
Experimental Evaluation of an Interferometric Light Microscopy Particle Counter for Titering and Characterization of Virus Preparations



Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Manufacturing, Titering and Characterization Methods
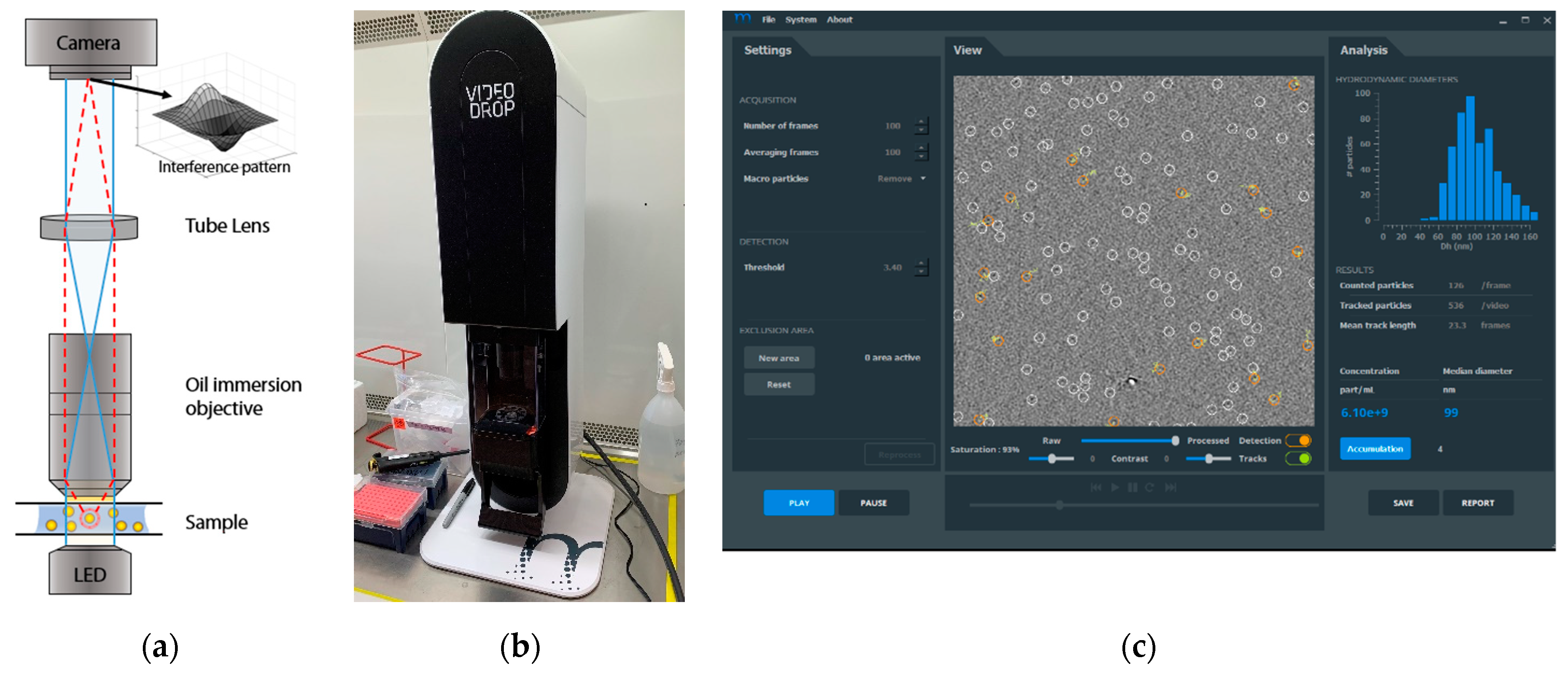
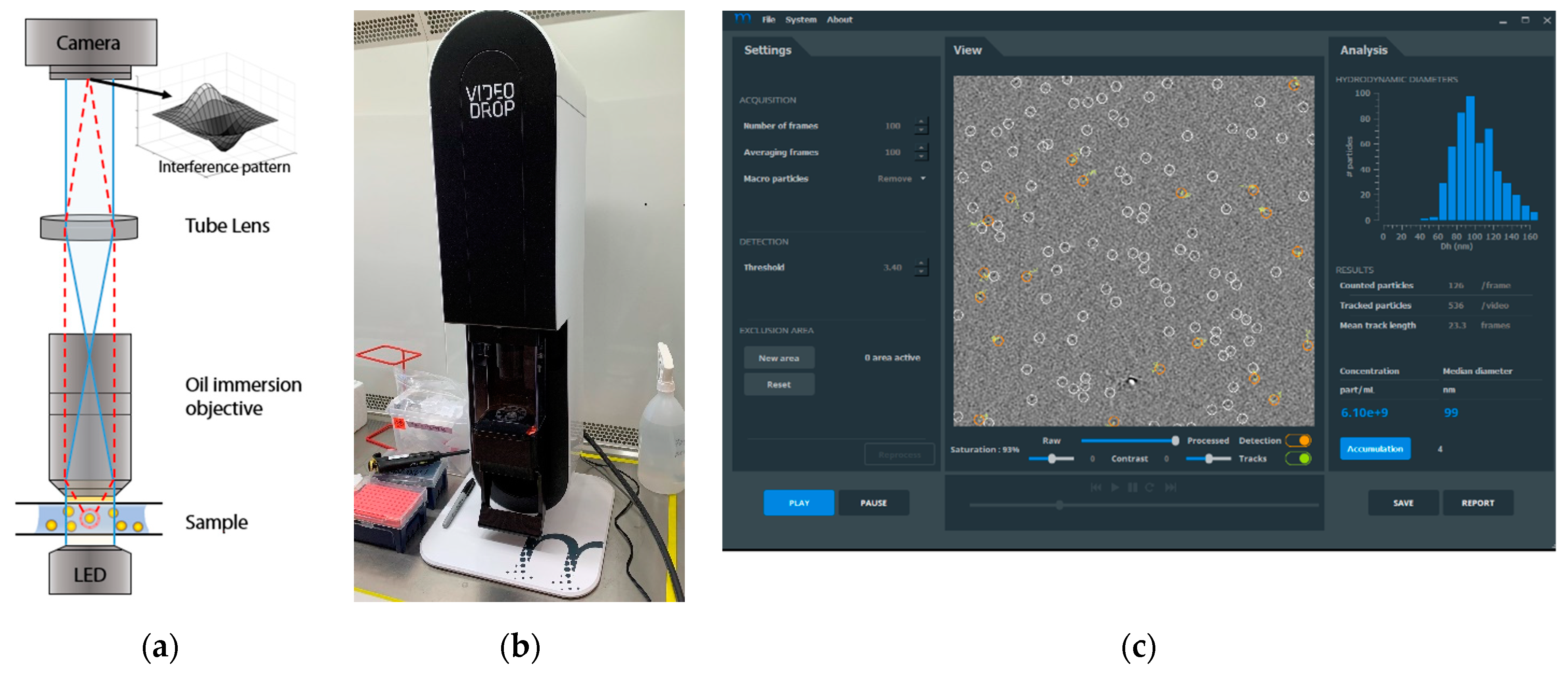
2.2. Videodrop Measurements
2.3. Calculations and Statistics
3. Results
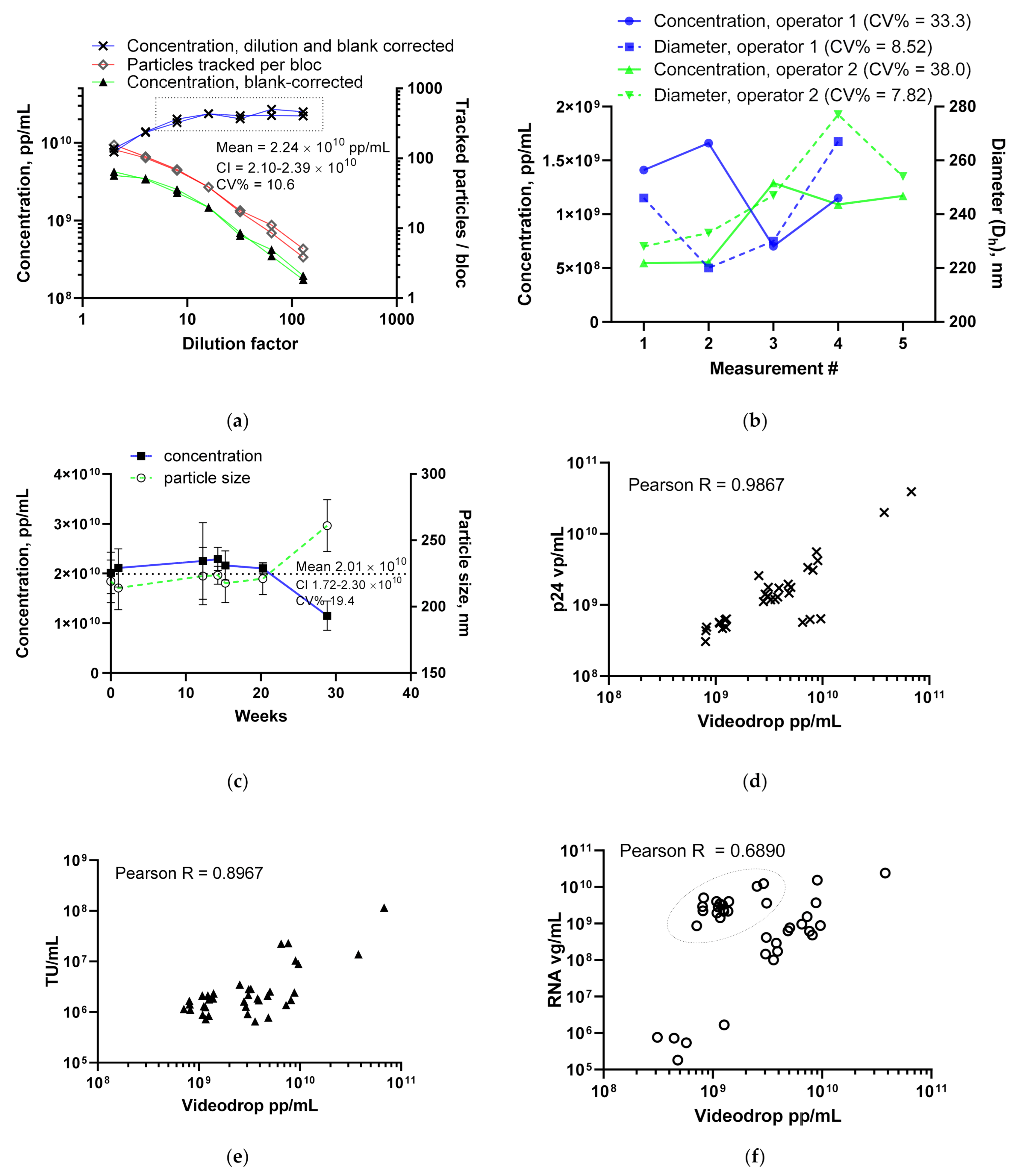
3.1. Lentivirus Particles/mL Assay Development
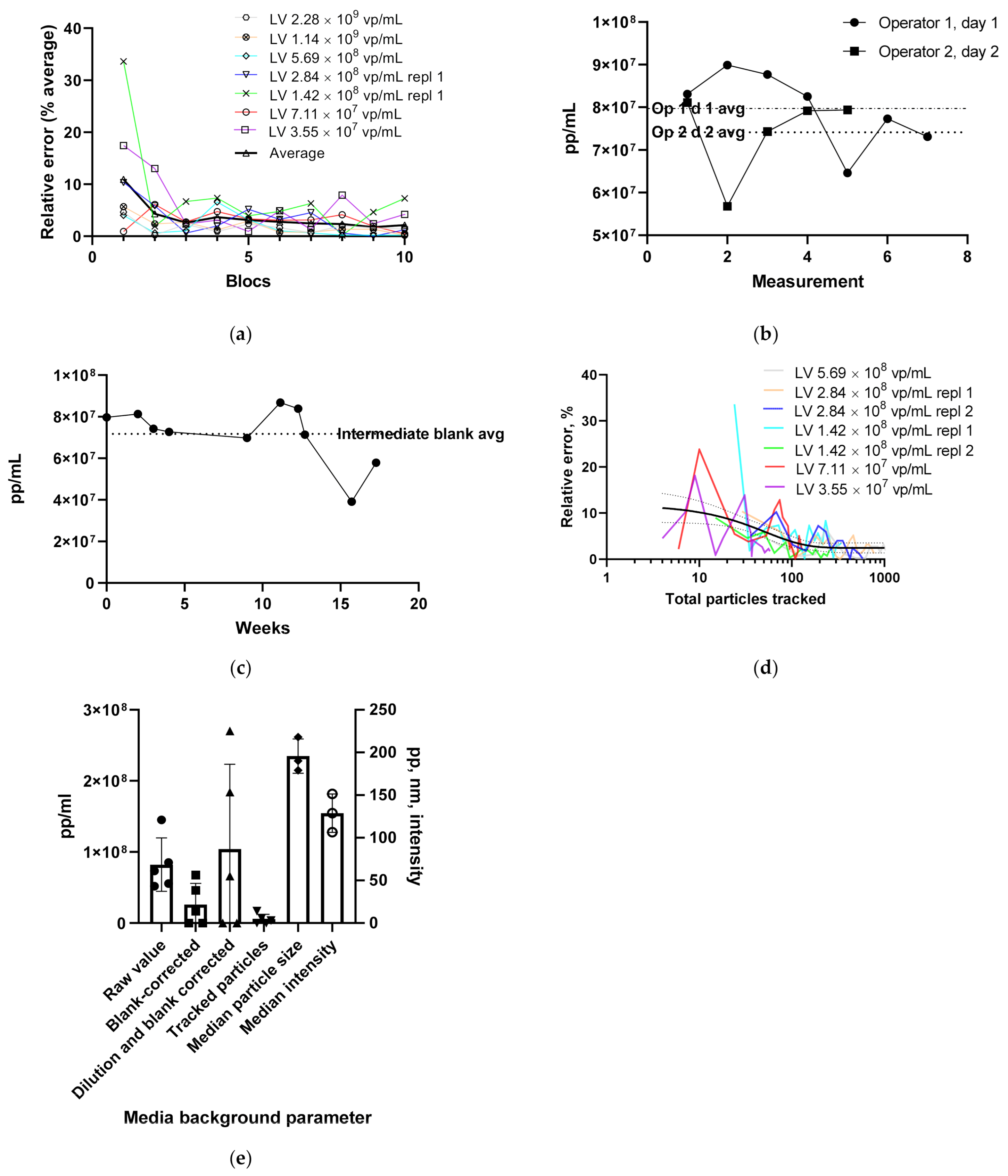
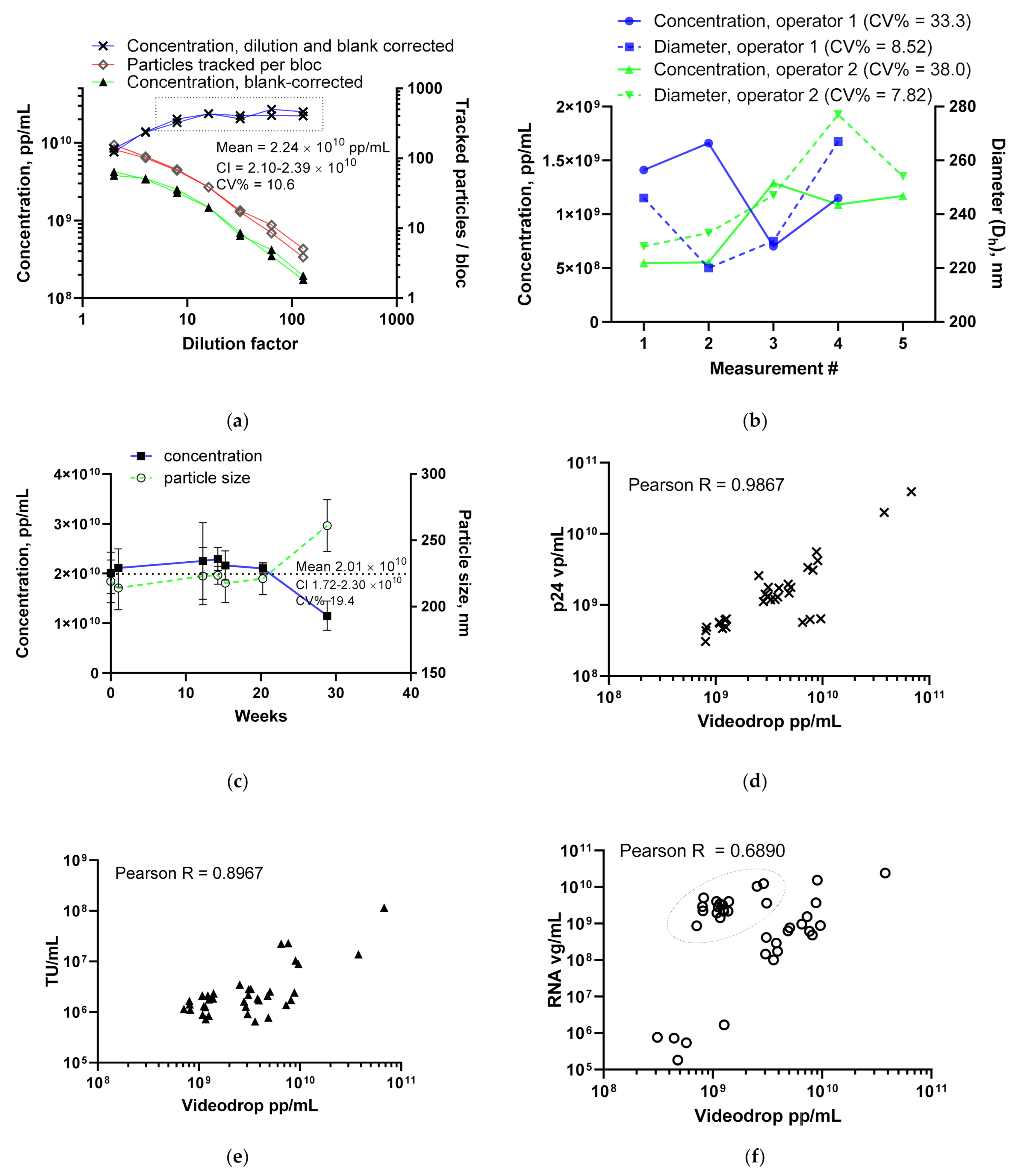
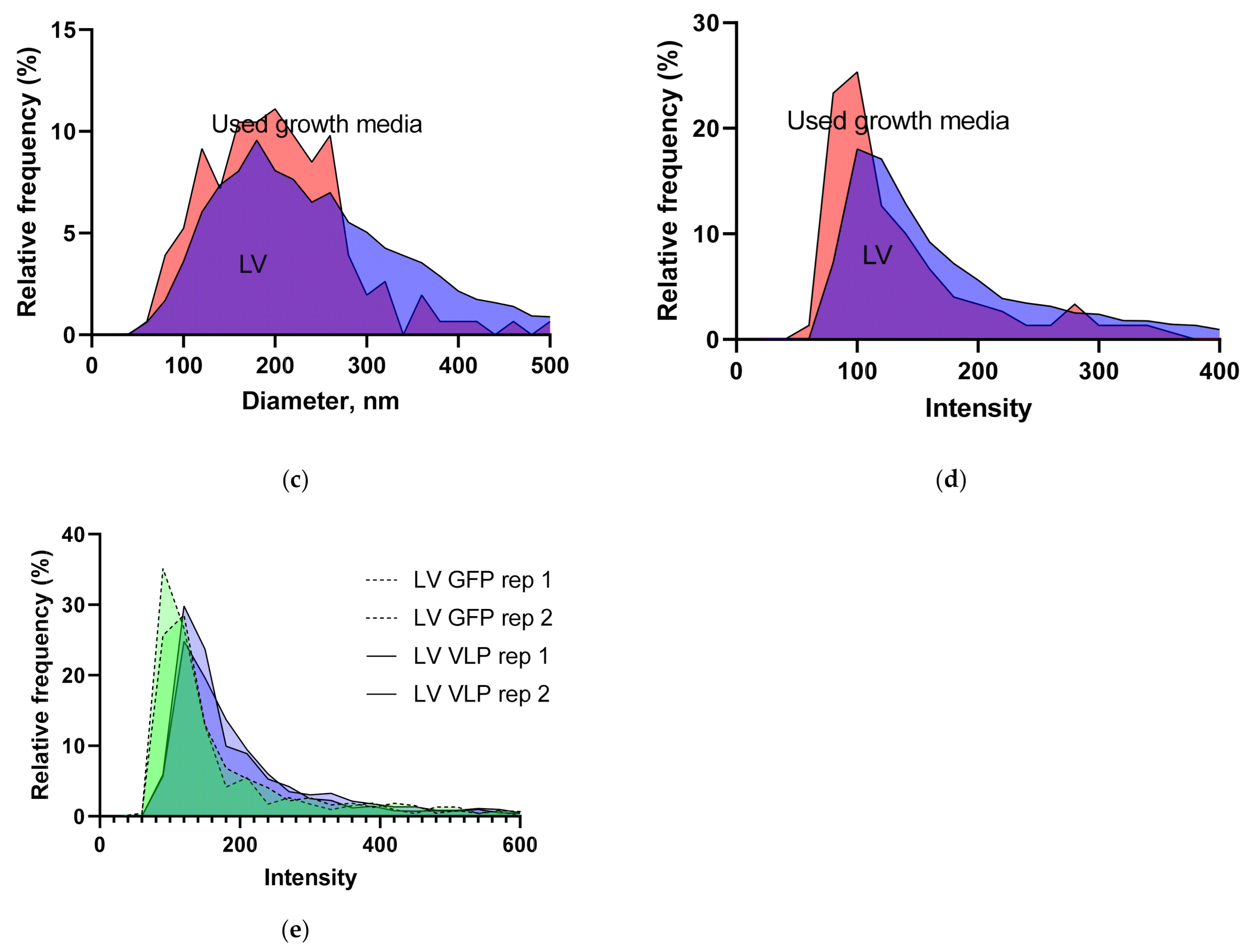
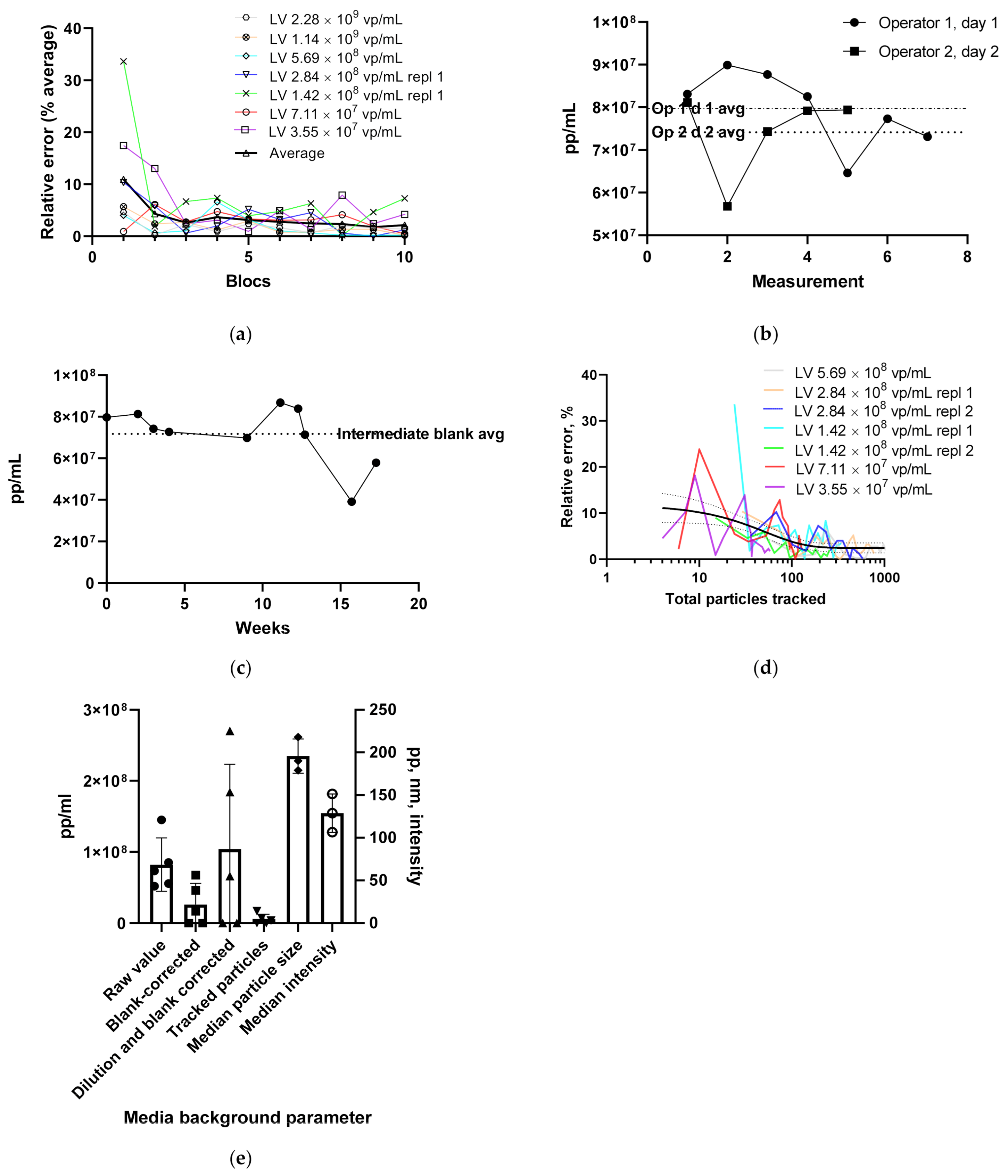
3.1.1. LV Dilution Curve and LV Control Intermediate Precision
3.1.2. Comparing the LV Assay against Traditional Titers
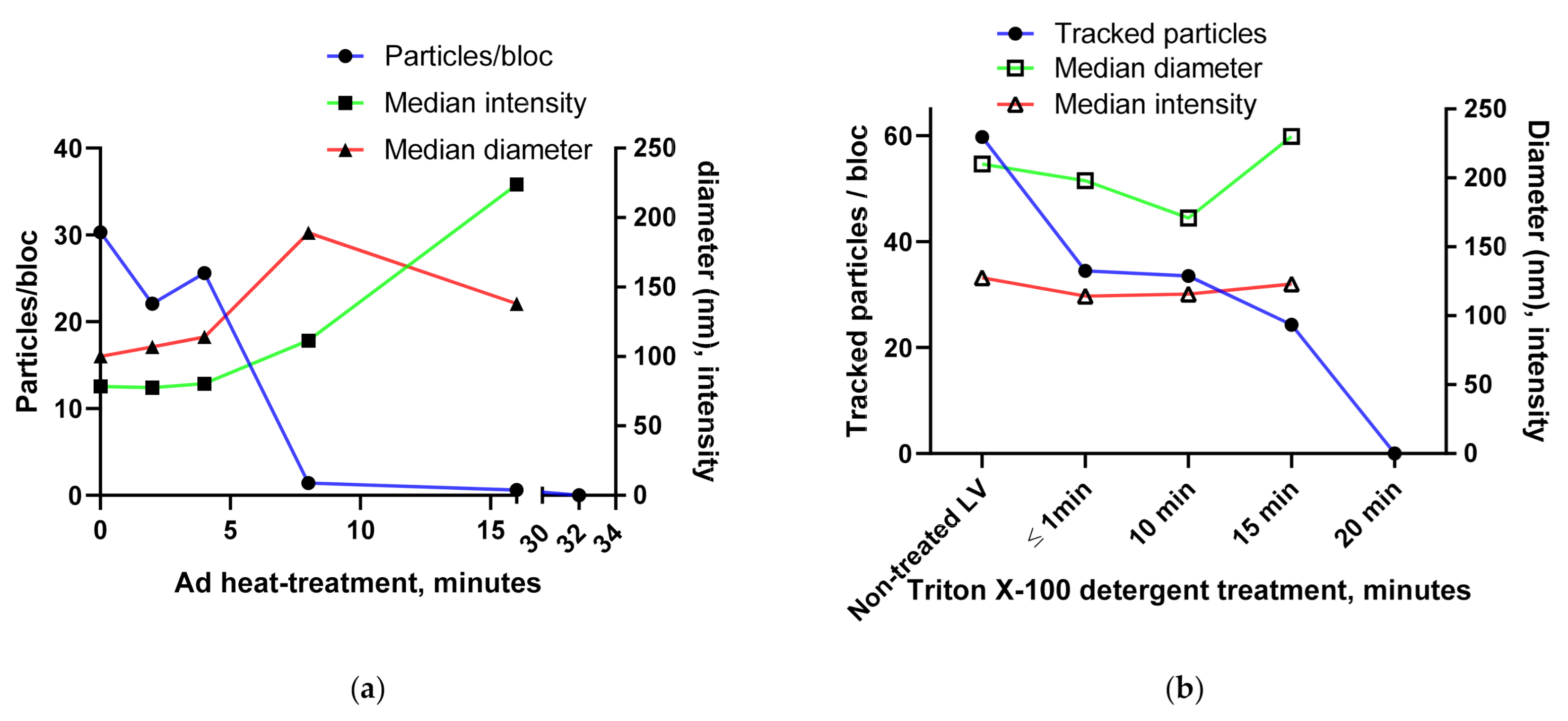
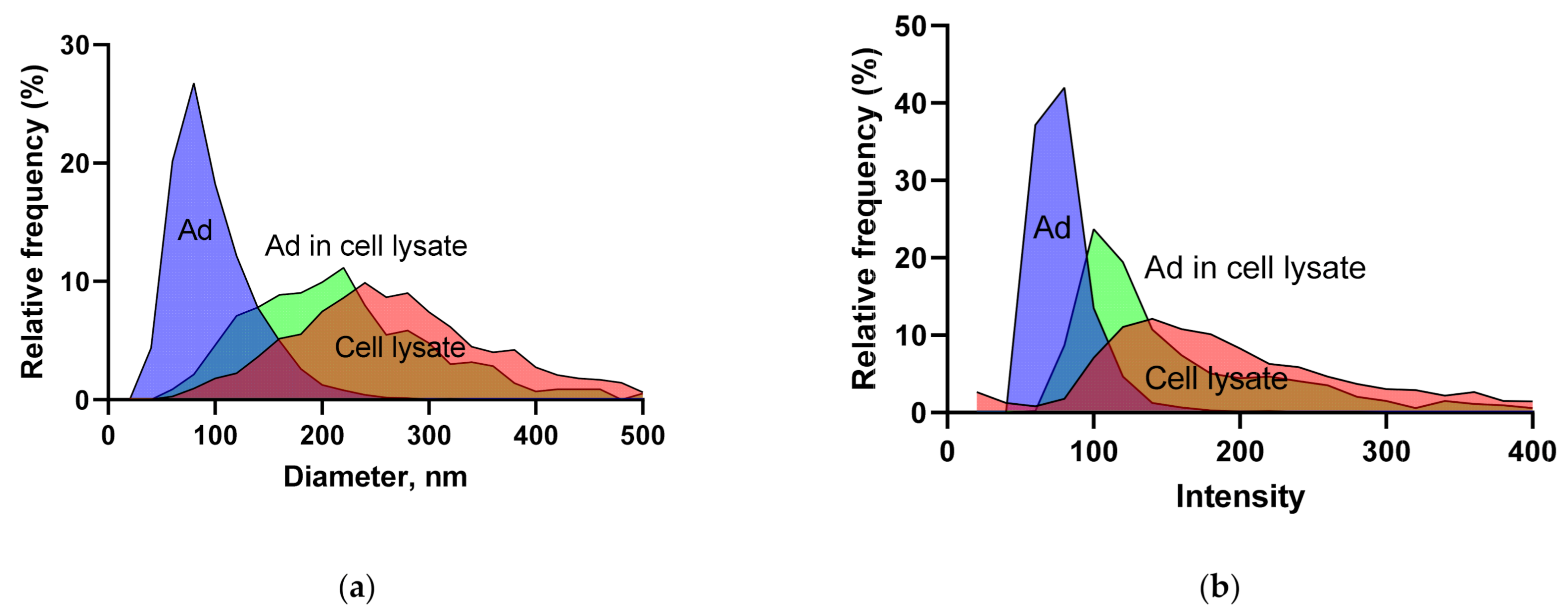
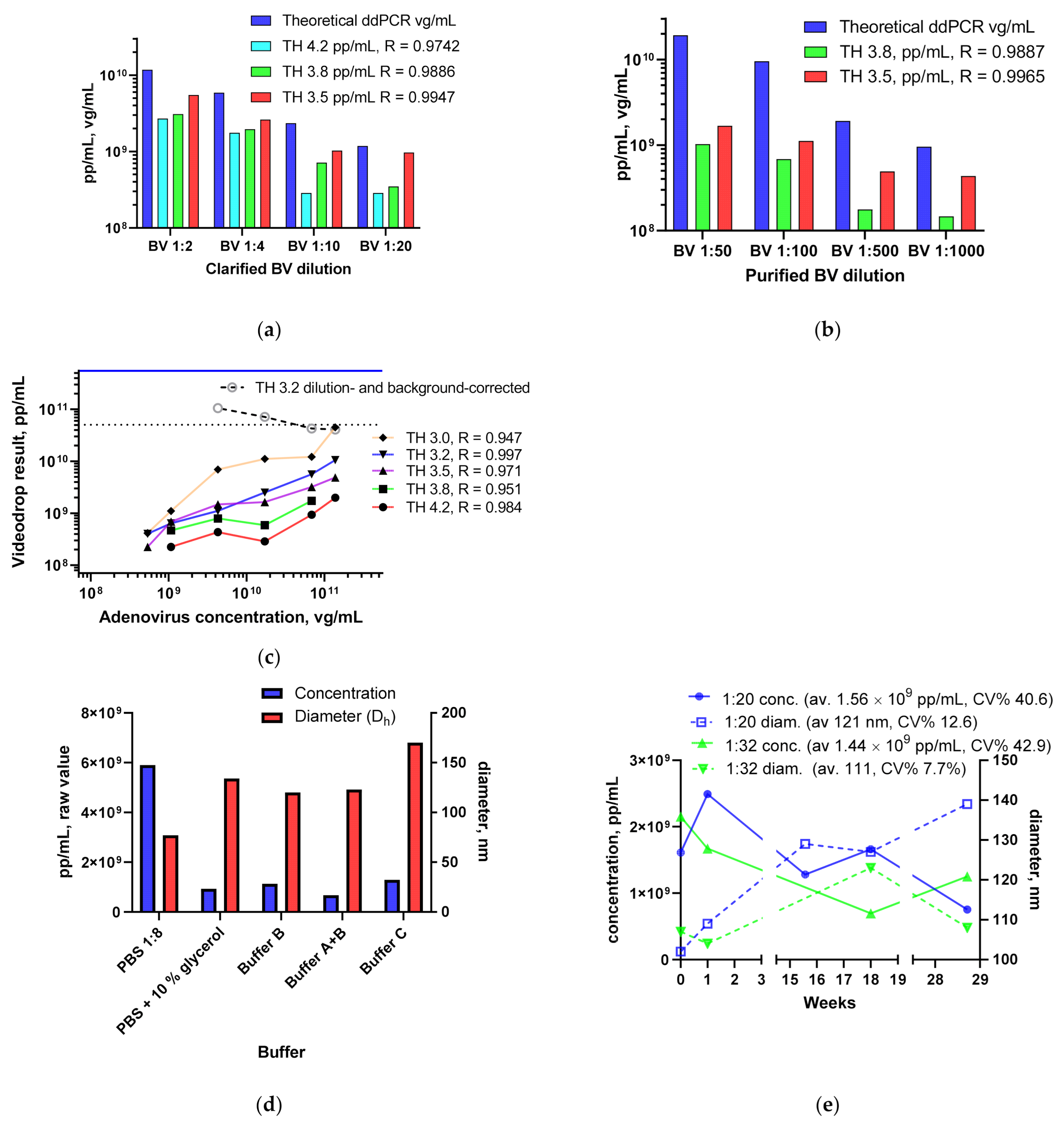
3.1.3. BV and Ad Tests
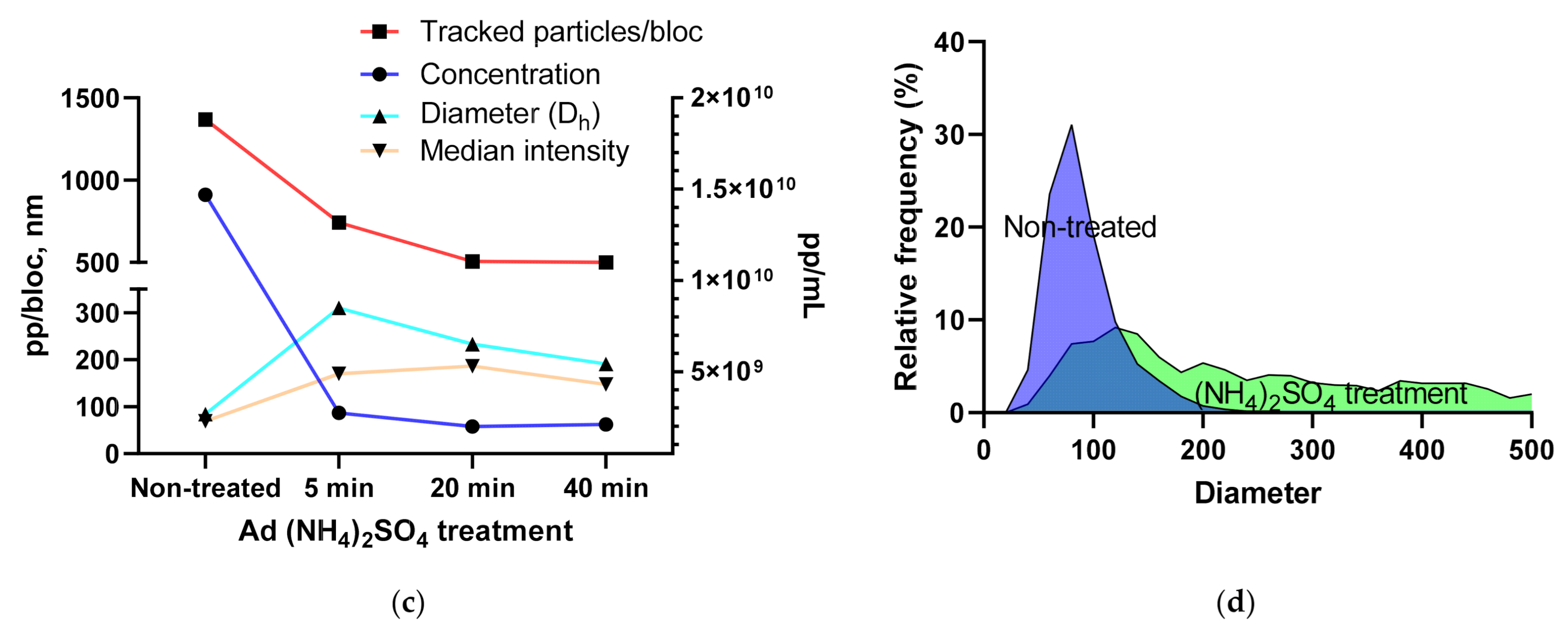
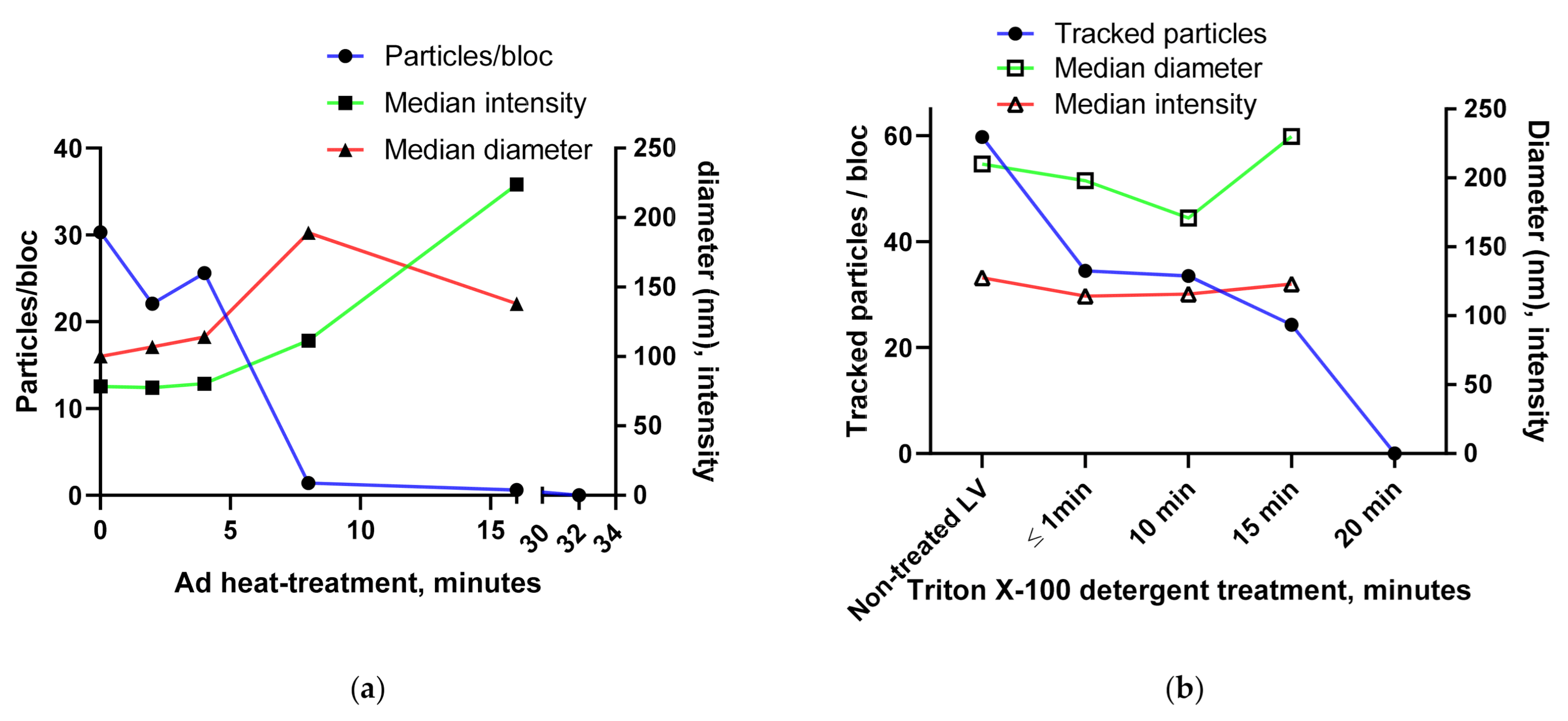
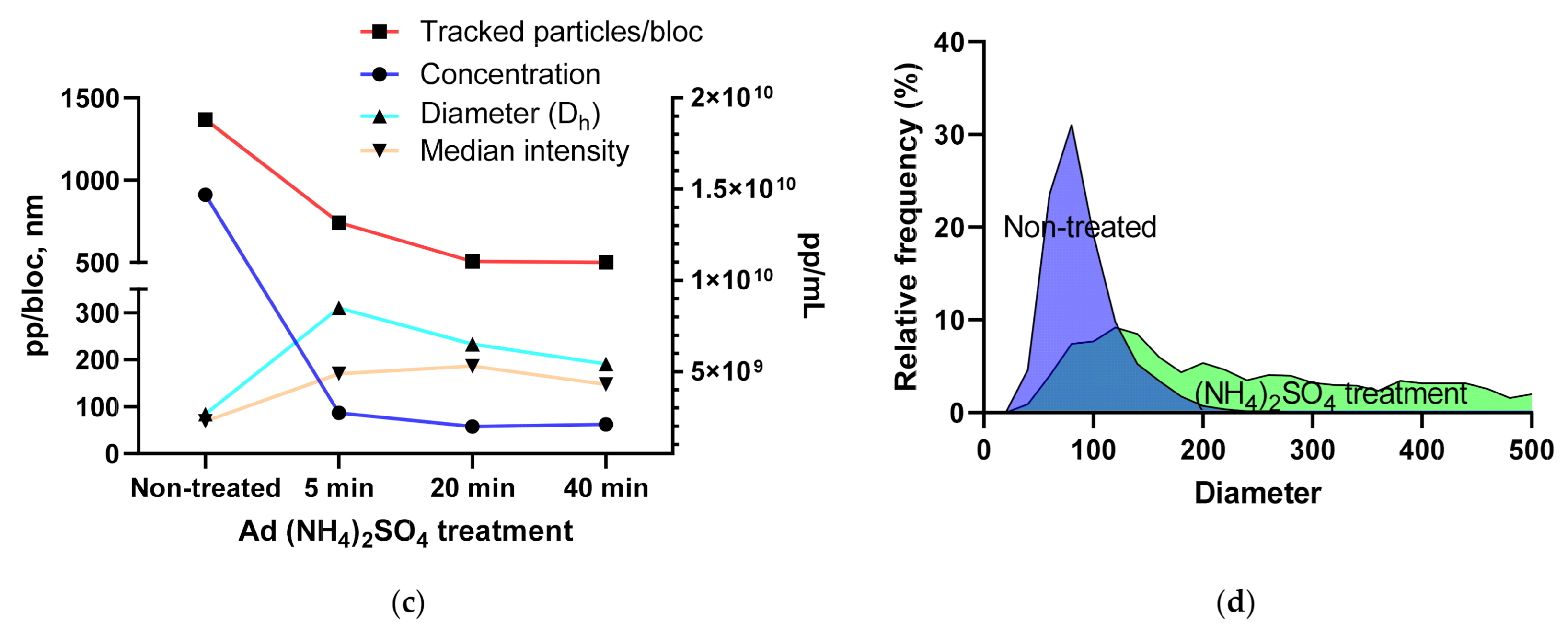
3.1.4. Using the Videodrop System for Detecting Virus Breakage or Aggregation
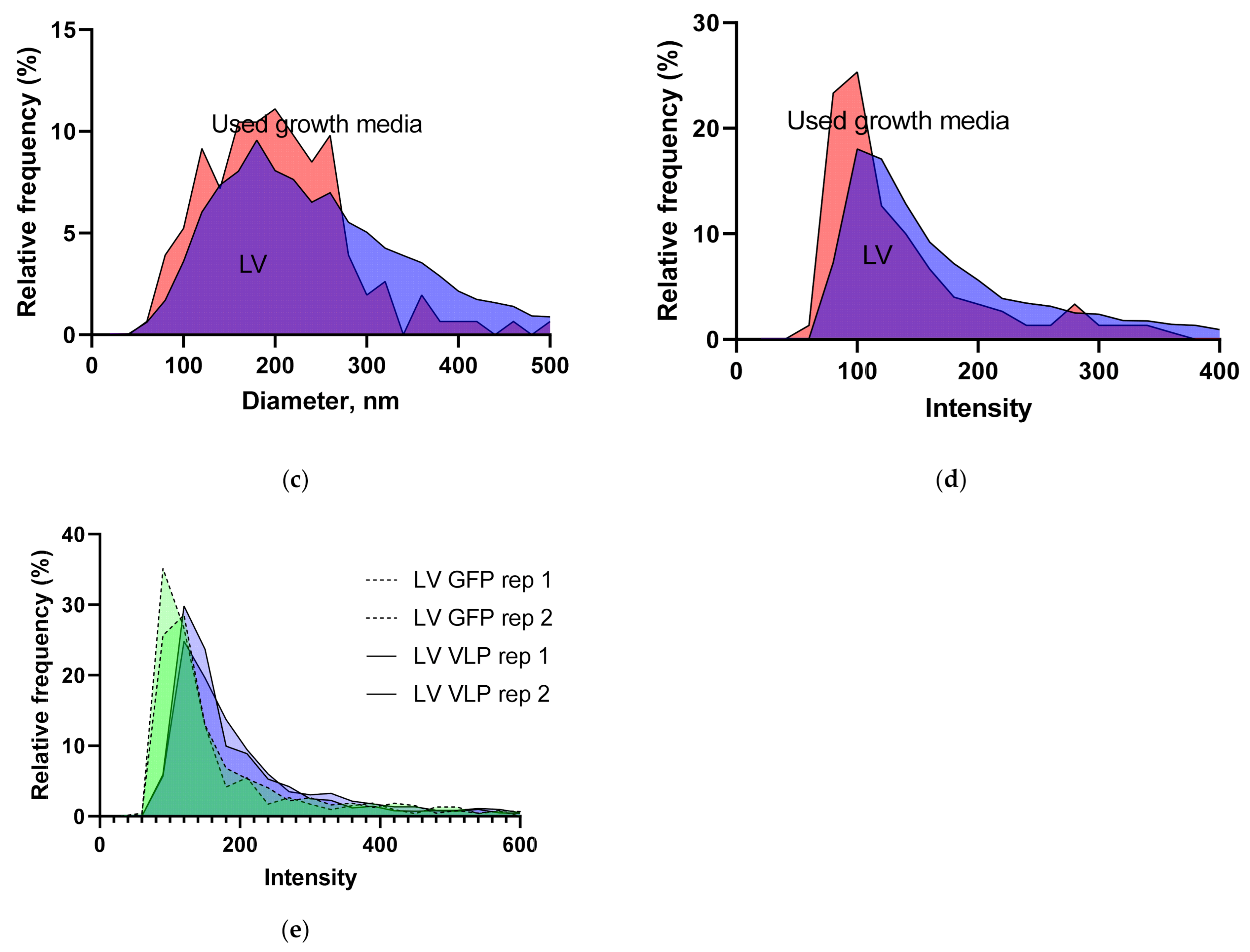
3.1.5. Intensity and Diameter Data
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Clark, M.F.; Adams, A.N.; Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of the Microplate Method of Enzyme-Linked Immunosorbent Assay for the Detection of Plant Viruses. J. Gen. Virol. 1977, 34, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Geraerts, M.; Willems, S.; Baekelandt, V.; Debyser, Z.; Gijsbers, R. Comparison of lentiviral vector titration methods. BMC Biotechnol. 2006, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabram, P.W.; Giroux, D.D.; Goudreau, A.M.; Gregory, R.J.; Horn, M.T.; Huyghe, B.G.; Liu, X.; Nunnally, M.H.; Sugarman, B.J.; Sutjipto, S. Analytical Anion-Exchange HPLC of Recombinant Type-5 Adenoviral Particles. Hum. Gene Ther. 1997, 8, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, I.; Larsen, B.; Gross, C.M.; Hermiston, T. High-performance liquid chromatography method for rapid assessment of viral particle number in crude adenoviral lysates of mixed serotype. Gene Ther. 2006, 14, 180–184. [Google Scholar] [CrossRef]
- Turkki, V.; Lesch, H.P.; Ryner, M.; Nilsson, J. Viral Vector Particle Integrity and Purity Analysis in Early Process Development. Bioprocess Int. 2017, 15, 34–37. [Google Scholar]
- Roingeard, P.; Raynal, P.-I.; Eymieux, S.; Blanchard, E. Virus detection by transmission electron microscopy: Still useful for diagnosis and a plus for biosafety. Rev. Med Virol. 2019, 29, e2019. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, Y.G.; McPherson, A. Atomic Force Microscopy in Imaging of Viruses and Virus-Infected Cells. Microbiol. Mol. Biol. Rev. 2011, 75, 268–285. [Google Scholar] [CrossRef] [Green Version]
- Filipe, V.; Hawe, A.; Jiskoot, W. Critical Evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the Measurement of Nanoparticles and Protein Aggregates. Pharm. Res. 2010, 27, 796–810. [Google Scholar] [CrossRef] [Green Version]
- Kramberger, P.; Ciringer, M.; Štrancar, A.; Peterka, M. Evaluation of nanoparticle tracking analysis for total virus particle determination. Virol. J. 2012, 9, 265. [Google Scholar] [CrossRef]
- Gast, M.; Sobek, H.; Mizaikoff, B. Nanoparticle Tracking of Adenovirus by Light Scattering and Fluorescence Detection. Hum. Gene Ther. Methods 2019, 30, 235–244. [Google Scholar] [CrossRef]
- Yang, L.; Yamamoto, T. Quantification of Virus Particles Using Nanopore-Based Resistive-Pulse Sensing Techniques. Front. Microbiol. 2016, 7, 1500. [Google Scholar] [CrossRef] [Green Version]
- Heider, S.; Muzard, J.; Zaruba, M.; Metzner, C. Integrated Method for Purification and Single-Particle Characterization of Lentiviral Vector Systems by Size Exclusion Chromatography and Tunable Resistive Pulse Sensing. Mol. Biotechnol. 2017, 59, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Hanne, J.; Zila, V.; Heilemann, M.; Müller, B.; Kräusslich, H.-G. Super-resolved insights into human immunodeficiency virus biology. FEBS Lett. 2016, 590, 1858–1876. [Google Scholar] [CrossRef]
- Lippé, R. Flow Virometry: A Powerful Tool to Functionally Characterize Viruses. J. Virol. 2017, 92, e01765-17. [Google Scholar] [CrossRef] [Green Version]
- Zamora, J.L.R.; Aguilar, H.C. Flow virometry as a tool to study viruses. Methods 2018, 134–135, 87–97. [Google Scholar] [CrossRef]
- Rossi, C.A.; Kearney, B.J.; Olschner, S.P.; Williams, P.L.; Robinson, C.G.; Heinrich, M.L.; Zovanyi, A.M.; Ingram, M.F.; Norwood, D.A.; Schoepp, R.J. Evaluation of ViroCyt® Virus Counter for Rapid Filovirus Quantitation. Viruses 2015, 7, 857–872. [Google Scholar] [CrossRef] [Green Version]
- Boccara, M.; Fedala, Y.; Bryan, C.V.; Bailly-Bechet, M.; Bowler, C.; Boccara, A.C. Full-field interferometry for counting and differentiating aquatic biotic nanoparticles: From laboratory to Tara Oceans. Biomed. Opt. Express 2016, 7, 3736–3746. [Google Scholar] [CrossRef] [Green Version]
- Roose-Amsaleg, C.; Fedala, Y.; Vénien-Bryan, C.; Garnier, J.; Boccara, A.-C.; Boccara, M. Utilization of interferometric light microscopy for the rapid analysis of virus abundance in a river. Res. Microbiol. 2017, 168, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Valkama, A.J.; Leinonen, H.M.; Lipponen, E.M.; Turkki, V.; Malinen, J.; Heikura, T.; Ylä-Herttuala, S.; Lesch, H.P. Optimization of lentiviral vector production for scale-up in fixed-bed bioreactor. Gene Ther. 2018, 25, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, H.M.; Lipponen, E.M.; Valkama, A.J.; Hynynen, H.; Oruetxebarria, I.; Turkki, V.; Olsson, V.; Kurkipuro, J.; Samaranayake, H.; Määttä, A.-M.; et al. Preclinical Proof-of-Concept, Analytical Development, and Commercial Scale Production of Lentiviral Vector in Adherent Cells. Mol. Ther. Methods Clin. Dev. 2019, 15, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Valkama, A.J.; Oruetxebarria, I.; Lipponen, E.M.; Leinonen, H.M.; Käyhty, P.; Hynynen, H.; Turkki, V.; Malinen, J.; Miinalainen, T.; Heikura, T.; et al. Development of Large-Scale Downstream Processing for Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2020, 17, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, H.M.; Lepola, S.; Lipponen, E.M.; Heikura, T.; Koponen, T.; Parker, N.R.; Ylä-Herttuala, S.; Lesch, H.P. Benchmarking of Scale-X Bioreactor System in Lentiviral and Adenoviral Vector Production. Hum. Gene Ther. 2020, 31, 376–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesch, H.P.; Heikkilä, K.M.; Lipponen, E.M.; Valonen, P.; Müller, A.; Räsänen, E.; Tuunanen, T.; Hassinen, M.M.; Parker, N.; Karhinen, M.; et al. Process Development of Adenoviral Vector Production in Fixed Bed Bioreactor: From Bench to Commercial Scale. Hum. Gene Ther. 2015, 26, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Lesch, H.P.; Turpeinen, S.; Niskanen, E.A.; Mähönen, A.J.; Airenne, K.J.; Ylä-Herttuala, S. Generation of Lentivirus Vectors Using Recombinant Baculoviruses. Gene Ther. 2008, 15, 1280–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Airenne, K.J.; Hiltunen, M.O.; Turunen, M.P.; Turunen, A.-M.; Laitinen, O.H.; Kulomaa, M.S.; Ylä-Herttuala, S. Baculovirus-mediated periadventitial gene transfer to rabbit carotid artery. Gene Ther. 2000, 7, 1499–1504. [Google Scholar] [CrossRef]
- Airenne, K.J.; Peltomaa, E.; Hytönen, V.P.; Laitinen, O.H.; Ylä-Herttuala, S. Improved generation of recombinant baculovirus genomes in Escherichia coli. Nucleic Acids Res. 2003, 31. [Google Scholar] [CrossRef]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Hutchins, B. Development of a Reference Material for Characterizing Adenovirus Vectors. Bioprocess. J. 2002, 1, 25–29. [Google Scholar] [CrossRef]
- Wang, Y.; Bergelson, S.; Feschenko, M. Determination of Lentiviral Infectious Titer by a Novel Droplet Digital PCR Method. Hum. Gene Ther. Methods 2018, 29, 96–103. [Google Scholar] [CrossRef]
- Motulsky, H.J.; Brown, R.E. Detecting outliers when fitting data with nonlinear regression—A new method based on robust nonlinear regression and the false discovery rate. BMC Bioinform. 2006, 7, 123. [Google Scholar] [CrossRef] [Green Version]
- Ukkonen, P.; Korpela, J.; Suni, J.; Hedman, K. Inactivation of human immunodeficiency virus in serum specimens as a safety measure for diagnostic immunoassays. Eur. J. Clin. Microbiol. Infect. Dis. 1988, 7, 518–523. [Google Scholar] [CrossRef]
- Jonges, M.; Liu, W.M.; Van Der Vries, E.; Jacobi, R.; Pronk, I.; Boog, C.; Koopmans, M.; Meijer, A.; Soethout, E. Influenza Virus Inactivation for Studies of Antigenicity and Phenotypic Neuraminidase Inhibitor Resistance Profiling. J. Clin. Microbiol. 2010, 48, 928–940. [Google Scholar] [CrossRef] [Green Version]
- Remy, M.; Alfter, M.; Chiem, M.-N.; Barbani, M.; Engler, O.; Suter-Riniker, F. Effective chemical virus inactivation of patient serum compatible with accurate serodiagnosis of infections. Clin. Microbiol. Infect. 2019, 25, 907.e7–907.e12. [Google Scholar] [CrossRef] [Green Version]
- Burova, E.; Ioffe, E. Chromatographic purification of recombinant adenoviral and adeno-associated viral vectors: Methods and implications. Gene Ther. 2005, 12, S5–S17. [Google Scholar] [CrossRef] [Green Version]
- Colby, W.W.; Shenk, T. Adenovirus type 5 virions can be assembled in vivo in the absence of detectable polypeptide IX. J. Virol. 1981, 39, 977–980. [Google Scholar] [CrossRef] [Green Version]
- Schagen, F.H.; Rademaker, H.J.; Rabelink, M.J.; van Ormondt, H.; Fallaux, F.J.; Van Der Eb, A.J.; Hoeben, R.C. Ammonium sulphate precipitation of recombinant adenovirus from culture medium: An easy method to increase the total virus yield. Gene Ther. 2000, 7, 1570–1574. [Google Scholar] [CrossRef] [Green Version]
- Shabram, P.W.; Vellekamp, G.; Ruan, Q.; Scandella, C. Purification of Adenovirus. In Adenoviral Vectors for Gene Therapy, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 197–227. [Google Scholar]
- Arroyo, J.O.; Cole, D.; Kukura, P. Interferometric scattering microscopy and its combination with single-molecule fluorescence imaging. Nat. Protoc. 2016, 11, 617–633. [Google Scholar] [CrossRef]
- González-Domínguez, I.; Puente-Massaguer, E.; Cervera, L.; Gòdia, F. Quality Assessment of Virus-Like Particles at Single Particle Level: A Comparative Study. Viruses 2020, 12, 223. [Google Scholar] [CrossRef] [Green Version]
- Vogel, R.; Savage, J.; Muzard, J.; Della Camera, G.; Vella, G.; Law, A.; Marchioni, M.; Mehn, D.; Geiss, O.; Peacock, B.; et al. Measuring particle concentration of multimodal synthetic reference materials and extracellular vesicles with orthogonal techniques: Who is up to the challenge? J. Extracell. Vesicles 2021, 10. [Google Scholar] [CrossRef]
- Heider, S.; Metzner, C. Quantitative real-time single particle analysis of virions. Virology 2014, 462–463, 199–206. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vector/Virus | Titer Type | Titer Value | CI | n | Method |
|---|---|---|---|---|---|
| LV internal control | vp/mL | 4.55 × 109 * | 4.16–4.93 × 109 | 46 | p24 ELISA |
| vg/mL | 8.29 × 109 | 6.85–9.74 × 109 | 49 ** | WPRE ddPCR | |
| TU/mL | 8.76 × 106 | 7.59–9.93 × 106 | 146 | qPCR | |
| Ad internal control | vp/mL | 5.0 × 1011 | NA (external data) | NA | HPLC |
| vg/mL | 5.48 × 1011 | 4.33–9.81 × 1011 | 22 | CMV ddPCR | |
| Purified BV | vg/mL | 9.58 × 1011 | 7.60 × 1011–1.16 × 1012 | 1 | WPRE ddPCR |
| Clarified BV | vg/mL | 2.35 × 1010 | 1.69–3.02 × 1010 | 1 | WPRE ddPCR |
| Vector/Virus | LV | Ad |
|---|---|---|
| Threshold setting | 4.2 | 3.2–3.5 |
| LOD, pp/mL (blank av + 3SD) * | 1.1 × 108 | 1.3 × 109 (TH 3.5) |
| Working range upper limit, pp/mL | 3 × 109 | N.D.† |
| Repeatability, single dilution (CV%) ‡ | 35.7 | N.D. |
| Repeatability, full assay (CV%) § | 14.0 | N.D. |
| Intermediate precision, full assay (CV%) ¶ | 19.4 | 40.6 |
| Method, Devices | Description | Range | Advantages | Disadvantages | References |
|---|---|---|---|---|---|
| Interferometric light microscopy (ILM) Videodrop (Myriade) | A drop (5–10 µL) of sample is illuminated by a visible light LED. Transmission brightfield microscope is used to measure the interferometric signal for NP detection. Brownian motion tracking for Dh. | >70 nm for biological NPs | Fast (≥3 min) and easy to use. Low sample volume Less affected by the screen effect No fluidics → easy start-up, cleaning and maintenance. Affordable device, low price-per-sample | High background noise Concentration limits max. 1 × 108–5 × 1010 pp/mL. For an assay with dilutions 1 × 108–7 × 109 or less (the Target Concentration method). Limited peer-reviewed literature available | [17,18] |
| Nanoparticle tracking analysis (NTA) NanoSight NS300, Nanosight LM10 (Malvern Panalytical Ltd.) | The sample (≥200 µL) is injected into a sample chamber and illuminated by a laser beam. NPs scatter the light, which is detected using a dark field microscope. Brownian motion tracking for Dh. | 30–1000 nm | Reasonably fast (≥5 min) High-resolution particle size distribution. Can be used label-free, but compatible with fluorescence labeling → Measurements from complex sample matrices. | Limited concentration range (107–109) and accuracy Large sample volume Screen effect | [8,9,10,39,40] |
| Tunable Resistive Pulse Sensing (TRPS) qNano qViro-X Exoid (IZON Science Ltd.) | The sample (40 µL) is pipetted into a flow cell. NPs suspended in electrolytes pass through a nanopore. A change in impedance is measured for each NP. Magnitude of the signal is used to count the the particle volume. Signal frequency is used to calculate the concentration. | 40 nm–20 μm | Accurate High concentration range 105–1011 particles/mL (size dependent) and very high resolving capacity. Affordable device Measures the actual particle diameter | Approx. 10 min per sample Membranes blocked by impurities | [11,40,41] |
| Flow virometry NanoAnalyzer (NanoFCM Inc) Virus Counter 3100 (Sartorius Stedim Biotech GmbH) +Standard cytometers optimized for virus detection | Flow-cytometer optimized for NPs: Sample is streamed through a sheat-fluid containing capillary. Single-particle flow is passed through laser light beams and the scattered light or fluorescent label is observed. Labels are typically used. | Generally ≥100–200 nm when using the scatter alone, ≥20 nm when using labels NanoAnalyzer: 40–1000 nm | NanoAnalyzer: Very fast, 1 min per measure. Very high resolving capacity. Low sample volume 10–100 µL Virus Counter 3100: Easy to operate, simple no-wash staining. Virus-specific kits, available. Universal Combo Dye for enveloped viruses. Linear dynamic range of 5 × 105–1 × 109 vp/mL. Simultaneous analysis of light scattering and fluorescent labeling | Problems associated with fluidics, clogging and bubbles. Daily calibration needed At least medium level technical expertise needed. Generally more expensive options. Virus Counter 3100: 30 min staining + <5 min detection. Higher price-per-sample. Kits available only for a limited number of viruses. No universal stain for non-enveloped viruses. Minimum sample volume 100 µL. | [14,15,16] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turkki, V.; Alppila, E.; Ylä-Herttuala, S.; Lesch, H.P. Experimental Evaluation of an Interferometric Light Microscopy Particle Counter for Titering and Characterization of Virus Preparations. Viruses 2021, 13, 939. https://doi.org/10.3390/v13050939
Turkki V, Alppila E, Ylä-Herttuala S, Lesch HP. Experimental Evaluation of an Interferometric Light Microscopy Particle Counter for Titering and Characterization of Virus Preparations. Viruses. 2021; 13(5):939. https://doi.org/10.3390/v13050939
Chicago/Turabian StyleTurkki, Vesa, Elisa Alppila, Seppo Ylä-Herttuala, and Hanna P. Lesch. 2021. "Experimental Evaluation of an Interferometric Light Microscopy Particle Counter for Titering and Characterization of Virus Preparations" Viruses 13, no. 5: 939. https://doi.org/10.3390/v13050939
APA StyleTurkki, V., Alppila, E., Ylä-Herttuala, S., & Lesch, H. P. (2021). Experimental Evaluation of an Interferometric Light Microscopy Particle Counter for Titering and Characterization of Virus Preparations. Viruses, 13(5), 939. https://doi.org/10.3390/v13050939