
Neuroprotective Function of High Glycolytic Activity in Astrocytes: Common Roles in Stroke and Neurodegenerative Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
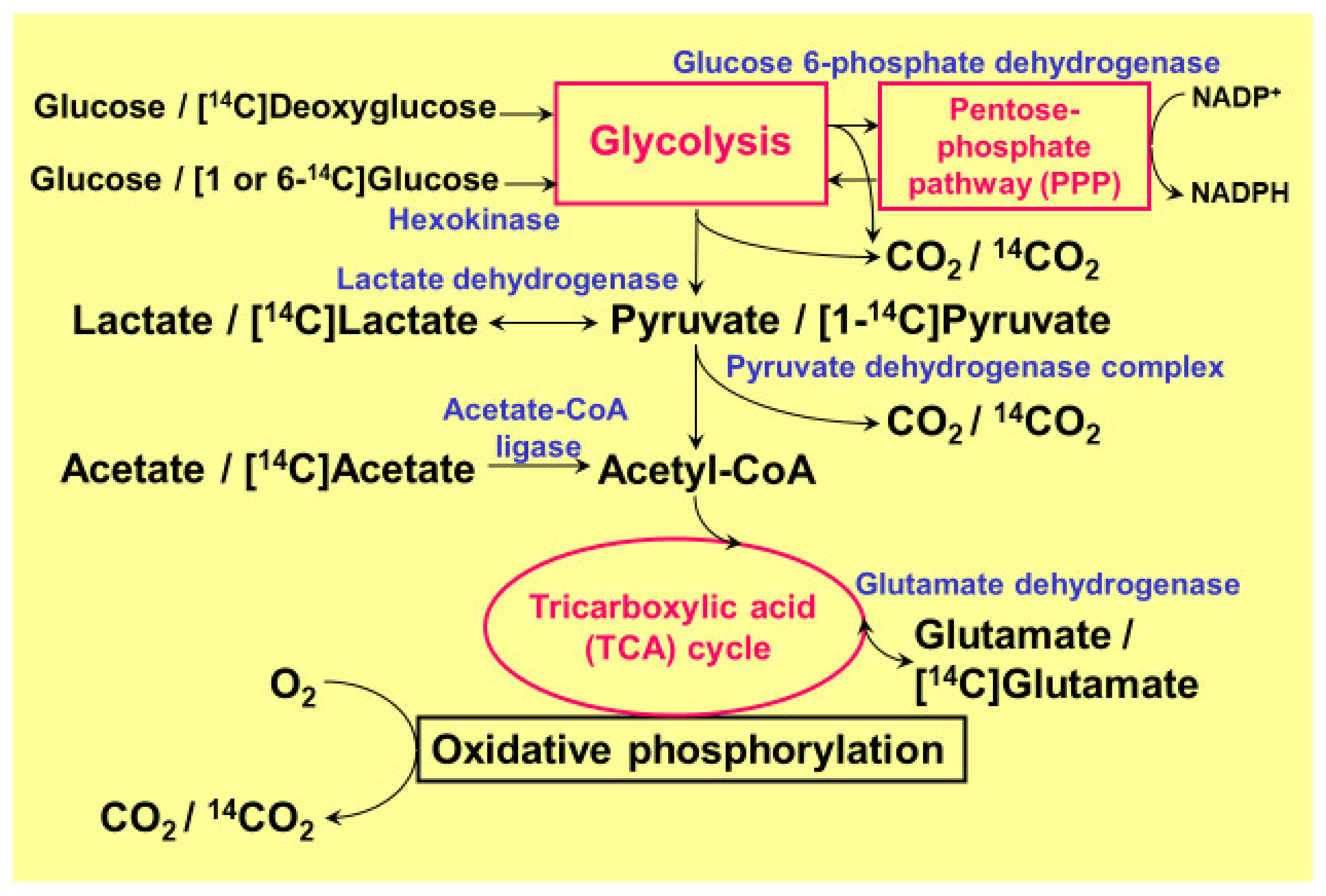
2. Pentose-Phosphate Pathway in Astrocytes
3. Activation of Astrocytic Pentose-Phosphate Pathway under Altered Supply of Glucose
4. Effects of Brain Ischemia on Astroglial PPP
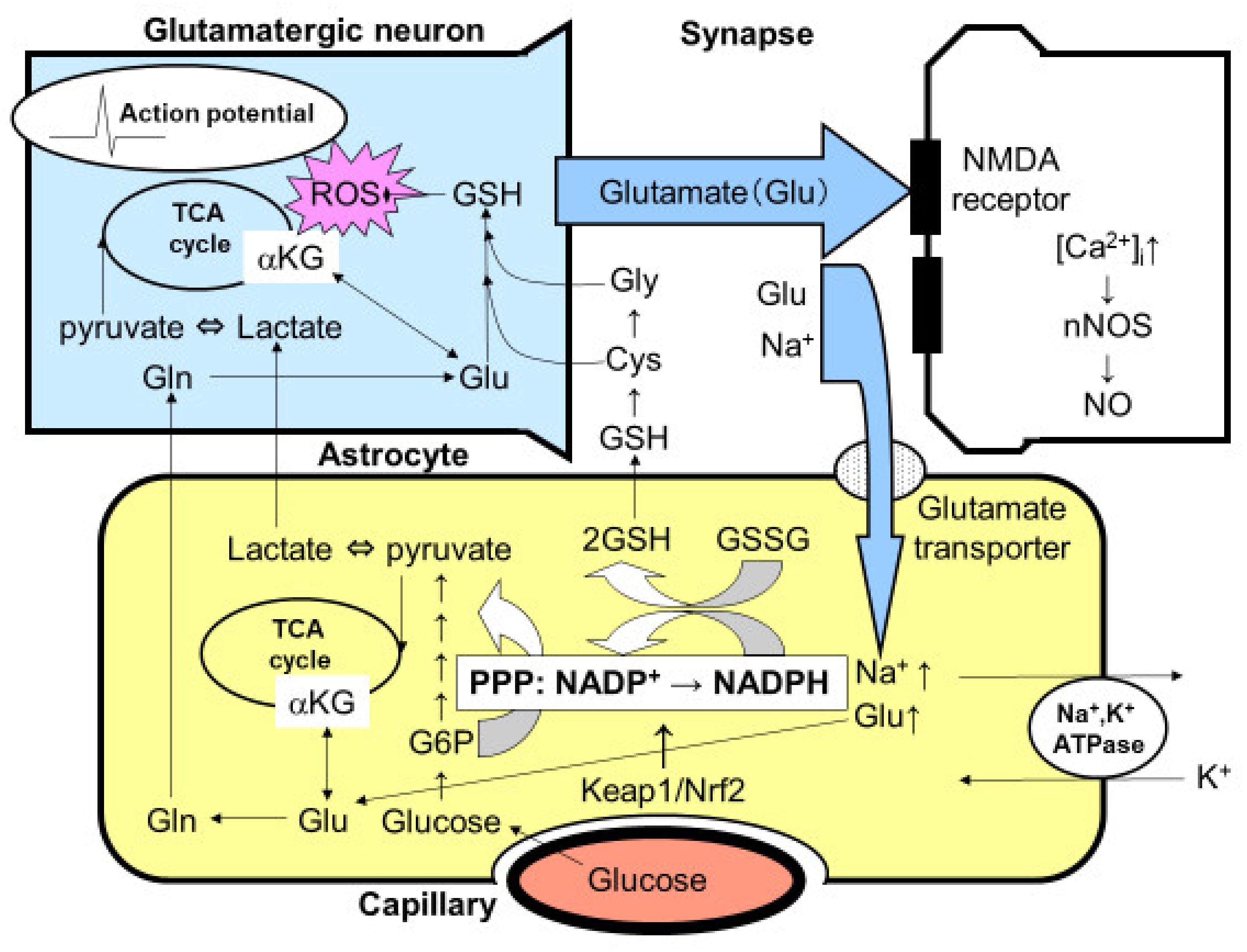
5. Glutamate Released from Neurons Triggers Increased PPP Flux in Astroglia
6. Fatty Acid Oxidation under Hypoxia and Reoxygenation
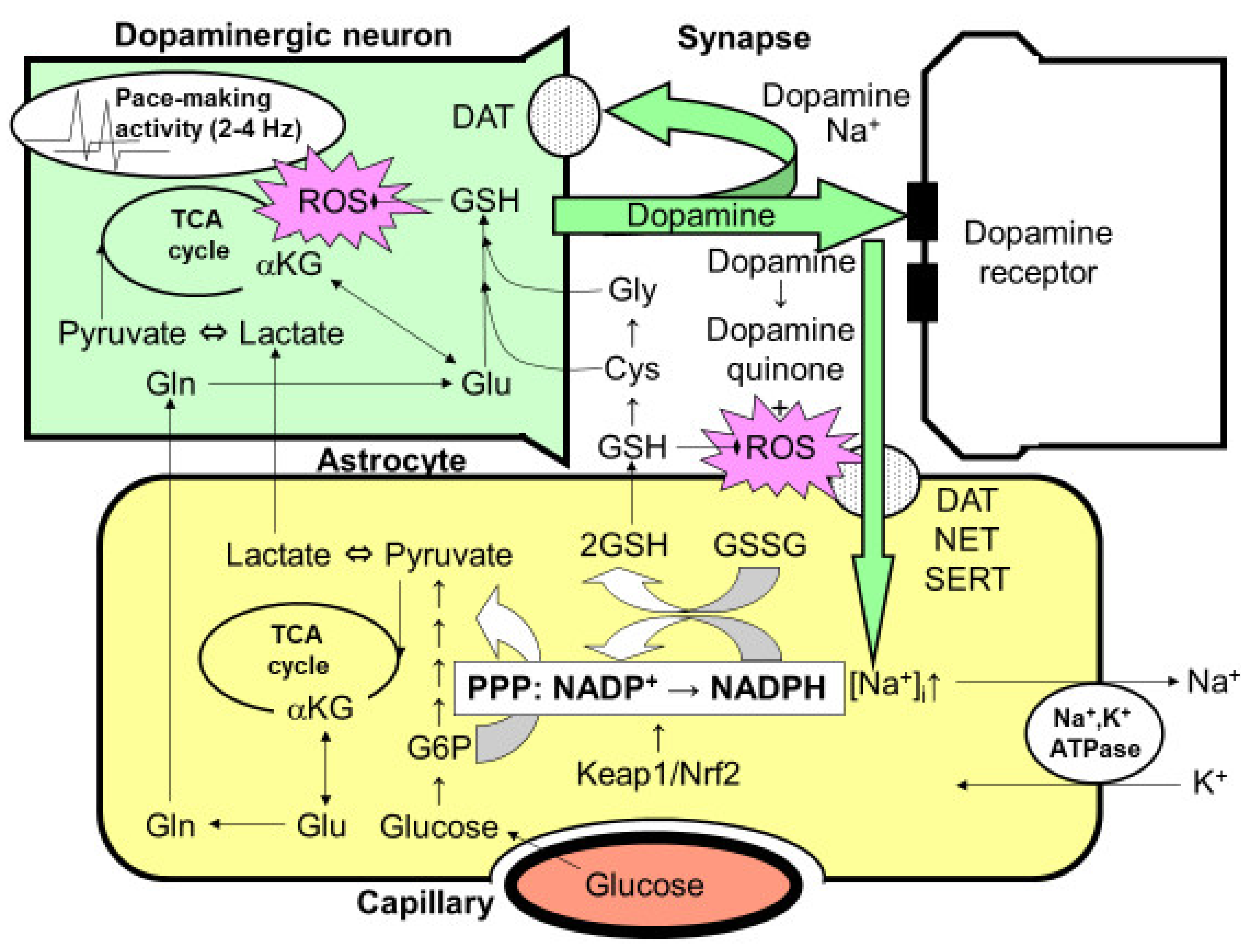
7. Activation of Pentose-Phosphate Pathway with Dopamine Metabolism
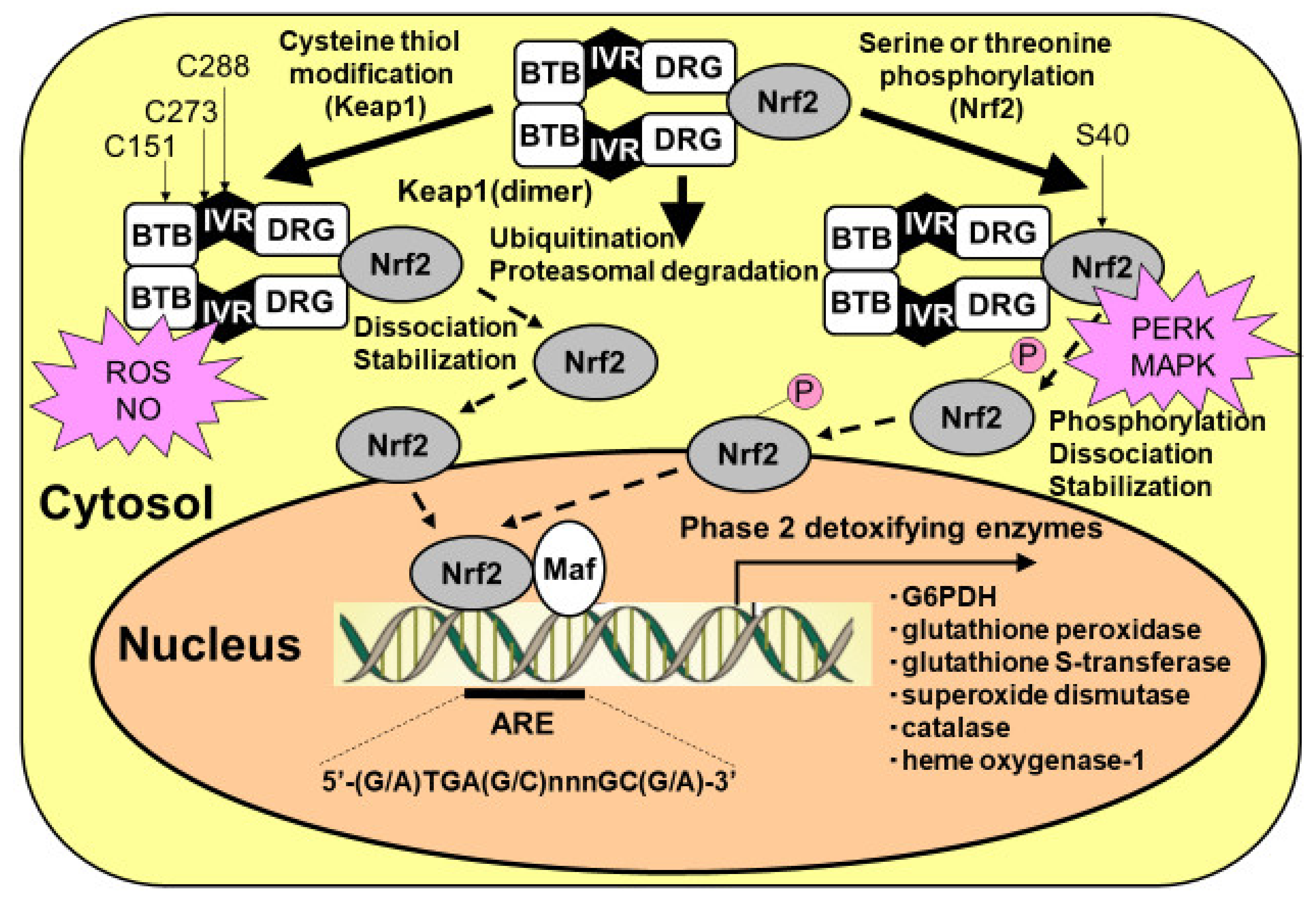
8. Regulation of PPP Flux by G6PDH Transcription by Nrf2
9. Dopamine-Induced Activation of Astroglial PPP
10. Toll-Like Receptor 4 and Keap1/Nrf2 under Ischemia
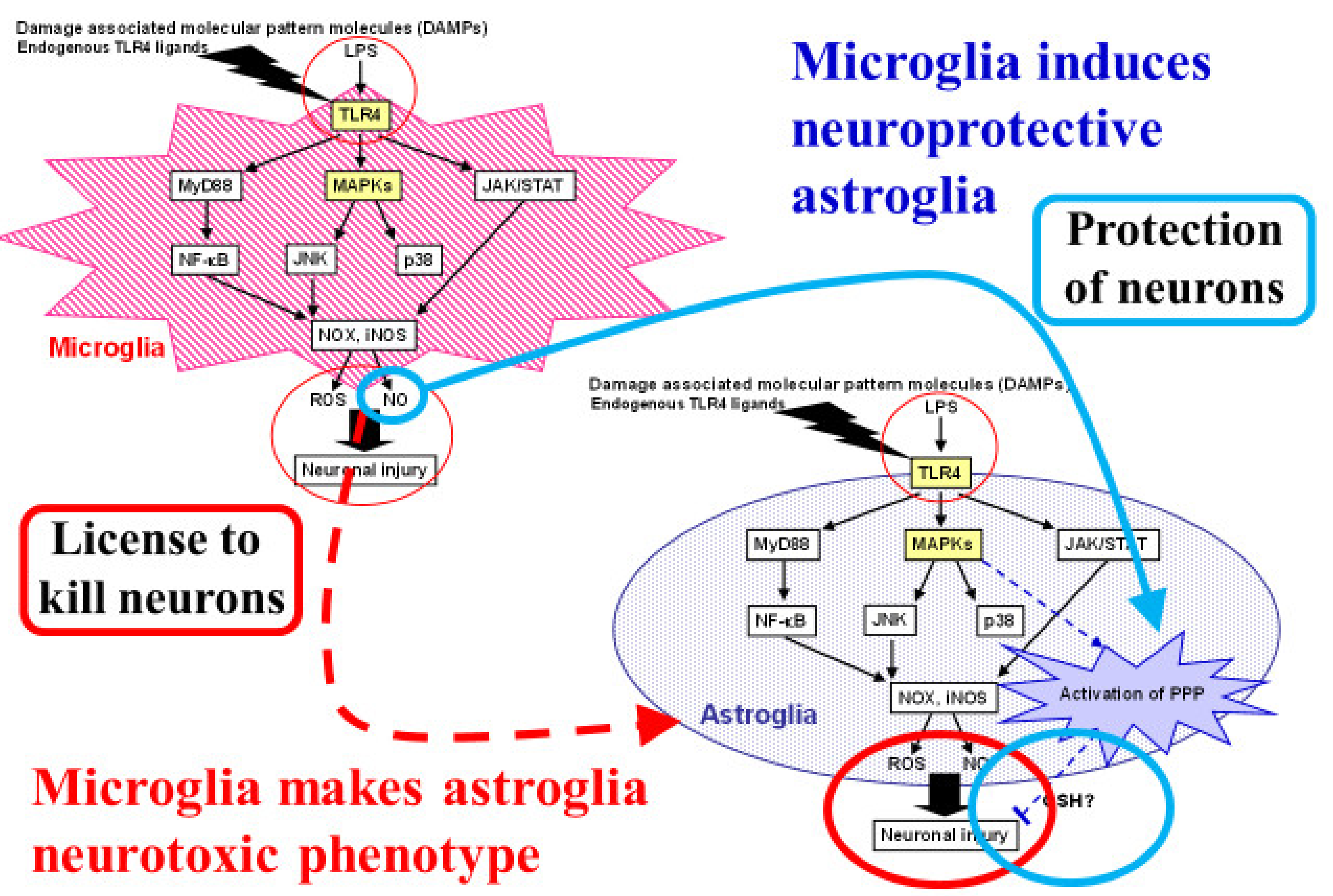
11. Interaction between Microglia and Astroglia
12. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviation
| AMPK | adenosine monophosphate-dependent kinase |
| ATP | adenosine triphosphate |
| ALS | amyotrophic lateral sclerosis |
| ARE | antioxidant response element |
| AQP4) | aquaporin 4 |
| BHB | β-hydroxybutyrate |
| CO2 | carbon dioxide |
| DAMPs | damage-associated molecular patterns |
| DMF | dimethyl fumarate |
| ER | endoplasmic reticulum |
| ECM | extracellular matrix |
| ERK1/2 | extracellular-signal regulated kinase 1/2 |
| G6P | glucose 6-phosphate |
| G6PDH | glucose 6-phosphate dehydrogenase |
| HBP | hexosamine biosynthetic pathway |
| iPSC | induced pluripotent stem cell |
| iNOS | inducible nitric oxide synthase |
| JAK/STAT | Janus kinases/signal transducers and activators of transcription |
| Keap1 | Kelch-like enoyl-CoA hydratase-associated protein 1 |
| LPS | lipopolysaccharide |
| MMP | matrix metalloproteinase |
| MAPK | mitogen-activated protein kinase |
| MCTs | monocarboxylate transporters |
| Myd88-NFκB | myeloid differentiation protein 88-nuclear factor-κB |
| NOX | NADPH oxidase |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| Nrf2 | nuclear factor erythroid 2 p45 subunit-related factor 2 |
| GSSG | oxidized form of glutathione |
| PPP | pentose-phosphate pathway |
| PPARγ | peroxisome-proliferator activated receptor γ |
| PKA | protein kinase-A |
| PERK | protein kinase RNA(PKR)-like ER kinase |
| PDHC | pyruvate dehydrogenase complex |
| RNSs | reactive nitrogen species |
| ROSs | reactive oxygen species |
| GSH | reduced form of glutathione |
| TLR4 | toll-like receptor 4 |
| TCA | tricarboxylic acid |
References
- Clarke, D.D.; Sokoloff, L. Circulation and energy metabolism of the brain. In Basic Neurochemistry: Molecular, Cellular, and Medical Aspects, 6th ed.; Siegel, G., Agranoff, B., Albers, R.W., Fisher, S., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999; pp. 637–669. [Google Scholar]
- Dienel, G.A. Energy metabolism in the brain. In From Molecules to Networks: An Introduction to Cellular and Molecular Neuroscience, 2nd ed.; Byrne, J.H., Roberts, J.L., Eds.; Academic Press: London, UK, 2009; pp. 49–110. [Google Scholar]
- Takahashi, S. Astrogliopathy as a loss of astroglial protective function against glycoxidative stress under hyperglycemia. Rinsho Shinkeigaku Clin. Neurol. 2012, 52, 41–51. [Google Scholar] [CrossRef]
- Takahashi, S. Metabolic compartmentalization between astroglia and neurons in physiological and pathophysiological conditions of neurovascular unit. Neuropathology 2020, 40, 121–137. [Google Scholar] [CrossRef]
- Takahashi, S. Lactate and ketone bodies act as energy substrates as well as signal molecules in the brain. In Psychology and Patho-physiological Outcomes of Eating; IntechOpen: London, UK, 2021; pp. 1–20. [Google Scholar]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M.; Farbood, Y.; Moghaddam, H.F. Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 2017, 38, 1167–1186. [Google Scholar] [CrossRef] [PubMed]
- Barthels, D.; Das, H. Current advances in ischemic stroke research and therapies. Biochim. Biophys. Acta. Mol. Basis. Dis. 2020, 1866, 165260. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Shinbo, S.; Asahi, A.; Imanaka, T. Very long chain fatty acid beta-oxidation in astrocytes: Contribution of the ABCD1-dependent and -independent pathways. Biol. Pharm. Bull. 2012, 35, 1972–1979. [Google Scholar] [CrossRef]
- Di Cesare Mannelli, L.; Zanardelli, M.; Micheli, L.; Ghelardini, C. PPAR-γ impairment alters peroxisome functionality in primary astrocyte cell cultures. Biomed. Res. Int. 2014, 2014, 546453. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet. Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Sun, M.S.; Jin, H.; Sun, X.; Huang, S.; Zhang, F.L.; Guo, Z.N.; Yang, Y. Free Radical Damage in Ischemia-Reperfusion Injury: An Obstacle in Acute Ischemic Stroke after Revascularization Therapy. Oxid. Med. Cell. Longev. 2018, 2018, 3804979. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Urzúa, S.; Rojas, I.; Líbano, L.; Rodrigo, R. Pathophysiology of Ischemic Stroke: Role of Oxidative Stress. Curr. Pharm. Des. 2020, 26, 4246–4260. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Tanaka, M.; Yuki, S.; Hirai, M.; Yamamoto, Y. How is edaravone effective against acute ischemic stroke and amyotrophic lateral sclerosis? J. Clin. Biochem. Nutr. 2018, 62, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Watanabe, K.; Yuki, S.; Akimoto, M.; Sakata, T.; Palumbo, J. Edaravone and its clinical development for amyotrophic lateral sclerosis. Amyotroph Lateral Scler Front. Degener. 2017, 18, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yuan, R.; Wang, Y.; Wei, C.; Zhang, S.; Yang, X.; Wu, B.; Liu, M. Early Prediction of Malignant Brain Edema after Ischemic Stroke. Stroke 2018, 49, 2918–2927. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kuroda, Y.; Yamashita, S.; Zhang, X.; Miyamoto, O.; Tamiya, T.; Nagao, S.; Xi, G.; Keep, R.F.; Itano, T. Edaravone attenuates brain edema and neurologic deficits in a rat model of acute intracerebral hemorrhage. Stroke 2008, 39, 463–469. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799.e19. [Google Scholar] [CrossRef]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys Acta Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef]
- Somjen, G.G. Nervenkitt: Notes on the history of the concept of neuroglia. Glia 1988, 1, 2–9. [Google Scholar] [CrossRef]
- Allen, N.J.; Barres, B.A. Neuroscience: Glia—More than just brain glue. Nature 2009, 457, 675–677. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Driscoll, B.F.; Law, M.J.; Sokoloff, L. Role of sodium and potassium ions in regulation of glucose metabolism in cultured astroglia. Proc. Natl. Acad. Sci. USA 1995, 92, 4616–4620. [Google Scholar] [CrossRef]
- Mazuel, L.; Blanc, J.; Repond, C.; Bouchaud, V.; Raffard, G.; Déglon, N.; Bonvento, G.; Pellerin, L.; Bouzier-Sore, A.K. A neuronal MCT2 knockdown in the rat somatosensory cortex reduces both the NMR lactate signal and the BOLD response during whisker stimulation. PLoS ONE 2017, 12, e0174990. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. The metabolic trinity, glucose-glycogen-lactate, links astrocytes and neurons in brain energetics, signaling, memory, and gene expression. Neurosci. Lett. 2017, 637, 18–25. [Google Scholar] [CrossRef]
- Dienel, G.A. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J. Neurosci. Res. 2017, 95, 2103–2125. [Google Scholar] [CrossRef]
- Pellerin, L. Neuroenergetics: Astrocytes Have a Sweet Spot for Glucose. Curr Biol. 2018, 28, R1258–R1260. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- Souza, D.G.; Almeida, R.F.; Souza, D.O.; Zimmer, E.R. The astrocyte biochemistry. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2019; Volume 95, pp. 142–150. [Google Scholar]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola de Freitas, A.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed]
- Bolaños, J.P. Bioenergetics and redox adaptations of astrocytes to neuronal activity. J. Neurochem. 2016, 139, 115–125. [Google Scholar] [CrossRef]
- Tang, B.L. Neuroprotection by glucose-6-phosphate dehydrogenase and the pentose phosphate pathway. J. Cell. Biochem. 2019, 120, 14285–14295. [Google Scholar] [CrossRef]
- Abe, T.; Takahashi, S.; Suzuki, N. Oxidative metabolism in cultured rat astroglia: Effects of reducing the glucose concentration in the culture medium and of D-aspartate or potassium stimulation. J. Cereb. Blood. Flow. Metab. 2006, 26, 153–160. [Google Scholar] [CrossRef]
- Takahashi, S. Energy metabolism of neurons and astroglia. Cereb. Blood Flow Metab. 2010, 21, 18–27. [Google Scholar]
- Takahashi, S. Astroglial protective mechanisms against ROS under brain ischemia. Rinsho Shinkeigaku 2011, 51, 1032–1035. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Izawa, Y.; Iizumi, T.; Abe, T.; Suzuki, N. Toll-like receptor 4 activation induced by LPS in combination with hypoxia increases PPP flux in astroglia and reduces oxidative stress. Cereb. Blood Flow Metab. 2012, 23, 66–74. [Google Scholar]
- Takahashi, S.; Izawa, Y.; Suzuki, N. Astroglial pentose phosphate pathway rates in response to high-glucose environments. ASN Neuro. 2012, 4, e00078. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Iizumi, T.; Mashima, K.; Abe, T.; Suzuki, N. Roles and regulation of ketogenesis in cultured astroglia and neurons under hypoxia and hypoglycemia. ASN Neuro 2014, 6, 1759091414550997. [Google Scholar] [CrossRef]
- Iizumi, T.; Takahashi, S.; Mashima, K.; Minami, K.; Izawa, Y.; Abe, T.; Hishiki, T.; Suematsu, M.; Kajimura, M.; Suzuki, N. A possible role of microglia-derived nitric oxide by lipopolysaccharide in activation of astroglial pentose-phosphate pathway via the Keap1/Nrf2 system. J. Neuroinflammation 2016, 13, 99. [Google Scholar] [CrossRef]
- Mashima, K.; Takahashi, S.; Minami, K.; Izawa, Y.; Abe, T.; Tsukada, N.; Hishiki, T.; Suematsu, M.; Kajimura, M.; Suzuki, N. Neuroprotective Role of Astroglia in Parkinson Disease by Reducing Oxidative Stress Through Dopamine-Induced Activation of Pentose-Phosphate Pathway. ASN Neuro 2018, 10, 1759091418775562. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Yadav, S. The glutathione cycle: Glutathione metabolism beyond the gamma-glutamyl cycle. IUBMB Life 2018, 70, 585–592. [Google Scholar] [CrossRef]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, D.; Megha, K.; Mishra, R.; Mandal, P.K. Glutathione in Brain: Overview of Its Conformations, Functions, Biochemical Characteristics, Quantitation and Potential Therapeutic Role in Brain Disorders. Neurochem. Res. 2020, 45, 1461–1480. [Google Scholar] [CrossRef]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Takahashi, S.; Abe, T.; Izawa, Y.; Suzuki, N. Effects of fluctuating glucose concentrations on oxidative metabolism of glucose in cultured neurons and astroglia. J. Diabetes Mellit. 2012, 2, 19–26. [Google Scholar] [CrossRef]
- Goyal, M.; Menon, B.K.; van Zwam, W.H.; Dippel, D.W.; Mitchell, P.J.; Demchuk, A.M.; Dávalos, A.; Majoie, C.B.; van der Lugt, A.; de Miquel, M.A.; et al. HERMES collaborators: Endovascular thrombectomy after large-vessel ischaemic stroke: A meta-analysis of individual patient data from five randomised trials. Lancet 2016, 387, 1723–1731. [Google Scholar] [CrossRef]
- Lo, E.H.; Dalkara, T.; Moskowitz, M.A. Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 2003, 4, 399–415. [Google Scholar] [CrossRef]
- Del Zoppo, G.J. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 2009, 158, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Del Zoppo, G.J. Relationship of neurovascular elements to neuron injury during ischemia. Cereb. Dis. 2009, 27, 65–76. [Google Scholar] [CrossRef]
- Takahashi, S. Treatment of acute ischemic stroke: Tissue clock and reperfusion. Masui 2012, 61, S11–S22. [Google Scholar]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Brown, J.E.; Bill, R.M.; Conner, A.C.; Conner, M.T. Hypothermia increases aquaporin 4 (AQP4) plasma membrane abundance in human primary cortical astrocytes via a calcium/transient receptor potential vanilloid 4 (TRPV4)- and calmodulin-mediated mechanism. Eur. J. Neurosci. 2017, 46, 2542–2547. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Bill, R.M.; Conner, A.C.; Heath, P.R.; Conner, M.T. Transcriptome Analysis of Gene Expression Provides New Insights into the Effect of Mild Therapeutic Hypothermia on Primary Human Cortical Astrocytes Cultured under Hypoxia. Front. Cell. Neurosci. 2017, 11, 386. [Google Scholar] [CrossRef] [PubMed]
- Yenari, M.A.; Han, H.S. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat. Rev. Neurosci. 2012, 13, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Binvignat, O.; Olloquequi, J. Excitotoxicity as a Target against Neurodegenerative Processes. Curr. Pharm. Des. 2020, 26, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.O.; Hsueh, K.; Park, H.T.; Sauceda, C.; Hwang, V.; Kumar, D.; Kim, S.; Rickert, E.; Mahata, S.; Webster, N.J.G. Astrocyte-Specific Deletion of Peroxisome-Proliferator Activated Receptor-gamma Impairs Glucose Metabolism and Estrous Cycling in Female Mice. J. Endocr. Soc. 2017, 1, 1332–1350. [Google Scholar] [CrossRef]
- Izawa, Y.; Takahashi, S.; Suzuki, N. Pioglitazone enhances pyruvate and lactate oxidation in cultured neurons but not in cultured astroglia. Brain Res. 2009, 1305, 64–73. [Google Scholar] [CrossRef]
- Hattoria, N.; Wanga, M.; Taka, H.; Fujimura, T.; Yoritaka, A.; Kubo, S.; Mochizuki, H. Toxic effects of dopamine metabolism in Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, S35–S38. [Google Scholar] [CrossRef]
- Jamwal, S.; Kumar, P. Insight into the Emerging Role of Striatal Neurotransmitters in the Pathophysiology of Parkinson’s Disease and Huntington’s Disease: A Review. Curr. Neuropharmacol. 2019, 17, 165–175. [Google Scholar] [CrossRef]
- Iovino, L.; Tremblay, M.E.; Civiero, L. Glutamate-induced excitotoxicity in Parkinson’s disease: The role of glial cells. J. Pharmacol. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef]
- Kunze, R. Dimethyl fumarate for ischemic stroke. Oncotarget 2017, 8, 14281–14282. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharm. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Scuderi, S.A.; Ardizzone, A.; Paterniti, I.; Esposito, E.; Campolo, M. Antioxidant and Anti-inflammatory Effect of Nrf2 Inducer Dimethyl Fumarate in Neurodegenerative Diseases. Antioxidants 2020, 9, 630. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. BEAM Study Investigators: Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. BEACON Trial Investigators: Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Yamawaki, K. Bardoxolone methyl: Drug development for diabetic kidney disease. Clin. Exp. Nephrol. 2020, 24, 857–864. [Google Scholar] [CrossRef]
- Takahashi, S.; Seki, M.; Suzuki, N. Roles of metabolic compartmentalization by astrocytes and neurons in the pathophysiology and treatment of Parkinson’s disease. Brain Nerve 2013, 65, 1497–1508. [Google Scholar]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, Z.; Xie, L.J.; Liu, G.F. Suppression of TLR4/NF-κB Signaling Pathway Improves Cerebral Ischemia-Reperfusion Injury in Rats. Mol. Neurobiol. 2018, 55, 4311–4319. [Google Scholar] [CrossRef]
- Famakin, B.M.; Vemuganti, R. Toll-Like Receptor 4 Signaling in Focal Cerebral Ischemia: A Focus on the Neurovascular Unit. Mol. Neurobiol. 2020, 57, 2690–2701. [Google Scholar] [CrossRef]
- Norden, D.M.; Trojanowski, P.J.; Villanueva, E.; Navarro, E.; Godbout, J.P. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016, 64, 300–316. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Shibata, K.; Yoshida, K.; Shigetomi, E.; Gachet, C.; Ikenaka, K.; Tanaka, K.F.; Koizumi, S. Transformation of Astrocytes to a Neuroprotective Phenotype by Microglia via P2Y(1) Receptor Downregulation. Cell Rep. 2017, 19, 1151–1164. [Google Scholar] [CrossRef]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef] [PubMed]
- Matejuk, A.; Ransohoff, R.M. Crosstalk between Astrocytes and Microglia: An Overview. Front Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Marsh, S.E.; Stevens, B. Microglia and Astrocytes in Disease: Dynamic Duo or Partners in Crime? Trends Immunol. 2020, 41, 820–835. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Leventoux, N.; Morimoto, S.; Imaizumi, K.; Sato, Y.; Takahashi, S.; Mashima, K.; Ishikawa, M.; Sonn, I.; Kondo, T.; Watanabe, H.; et al. Human Astrocytes Model Derived from Induced Pluripotent Stem Cells. Cells 2020, 9, 2680. [Google Scholar] [CrossRef]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood-brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; de Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8, 573775. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, S.N.; Xu, T.Y.; Miao, Z.W.; Su, D.F.; Miao, C.Y. Organoid technology for brain and therapeutics research. CNS Neurosci. Ther. 2017, 23, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.N.; Wang, Z.; Xu, T.Y.; Cheng, M.H.; Li, W.L.; Miao, C.Y. Cerebral Organoids Repair Ischemic Stroke Brain Injury. Transl. Stroke Res. 2020, 11, 983–1000. [Google Scholar] [CrossRef] [PubMed]
- Cambron, M.; D’Haeseleer, M.; Laureys, G.; Clinckers, R.; Debruyne, J.; De Keyser, J. White-matter astrocytes, axonal energy metabolism, and axonal degeneration in multiple sclerosis. J. Cereb. Blood Flow Metab. 2012, 32, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.M.; Lee, Y.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of axons. Trends Cell Biol. 2013, 23, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Invest. 2017, 127, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.; Yasuda, D.; Fujimori, K.; Morimoto, S.; Takahashi, S. Ropinirole, a New ALS Drug Candidate Developed Using iPSCs. Trends Pharmacol. Sci. 2020, 41, 99–109. [Google Scholar] [CrossRef]
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering 2021, 8, 30. [Google Scholar] [CrossRef]
- Salman, M.M.; Al-Obaidi, Z.; Kitchen, P.; Loreto, A.; Bill, R.M.; Wade-Martins, R. Advances in Applying Computer-Aided Drug Design for Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4688. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, S. Neuroprotective Function of High Glycolytic Activity in Astrocytes: Common Roles in Stroke and Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 6568. https://doi.org/10.3390/ijms22126568
Takahashi S. Neuroprotective Function of High Glycolytic Activity in Astrocytes: Common Roles in Stroke and Neurodegenerative Diseases. International Journal of Molecular Sciences. 2021; 22(12):6568. https://doi.org/10.3390/ijms22126568
Chicago/Turabian StyleTakahashi, Shinichi. 2021. "Neuroprotective Function of High Glycolytic Activity in Astrocytes: Common Roles in Stroke and Neurodegenerative Diseases" International Journal of Molecular Sciences 22, no. 12: 6568. https://doi.org/10.3390/ijms22126568
APA StyleTakahashi, S. (2021). Neuroprotective Function of High Glycolytic Activity in Astrocytes: Common Roles in Stroke and Neurodegenerative Diseases. International Journal of Molecular Sciences, 22(12), 6568. https://doi.org/10.3390/ijms22126568