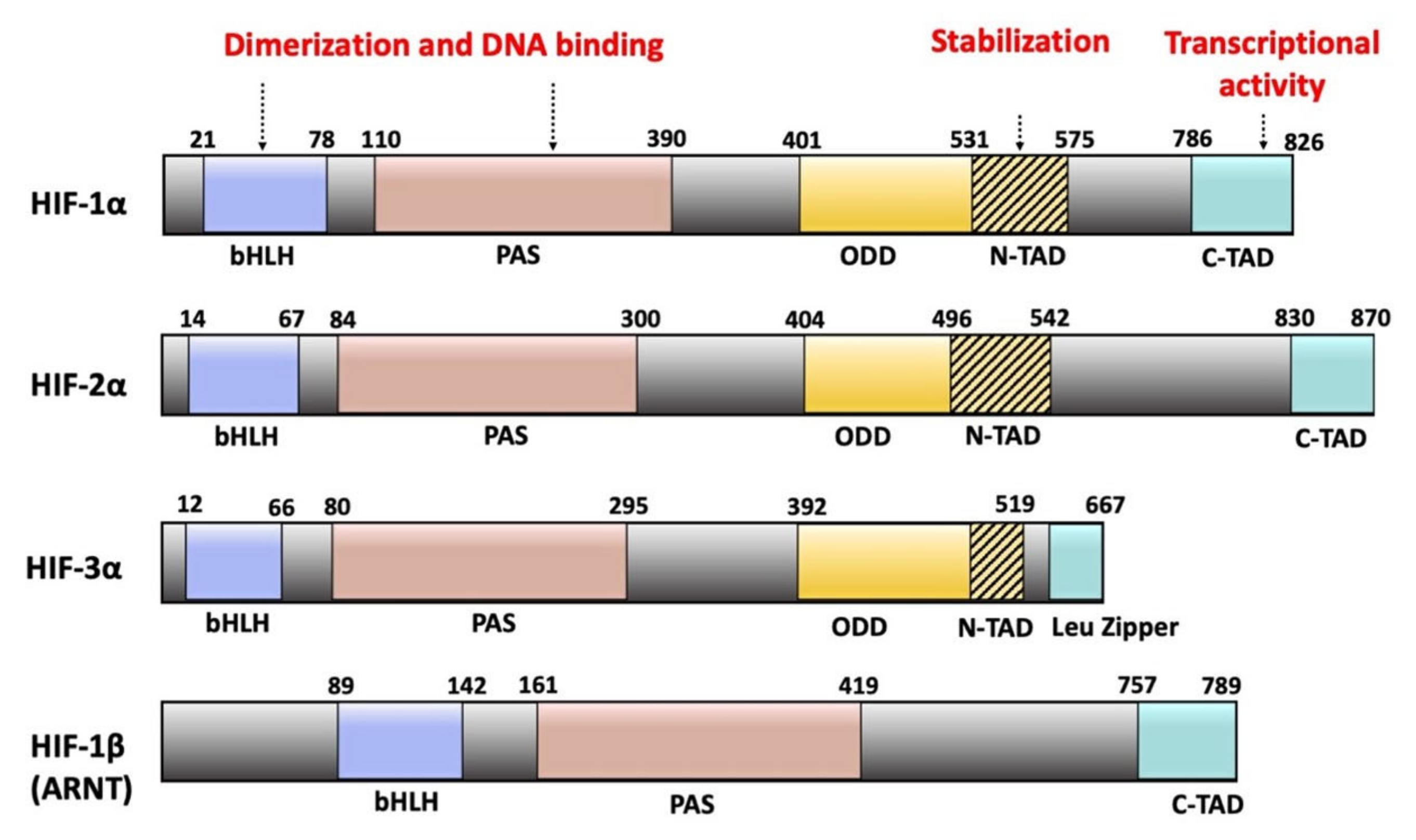{kind=link}
{kind=link}
{kind=link}
Table 1.
Effect of hypoxia on RAGE ligand levels or RAGE/ligand signaling.
| Ligands | Cancer Type | Pathological Response | Reference |
|---|---|---|---|
| HMGB1 | Melanoma | Under hypoxia, melanoma cells release HMGB1, which promotes melanoma growth and metastasis | [150] |
| Hepatocellular carcinoma | HMGB1 released under hypoxia promotes cellular invasion and metastasis | [177,179] | |
| Glioblastoma | HMGB1 released under hypoxia promotes cell proliferation and invasion | [180] | |
| S100B | Colon adenocarcinoma | Stimulation of RAGE by S100B activates HIF1-α and the expression of HIF-1α dependent genes | [190] |
| S100P | Hepatocellular carcinoma | Hypoxia upregulates S100P and further promotes metastasis of HCC | [196] |
| S100A4 | Colorectal Cancer | Activation of RAGE by S100A4 results in increased levels of HIF-1α | [197] |
| Gastric, ovarian cancer | Hypoxia upregulates S100A4 | [205] | |
| S100A7 | Breast Cancer | Activation of RAGE by S100A7 results in increased angiogenesis | [13] |
| S100A8/A9 | Prostate Cancer | Hypoxia upregulates S100A8 and S100A9 transcript and protein levels | [223] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Taneja, S.; Vetter, S.W.; Leclerc, E. Hypoxia and the Receptor for Advanced Glycation End Products (RAGE) Signaling in Cancer. Int. J. Mol. Sci. 2021, 22, 8153. https://doi.org/10.3390/ijms22158153
AMA Style
Taneja S, Vetter SW, Leclerc E. Hypoxia and the Receptor for Advanced Glycation End Products (RAGE) Signaling in Cancer. International Journal of Molecular Sciences. 2021; 22(15):8153. https://doi.org/10.3390/ijms22158153
Chicago/Turabian StyleTaneja, Sakshi, Stefan W. Vetter, and Estelle Leclerc. 2021. "Hypoxia and the Receptor for Advanced Glycation End Products (RAGE) Signaling in Cancer" International Journal of Molecular Sciences 22, no. 15: 8153. https://doi.org/10.3390/ijms22158153
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.