
iPLA2β Contributes to ER Stress-Induced Apoptosis during Myocardial Ischemia/Reperfusion Injury

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Primary Neonatal Rat Ventricular Myocyte (NRVM) Isolation, Culture and Transfection
2.3. Simulated Ischemia/Reperfusion (sI/R) and Cell Death Assay
2.4. Generation of iPLA2β Knockout (KO) Mice
2.5. Mouse Genotyping
2.6. Cardiac Ischemia/Reperfusion
2.7. Echocardiography
2.8. TUNEL Staining
2.9. Collection of Human Blood Samples
2.10. ELISA Assay
2.11. Immunoblotting
2.12. Immunofluorescence Staining
2.13. Flow Cytometry
2.14. RNA Isolation and Realtime PCR
2.15. Cell Surface Protein Biotinylation and Detection
2.16. Assay for [Ca2+]i Levels
2.17. Statistics
3. Results
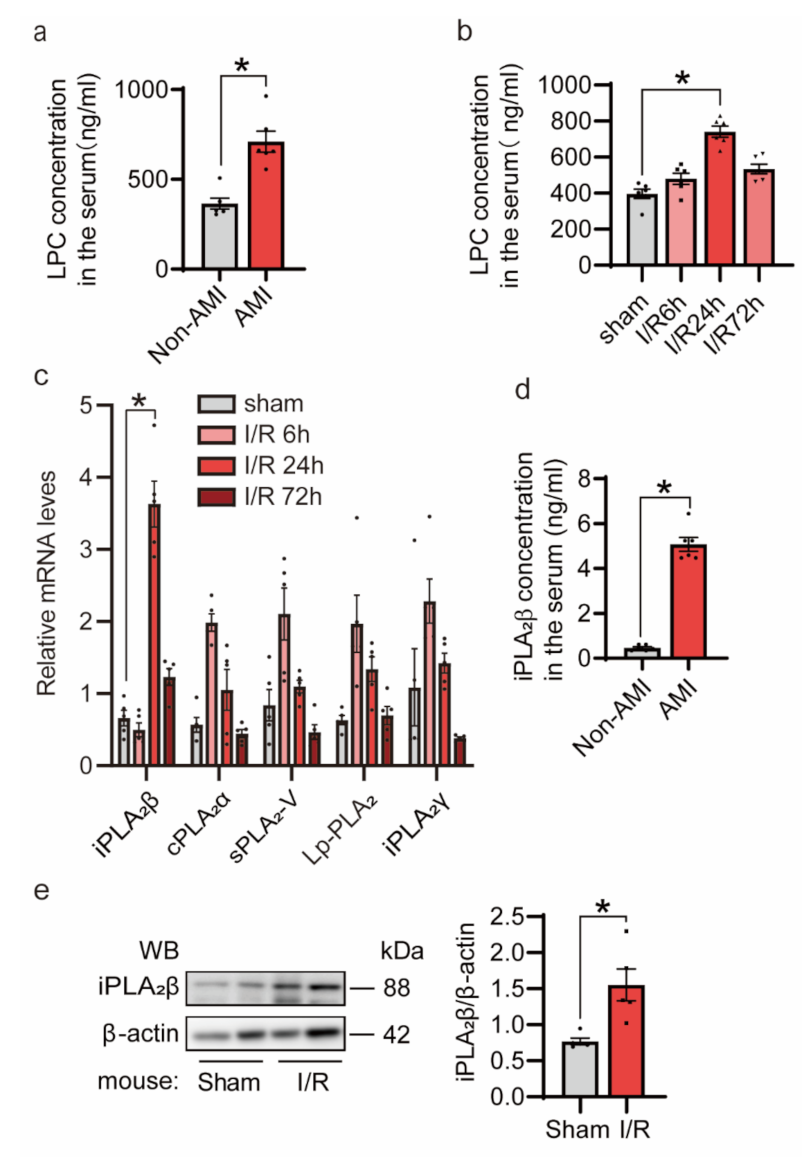
3.1. Myocardial Ischemia/Reperfusion (I/R) Injury Induces iPLA2β In Vivo
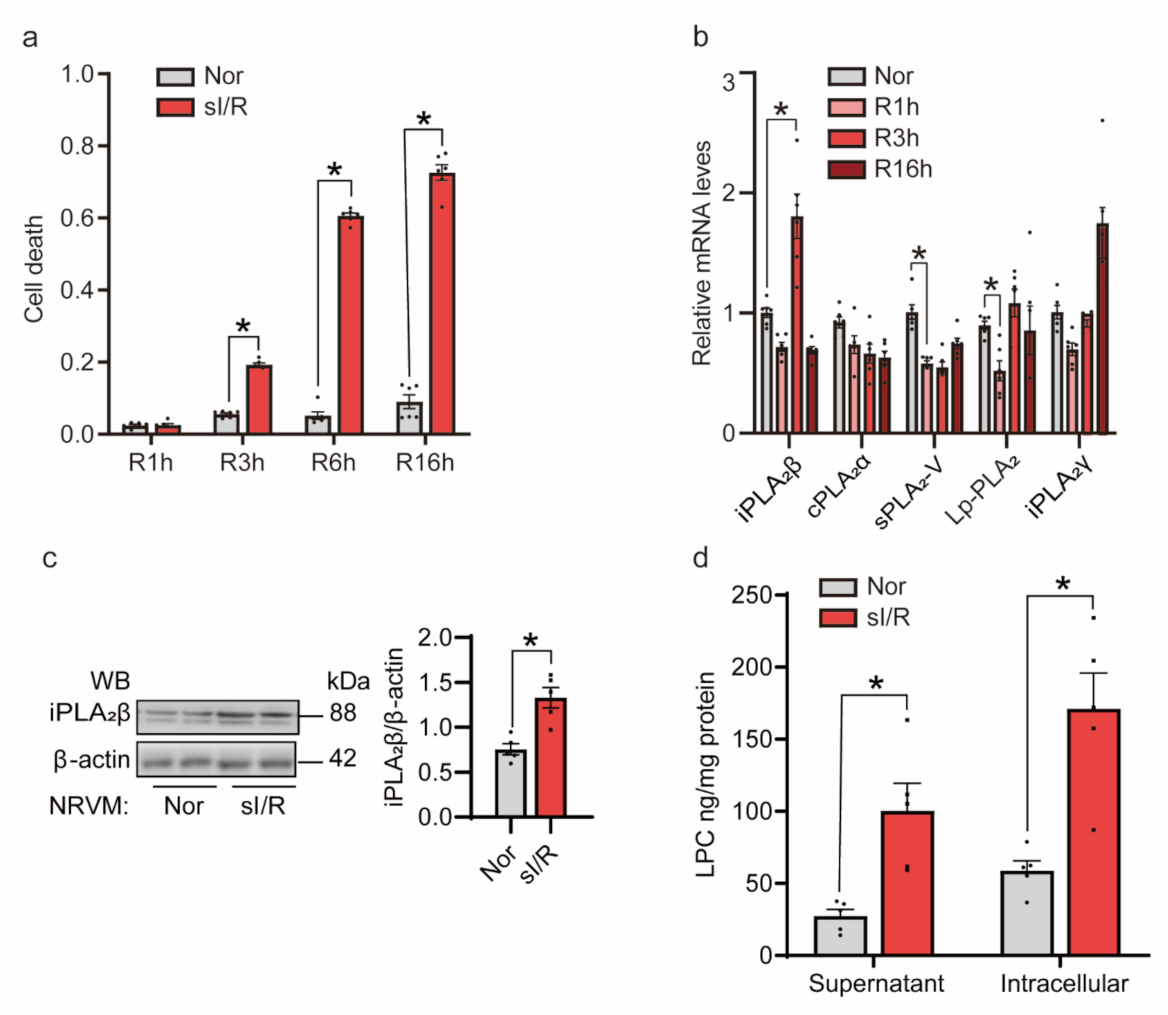
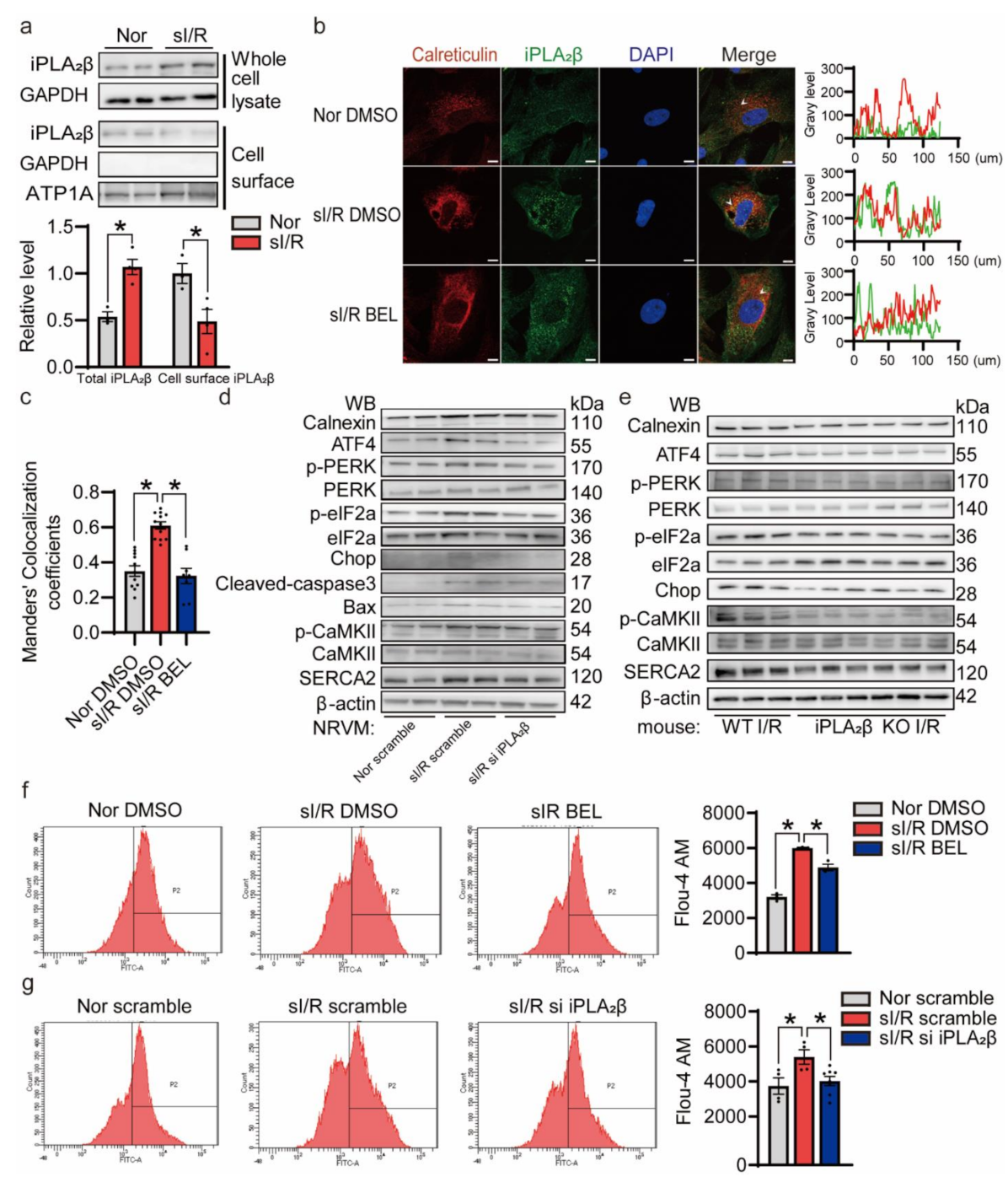
3.2. sI/R Induced iPLA2β In Vitro
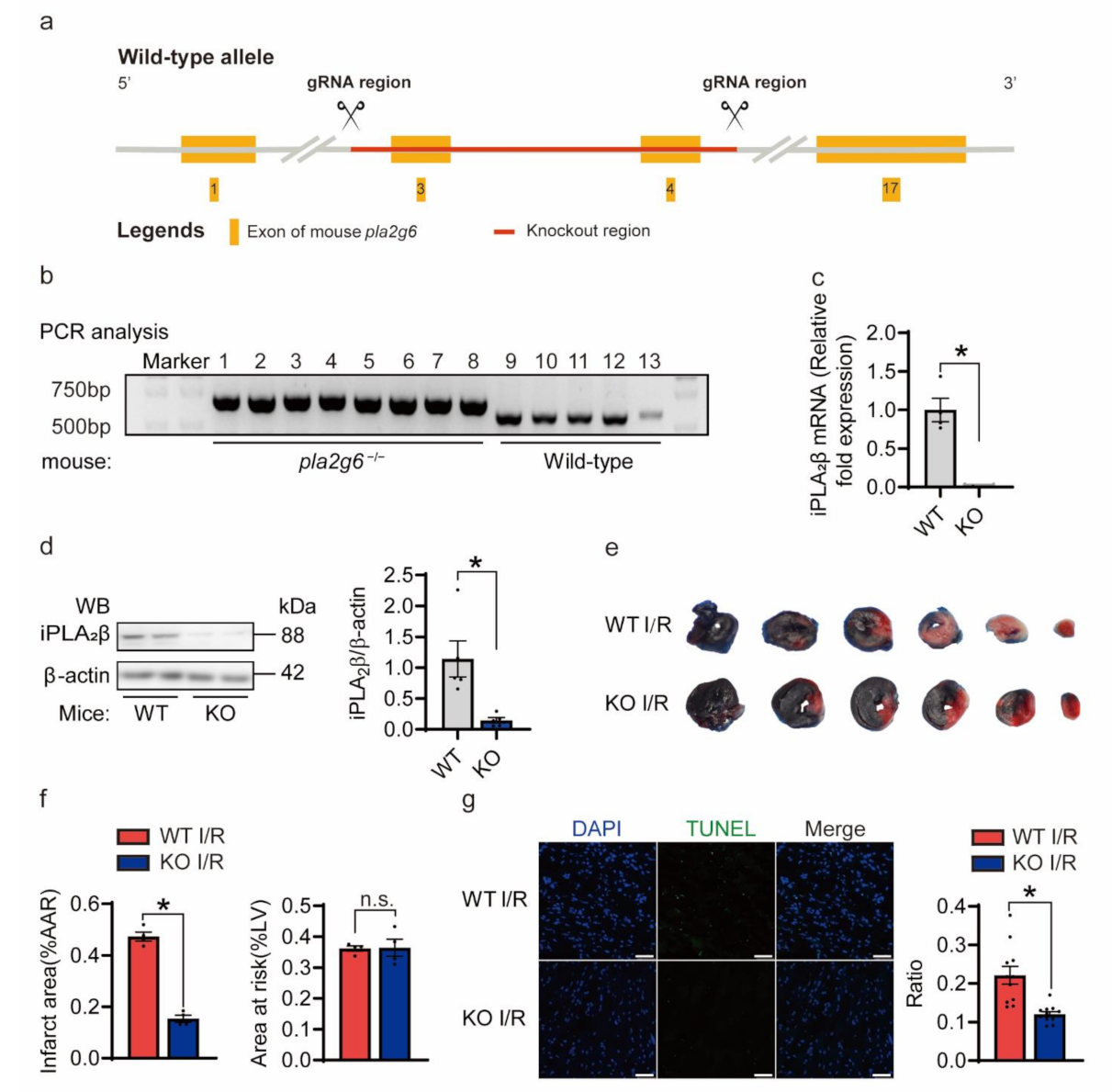
3.3. Knockout of iPLA2β Alleviated Heart I/R Injury
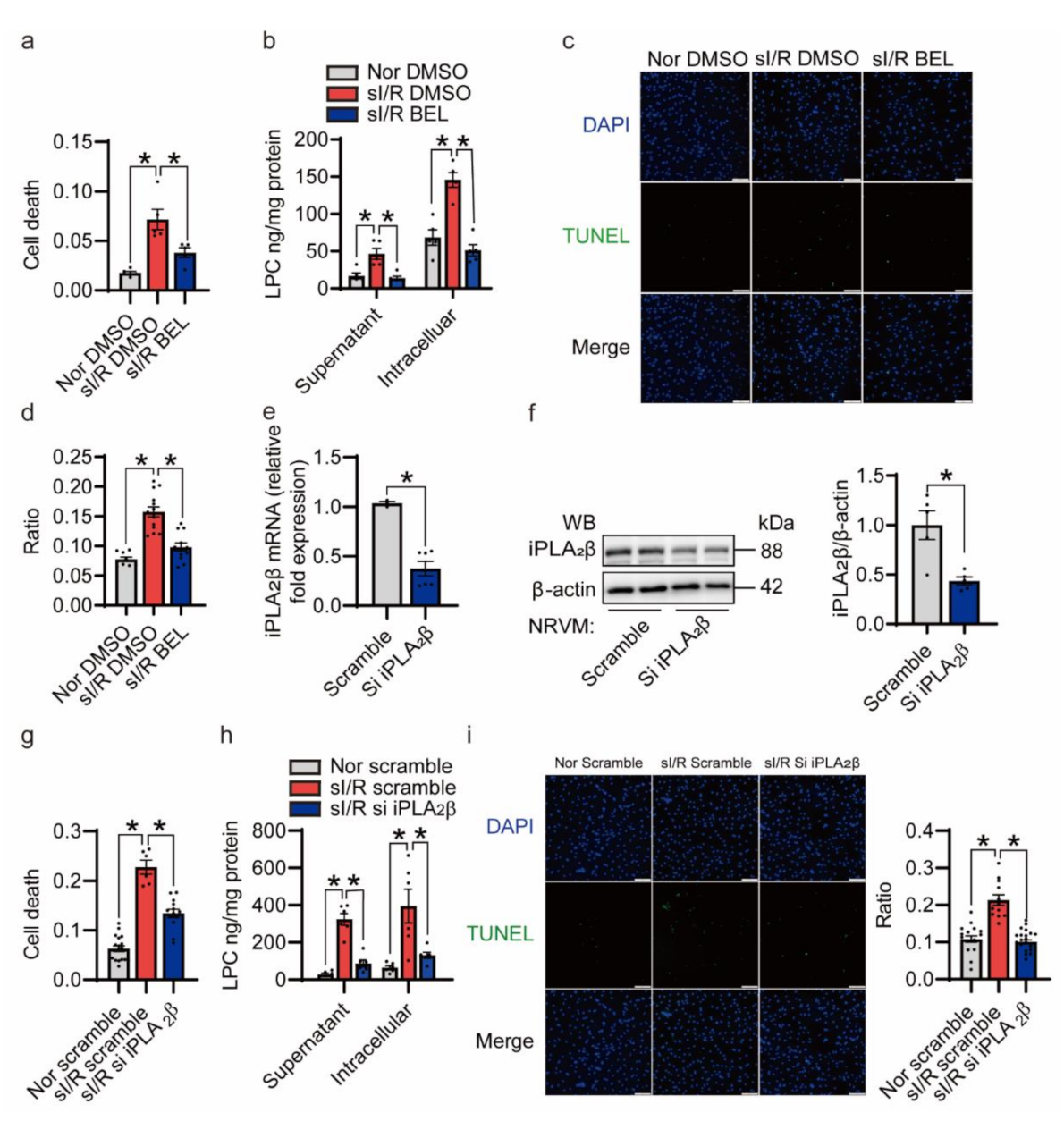
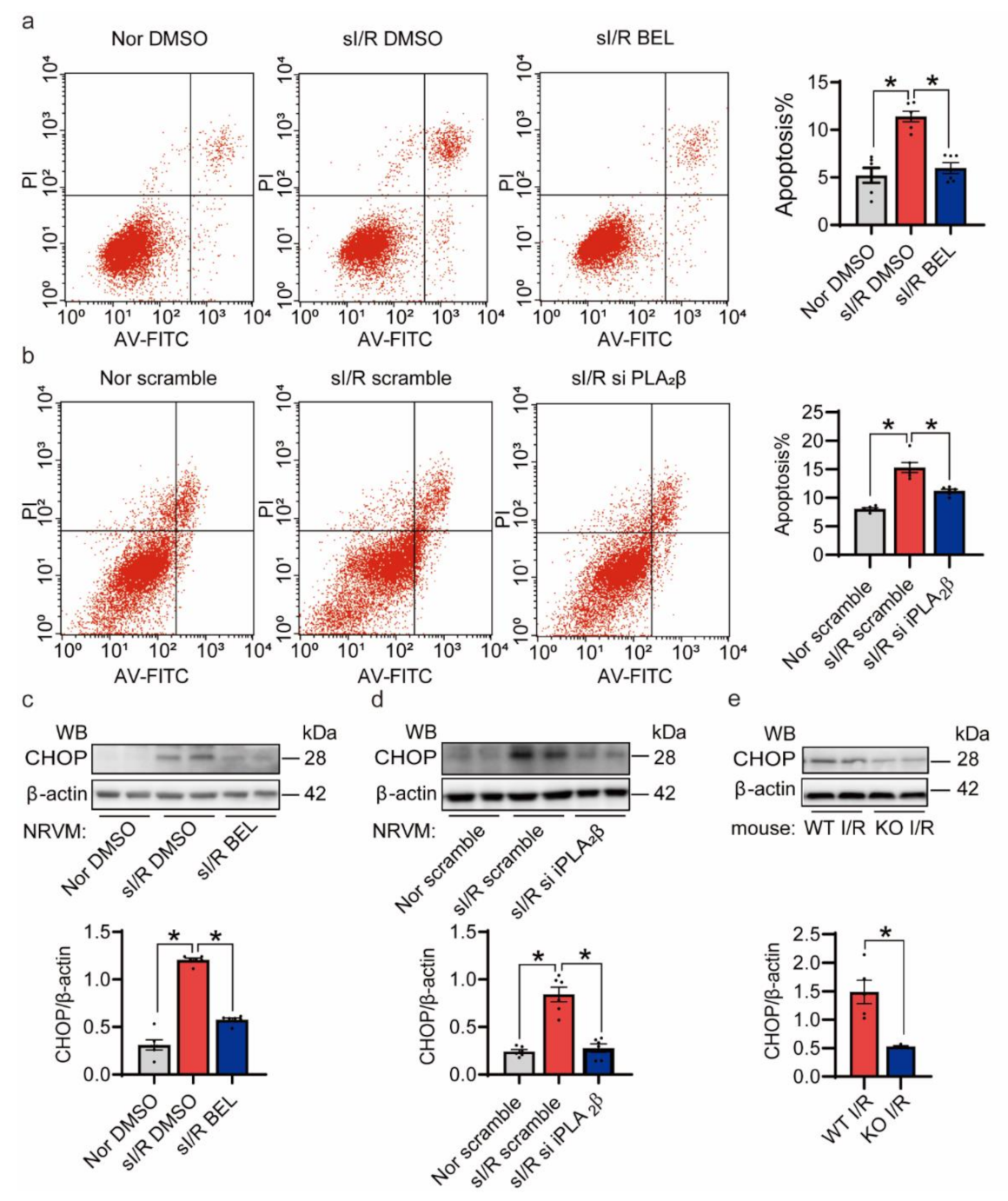
3.4. Inhibition of iPLA2β Ameliorates NRVMs Death Triggered by sI/R In Vitro
3.5. The Reduction of I/R Injury Is Mainly Achieved by Reducing Apoptosis
3.6. iPLA2β Translocation to the ER Aggravates ER Stress-Induced Apoptosis during I/R
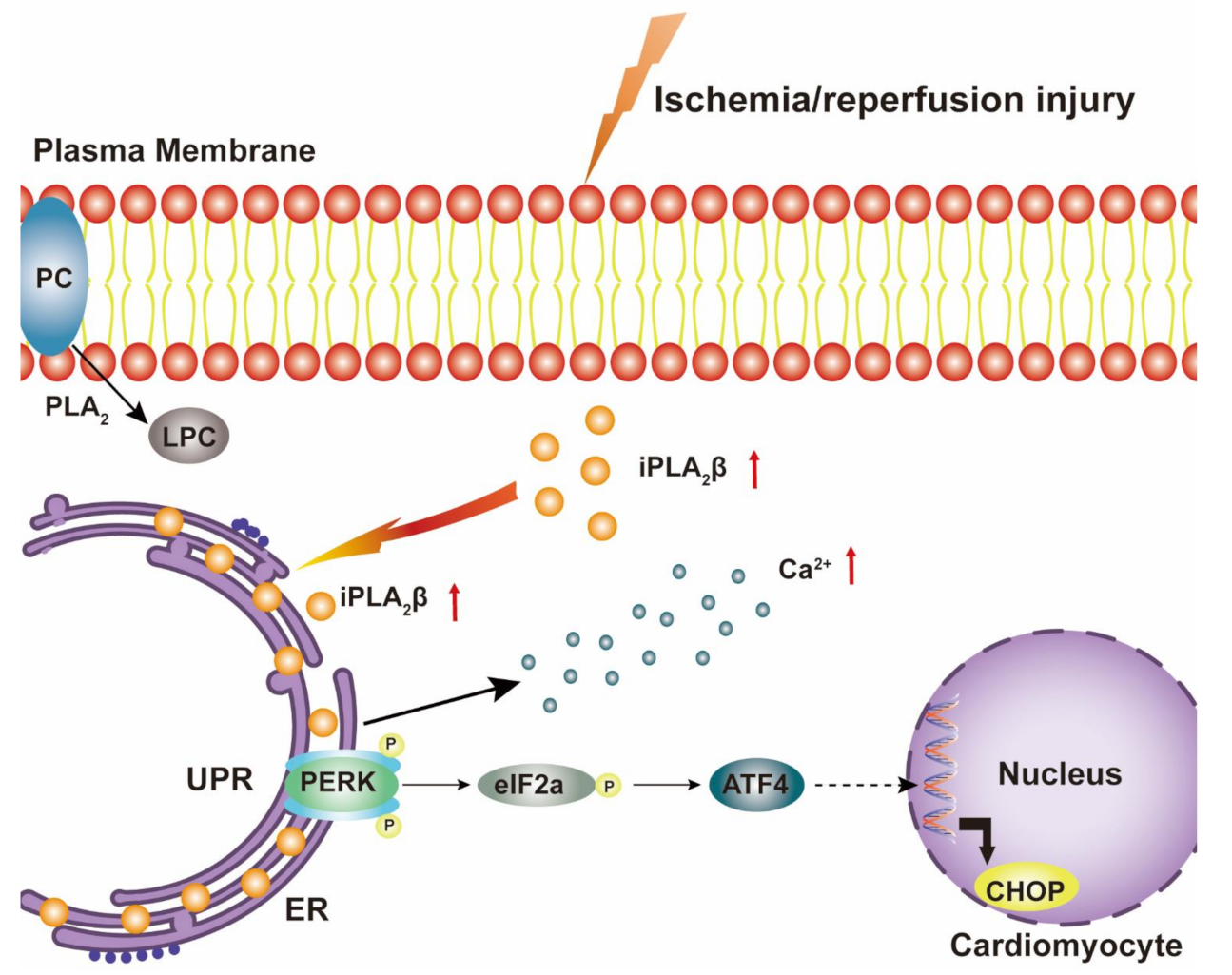
4. Discussion
5. Limitation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Chilian, W.; Crea, F.; Davidson, S.M.; Ferdinandy, P.; Garcia-Dorado, D.; Van Royen, N.; Schulz, R.; Heusch, G. The coronary circulation in acute myocardial ischaemia/reperfusion injury: A target for cardioprotection. Cardiovasc. Res. 2019, 115, 1143–1155. [Google Scholar] [CrossRef]
- Yellon, D.; Hausenloy, D.J. Myocardial Reperfusion Injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The Mammalian Unfolded Protein Response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.V.; Deng, Y.; Gao, N.; Pedrozo, Z.; Li, D.L.; Morales, C.R.; Criollo, A.; Luo, X.; Tan, W.; Jiang, N.; et al. Spliced X-Box Binding Protein 1 Couples the Unfolded Protein Response to Hexosamine Biosynthetic Pathway. Cell 2014, 156, 1179–1192. [Google Scholar] [CrossRef]
- Meyer, B.A.; Doroudgar, S. ER Stress-Induced Secretion of Proteins and Their Extracellular Functions in the Heart. Cells 2020, 9, 2066. [Google Scholar] [CrossRef] [PubMed]
- Arnsdorf, M.F.; Sawicki, G.J. The effects of lysophosphatidylcholine, a toxic metabolite of ischemia, on the components of cardiac excitability in sheep Purkinje fibers. Circ. Res. 1981, 49, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, K.M.; Scarabelli, T.M.; Turtle, L.; Chanalaris, A.; Townsend, P.A.; Carroll, C.J.; Hubank, M.; Stephanou, A.; Knight, R.A.; Latchman, D.S. Urocortin protects cardiac myocytes from ischemia/reperfusion injury by attenuating calcium insensitive phospholipase A 2 gene expression. FASEB J. 2003, 17, 2313–2315. [Google Scholar] [CrossRef]
- Shinzawa, K.; Tsujimoto, Y. PLA2 activity is required for nuclear shrinkage in caspase-independent cell death. J. Cell Biol. 2003, 163, 1219–1230. [Google Scholar] [CrossRef]
- Ghosh, M.; Tucker, D.E.; Burchett, S.A.; Leslie, C.C. Properties of the Group IV phospholipase A2 family. Prog. Lipid Res. 2006, 45, 487–510. [Google Scholar] [CrossRef]
- Lei, X.; Bone, R.N.; Ali, T.; Zhang, S.; Bohrer, A.; Tse, H.M.; Bidasee, K.R.; Ramanadham, S. Evidence of Contribution of iPLA2β-Mediated Events During Islet β-Cell Apoptosis Due to Proinflammatory Cytokines Suggests a Role for iPLA2β in T1D Development. Endocrinology 2014, 155, 3352–3364. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Ali, T.; Ashley, J.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Bao, S.; Lei, X.; Jin, C.; Zhang, S.; Turk, J. Evidence for proteolytic processing and stimulated organelle redistribution of iPLA(2)beta. Biochim. Biophys. Acta 2010, 1801, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Malley, K.R.; Koroleva, O.; Miller, I.; Sanishvili, R.; Jenkins, C.M.; Gross, R.W.; Korolev, S. The structure of iPLA2β reveals dimeric active sites and suggests mechanisms of regulation and localization. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.-J.; Li, P.-Y.; Wang, C.; Xin, Y.; Lu, W.-W.; Zhang, X.-X.; Zuo, S.; Ma, C.-S.; Tang, C.-S.; Nie, S.-P.; et al. Inhibition of endoplasmic reticulum stress by neuregulin-1 protects against myocardial ischemia/reperfusion injury. Peptides 2017, 88, 196–207. [Google Scholar] [CrossRef]
- Ngoh, G.A.; Facundo, H.T.; Hamid, T.; Dillmann, W.; Zachara, N.E.; Jones, S.P. Unique Hexosaminidase Reduces Metabolic Survival Signal and Sensitizes Cardiac Myocytes to Hypoxia/Reoxygenation Injury. Circ. Res. 2009, 104, 41–49. [Google Scholar] [CrossRef]
- Wang, J.X.; Zhang, X.J.; Li, Q.; Wang, K.; Wang, Y.; Jiao, J.Q. MicroRNA-103/107 Regulate Programmed Necrosis and Myocardial Ischemia/Reperfusion Injury Through Targeting FADD. Circ. Res. 2015, 117, 352–363. [Google Scholar] [CrossRef]
- Zinchuk, V.; Grossenbacher-Zinchuk, O. Quantitative colocalization analysis of confocal fluorescence microscopy images. Curr. Protocols Cell Biol. 2008, 39, 4–19. [Google Scholar] [CrossRef]
- Sobel, B.E.; Corr, P.B.; Robison, A.K.; Goldstein, R.A.; Witkowski, F.X.; Klein, M.S. Accumulation of lysophosphoglycerides with arrhythmogenic properties in ischemic myocardium. J. Clin. Investig. 1978, 62, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Corr, P.B.; Gross, R.W.E.; Sobel, B. Amphipathic metabolites and membrane dysfunction in ischemic myocardium. Circ. Res. 1984, 55, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Bai, N.; So, J.; Laher, I.; MacLeod, K.; Rodrigues, B. The ischemic metabolite lysophosphatidylcholine increases rat coronary arterial tone by endothelium-dependent mechanisms. J. Mol. Cell. Cardiol. 2009, 47, 112–120. [Google Scholar] [CrossRef]
- Anwar, K.; Voloshyna, I.; Littlefield, M.J.; Carsons, S.E.; Wirkowski, P.A.; Jaber, N.L.; Sohn, A.; Eapen, S.; Reiss, A.B. COX-2 Inhibition and Inhibition of Cytosolic Phospholipase A2 Increase CD36 Expression and Foam Cell Formation in THP-1 Cells. Lipids 2010, 46, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S. Future Role for Selective Phospholipase A2 Inhibitors in the Prevention of Atherosclerotic Cardiovascular Disease. Cardiovasc. Drugs Ther. 2009, 23, 93–101. [Google Scholar] [CrossRef]
- Mallat, Z.; Steg, P.G.; Benessiano, J.; Tanguy, M.-L.; Fox, K.A.; Collet, J.-P.; Dabbous, O.H.; Henry, P.; Carruthers, K.F.; Dauphin, A.; et al. Circulating Secretory Phospholipase A2 Activity Predicts Recurrent Events in Patients With Severe Acute Coronary Syndromes. J. Am. Coll. Cardiol. 2005, 46, 1249–1257. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zalewski, A.; Macphee, C. Role of Lipoprotein-Associated Phospholipase A2 in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 923–931. [Google Scholar] [CrossRef]
- Mancuso, D.J.; Abendschein, D.R.; Jenkins, C.M.; Han, X.; Saffitz, J.E.; Schuessler, R.B.; Gross, R.W. Cardiac ischemia activates calcium-independent phospholipase A(2)beta, precipitating ventricular tachyarrhythmias in transgenic mice—Rescue of the lethal electrophysiologic phenotype by mechanism-based inhibition. J. Biol. Chem. 2003, 278, 22231–22236. [Google Scholar] [CrossRef]
- Moon, S.H.; Mancuso, D.J.; Sims, H.F.; Liu, X.; Nguyen, A.L.; Yang, K.; Guan, S.; Dilthey, B.G.; Jenkins, C.M.; Weinheimer, C.J.; et al. Cardiac Myocyte-specific Knock-out of Calcium-independent Phospholipase A2γ (iPLA2γ) Decreases Oxidized Fatty Acids during Ischemia/Reperfusion and Reduces Infarct Size. J. Biol. Chem. 2016, 291, 19687–19700. [Google Scholar] [CrossRef]
- Fuentes, L.; Pérez, R.; Nieto, M.L.; Balsinde, J.; Balboa, M.A. Bromoenol Lactone Promotes Cell Death by a Mechanism Involving Phosphatidate Phosphohydrolase-1 Rather than Calcium-independent Phospholipase A2. J. Biol. Chem. 2003, 278, 44683–44690. [Google Scholar] [CrossRef]
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell Death in the Pathogenesis of Heart Disease: Mechanisms and Significance. Annu. Rev. Physiol. 2010, 72, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xia, Z.; Chen, Y.; Qi, D.; Zheng, H. Mechanism and Therapies of Oxidative Stress-Mediated Cell Death in Ischemia Reperfusion Injury. Oxidative Med. Cell. Longev. 2018, 2018, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Aghaei, M.; Motallebnezhad, M.; Ghorghanlu, S.; Jabbari, A.; Enayati, A.; Rajaei, M.; Pourabouk, M.; Moradi, A.; Alizadeh, A.M.; Khori, V. Targeting autophagy in cardiac ischemia/reperfusion injury: A novel therapeutic strategy. J. Cell. Physiol. 2019, 234, 16768–16778. [Google Scholar] [CrossRef]
- Cheng, Y.; Xia, Z.; Han, Y.; Rong, J. Plant Natural Product Formononetin Protects Rat Cardiomyocyte H9c2 Cells against Oxygen Glucose Deprivation and Reoxygenation via Inhibiting ROS Formation and Promoting GSK-3βPhosphorylation. Oxidative Med. Cell. Longev. 2016, 2016, 1–11. [Google Scholar] [CrossRef]
- Dong, Y.; Chen, H.; Gao, J.; Liu, Y.; Li, J.; Wang, J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J. Mol. Cell. Cardiol. 2019, 136, 27–41. [Google Scholar] [CrossRef]
- Minamino, T.; Komuro, I.; Kitakaze, M. Endoplasmic Reticulum Stress As a Therapeutic Target in Cardiovascular Disease. Circ. Res. 2010, 107, 1071–1082. [Google Scholar] [CrossRef]
- Bi, X.; Zhang, G.; Wang, X.; Nguyen, C.Y.; May, H.; Li, X.; Al-Hashimi, A.A.; Austin, R.C.; Gillette, T.G.; Fu, G.S.; et al. Endoplasmic Reticulum Chaperone GRP78 Protects Heart From Ischemia/Reperfusion Injury Through Akt Activation. Circ. Res. 2018, 122, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Hu, S.; Jin, Q.; Li, D.; Tian, F.; Toan, S.; Li, Y.; Zhou, H.; Chen, Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018, 16, 157–168. [Google Scholar] [CrossRef]
- Bao, S.; Li, Y.; Lei, X.; Wohltmann, M.; Jin, W.; Bohrer, A.; Semenkovich, C.F.; Ramanadham, S.; Tabas, I.; Turk, J. Attenuated Free Cholesterol Loading-induced Apoptosis but Preserved Phospholipid Composition of Peritoneal Macrophages from Mice That Do Not Express Group VIA Phospholipase A2. J. Biol. Chem. 2007, 282, 27100–27114. [Google Scholar] [CrossRef]
- Atsumi, G.-I.; Tajima, M.; Hadano, A.; Nakatani, Y.; Murakami, M.; Kudo, I. Fas-induced Arachidonic Acid Release Is Mediated by Ca2+-independent Phospholipase A2 but Not Cytosolic Phospholipase A2, Which Undergoes Proteolytic Inactivation. J. Biol. Chem. 1998, 273, 13870–13877. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-Y.; Tyurin, V.A.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Anthonymuthu, T.S.; Zhai, Y.-J.; Pan, M.-H.; Gong, H.-B.; Lu, D.-H.; Sun, J.; et al. Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol. 2021, 1–12. [Google Scholar] [CrossRef]
- Abeele, F.V.; Zholos, A.; Bidaux, G.; Shuba, Y.; Thebault, S.; Beck, B.; Flourakis, M.; Panchin, Y.; Skryma, R.; Prevarskaya, N. Ca2+-independent phospholipase A2-dependent gating of TRPM8 by lysophospholipids. J. Biol. Chem. 2006, 281, 40174–40182. [Google Scholar] [CrossRef] [PubMed]
- Beck, G.; Sugiura, Y.; Shinzawa, K.; Kato, S.; Setou, M.; Tsujimoto, Y.; Sakoda, S.; Sumi-Akamaru, H. Neuroaxonal Dystrophy in Calcium-Independent Phospholipase A2β Deficiency Results from Insufficient Remodeling and Degeneration of Mitochondrial and Presynaptic Membranes. J. Neurosci. 2011, 31, 11411–11420. [Google Scholar] [CrossRef]
- Meyer, E.; Kurian, M.A.; Hayflick, S.J. Neurodegeneration with Brain Iron Accumulation: Genetic Diversity and Pathophysiological Mechanisms. Annu. Rev. Genom. Hum. Genet. 2015, 16, 257–279. [Google Scholar] [CrossRef]
- Morgan, N.V.; Westaway, S.K.; Morton, J.E.; Gregory, A.; Gissen, P.; Sonek, S. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat. Genet. 2006, 38, 752–754. [Google Scholar] [CrossRef]
- Kruer, M.C.; Paisán-Ruiz, C.; Boddaert, N.; Yoon, M.Y.; Hama, H.; Ms, A.G.; Malandrini, A.; Woltjer, R.L.; Munnich, A.; Gobin, S.; et al. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann. Neurol. 2010, 68, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Hardy, J.; Bhatia, K.P. Syndromes of neurodegeneration with brain iron accumulation (NBIA): An update on clinical presentations, histological and genetic underpinnings, and treatment considerations. Mov. Disord. 2011, 27, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yen, A.; Rymarczyk, G.; Asai, H.; Trengrove, C.; Aziz, N.; Kirber, M.T.; Mostoslavsky, G.; Ikezu, T.; Wolozin, B.; et al. Impairment of PARK14-dependent Ca2+ signalling is a novel determinant of Parkinson’s disease. Nat. Commun. 2016, 7, 10332. [Google Scholar] [CrossRef] [PubMed]
- Guidubaldi, A.; Piano, C.; Santorelli, F.M.; Silvestri, G.; Petracca, M.; Tessa, A.; Bentivoglio, A.R. Novel mutations in SPG11 cause hereditary spastic paraplegia associated with early-onset levodopa-responsive Parkinsonism. Mov. Disord. 2011, 26, 553–556. [Google Scholar] [CrossRef]
- Bao, S.; Jin, C.; Zhang, S.; Turk, J.; Ma, Z.; Ramanadham, S. Beta-cell calcium-independent group VIA phospholipase A(2) (iPLA(2)beta): Tracking iPLA(2)beta movements in response to stimulation with insulin secretagogues in INS-1 cells. Diabetes 2004, 53, S186–S189. [Google Scholar] [CrossRef]
- Seleznev, K.; Zhao, C.; Zhang, X.H.; Song, K.; Ma, Z.A. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J. Biol. Chem. 2006, 281, 22275–22288. [Google Scholar] [CrossRef]
- Ramanadham, S.; Song, H.; Bao, S.; Hsu, F.F.; Zhang, S.; Ma, Z.; Jin, C.; Turk, J. Islet complex lipids: Involvement in the actions of group VIA calcium-independent phospholipase A(2) in beta-cells. Diabetes 2004, 53, S179–S185. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.-D.; Poulsen, K.A.; Lambert, I.H.; Pedersen, S.F. HL-1 mouse cardiomyocyte injury and death after simulated ischemia and reperfusion: Roles of pH, Ca2+-independent phospholipase A2, and Na+/H+ exchange. Am. J. Physiol. Physiol. 2009, 296, C1227–C1242. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Blasé, J.R.; Hoft, D.F.; Marentette, J.O.; Turk, J.; McHowat, J. Mice with Genetic Deletion of Group VIA Phospholipase A2β Exhibit Impaired Macrophage Function and Increased Parasite Load in Trypanosoma cruzi-Induced Myocarditis. Infect. Immun. 2016, 84, 1137–1142. [Google Scholar] [CrossRef]
- Kan, H.; Xie, Z.; Finkel, M.S. iPLA2 inhibitor blocks negative inotropic effect of HIV gp120 on cardiac myocytes. J. Mol. Cell. Cardiol. 2006, 40, 131–137. [Google Scholar] [CrossRef]
- Su, X.; Han, X.; Mancuso, D.J.; Abendschein, A.D.R.; Gross, R.W. Accumulation of Long-Chain Acylcarnitine and 3-Hydroxy Acylcarnitine Molecular Species in Diabetic Myocardium: Identification of Alterations in Mitochondrial Fatty Acid Processing in Diabetic Myocardium by Shotgun Lipidomics†. Biochemistry 2005, 44, 5234–5245. [Google Scholar] [CrossRef]
- Smani, T.; Zakharov, S.I.; Leno, E.; Csutora, P.; Trepakova, E.S.; Bolotina, V.M. Ca2+-independent Phospholipase A2 Is a Novel Determinant of Store-operated Ca2+ Entry. J. Biol. Chem. 2003, 278, 11909–11915. [Google Scholar] [CrossRef]
- Singaravelu, K.; Lohr, C.; Deitmer, J.W. Regulation of Store-Operated Calcium Entry by Calcium-Independent Phospholipase A2 in Rat Cerebellar Astrocytes. J. Neurosci. 2006, 26, 9579–9592. [Google Scholar] [CrossRef]
- Boittin, F.-X.; Petermann, O.; Hirn, C.; Mittaud, P.; Dorchies, O.M.; Roulet, E.; Ruegg, U. Ca2+-independent phospholipase A2 enhances store-operated Ca2+ entry in dystrophic skeletal muscle fibers. J. Cell Sci. 2006, 119, 3733–3742. [Google Scholar] [CrossRef]
- Ross, K.; Whitaker, M.; Reynolds, N.J. Agonist-induced calcium entry correlates with STIM1 translocation. J. Cell. Physiol. 2007, 211, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Boittin, F.-X.; Gribi, F.; Serir, K.; Bény, J.-L. Ca2+-independent PLA2 controls endothelial store-operated Ca2+ entry and vascular tone in intact aorta. Am. J. Physiol. Circ. Physiol. 2008, 295, H2466–H2474. [Google Scholar] [CrossRef]
- Ming, Y.; Zhu, X.; Tuma-Kellner, S.; Ganzha, A.; Liebisch, G.; Gan-Schreier, H.; Chamulitrat, W. iPla2β Deficiency Suppresses Hepatic ER UPR, Fxr, and Phospholipids in Mice Fed with MCD Diet, Resulting in Exacerbated Hepatic Bile Acids and Biliary Cell Proliferation. Cells 2019, 8, 879. [Google Scholar] [CrossRef] [PubMed]
- Fun, X.H.; Thibault, G. Lipid bilayer stress and proteotoxic stress-induced unfolded protein response deploy divergent transcriptional and non-transcriptional programmes. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1865, 158449. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Shi, Y.; Surendranath, A.R.; Jalal, N.; Pathak, J.L.; Subramaniyam, S. Role of Calcium-Independent Phospholipase A2 VIA in Mediating Neurological Disorder and Cancer. Trans. Tianjin Univ. 2017, 23, 1–10. [Google Scholar] [CrossRef]
- Mori, A.; Hatano, T.; Inoshita, T.; Shiba-Fukushima, K.; Koinuma, T.; Meng, H.; Kubo, S.-I.; Spratt, S.; Cui, C.; Yamashita, C.; et al. Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and α-synuclein stability through membrane remodeling. Proc. Natl. Acad. Sci. USA 2019, 116, 20689–20699. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S.; Cochemé, H.M.; Khan, S.; Asghari, S.; Bhatia, K.P.; et al. Loss ofPLA2G6leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 2015, 138, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.D.; Gottlieb, R.A. Inhibition of mitochondrial calcium-independent phospholipase A2 (iPLA2) attenuates mitochondrial phospholipid loss and is cardioprotective. Biochem. J. 2002, 362, 23–32. [Google Scholar] [CrossRef]
- Dohnal, V.; Dvořák, V.; Malíř, F.; Ostrý, V.; Roubal, T. A comparison of ELISA and HPLC methods for determination of ochratoxin A in human blood serum in the Czech Republic. Food Chem. Toxicol. 2013, 62, 427–431. [Google Scholar] [CrossRef]
- Beyene, A.M.; Du, X.; Schrunk, D.E.; Ensley, S.; Rumbeiha, W.K. High-performance liquid chromatography and Enzyme-Linked Immunosorbent Assay techniques for detection and quantification of aflatoxin B1 in feed samples: A comparative study. BMC Res. Notes 2019, 12, 1–6. [Google Scholar] [CrossRef]
- Omar, S.S.; Haddad, M.A.; Parisi, S. Validation of HPLC and Enzyme-Linked Immunosorbent Assay (ELISA) Techniques for Detection and Quantification of Aflatoxins in Different Food Samples. Foods 2020, 9, 661. [Google Scholar] [CrossRef]
- Huang, Q.; Lei, H.; Dong, M.; Tang, H.; Wang, Y. Quantitative analysis of 10 classes of phospholipids by ultrahigh-performance liquid chromatography tandem triple-quadrupole mass spectrometry. Analyst 2019, 144, 3980–3987. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Ruwhof, C.; van der Laarse, A. Mechanical stress-induced cardiac hypertrophy: Mechanisms and signal transduction pathways. Cardiovasc. Res. 2000, 47, 23–37. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non- AMI Group (n = 6) | AMI Group (n = 6) | |
|---|---|---|
| Age (years) | 54.83 ± 4.79 | 55.00 ± 8.72 |
| Gender (female: male) | 3:3 | 2:4 |
| BMI | 21.32 ± 2.32 | 24.77 ± 5.73 |
| Hypertension (Yes: No) | 2:4 | 3:3 |
| Diabetes (Yes: No) | 3:3 | 1:5 |
| Smoke (Yes: No) | 2:4 | 4:2 |
| SBP (mmHg) | 119.00 ± 11.05 | 121.83 ± 17.27 |
| DBP (mmHg) | 67.50 ± 10.31 | 77.50 ± 17.41 |
| TG (mmol/L) | 1.59 ± 1.16 | 1.40 ± 0.57 |
| CHO (mmol/L) | 5.13 ± 1.20 | 4.03 ± 1.11 |
| HDL (mmol/L) | 1.02 ± 0.15 | 1.04 ± 0.14 |
| LDL (mmol/L) | 2.25 ± 1.15 | 1.87 ± 0.62 |
| Parameter | WT I/R (n = 5) | KO I/R (n = 5) |
|---|---|---|
| IVS (mm) | 1.00 ± 0.17 | 0.85 ± 0.96 |
| LVPWs (mm) | 1.08 ± 0.14 | 1.07 ± 0.22 |
| LVIDd (mm) | 3.65 ± 0.26 | 3.05 ± 0.41 * |
| LVIDs (mm) | 3.22 ± 0.26 | 2.40 ± 0.37 * |
| FS (%) | 11.79 ± 2.69 | 21.46 ± 2.26 * |
| EF (%) | 26.02 ± 5.54 | 44.91 ± 4.26 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, T.; Lin, J.; Gong, Y.; Bi, X.; Hu, S.; Lv, Q.; Chen, J.; Li, X.; Chen, J.; Zhang, W.; et al. iPLA2β Contributes to ER Stress-Induced Apoptosis during Myocardial Ischemia/Reperfusion Injury. Cells 2021, 10, 1446. https://doi.org/10.3390/cells10061446
Jin T, Lin J, Gong Y, Bi X, Hu S, Lv Q, Chen J, Li X, Chen J, Zhang W, et al. iPLA2β Contributes to ER Stress-Induced Apoptosis during Myocardial Ischemia/Reperfusion Injury. Cells. 2021; 10(6):1446. https://doi.org/10.3390/cells10061446
Chicago/Turabian StyleJin, Tingting, Jun Lin, Yingchao Gong, Xukun Bi, Shasha Hu, Qingbo Lv, Jiaweng Chen, Xiaoting Li, Jiaqi Chen, Wenbin Zhang, and et al. 2021. "iPLA2β Contributes to ER Stress-Induced Apoptosis during Myocardial Ischemia/Reperfusion Injury" Cells 10, no. 6: 1446. https://doi.org/10.3390/cells10061446
APA StyleJin, T., Lin, J., Gong, Y., Bi, X., Hu, S., Lv, Q., Chen, J., Li, X., Chen, J., Zhang, W., Wang, M., & Fu, G. (2021). iPLA2β Contributes to ER Stress-Induced Apoptosis during Myocardial Ischemia/Reperfusion Injury. Cells, 10(6), 1446. https://doi.org/10.3390/cells10061446