
Coxsackievirus B3—Its Potential as an Oncolytic Virus


Abstract
1. Introduction
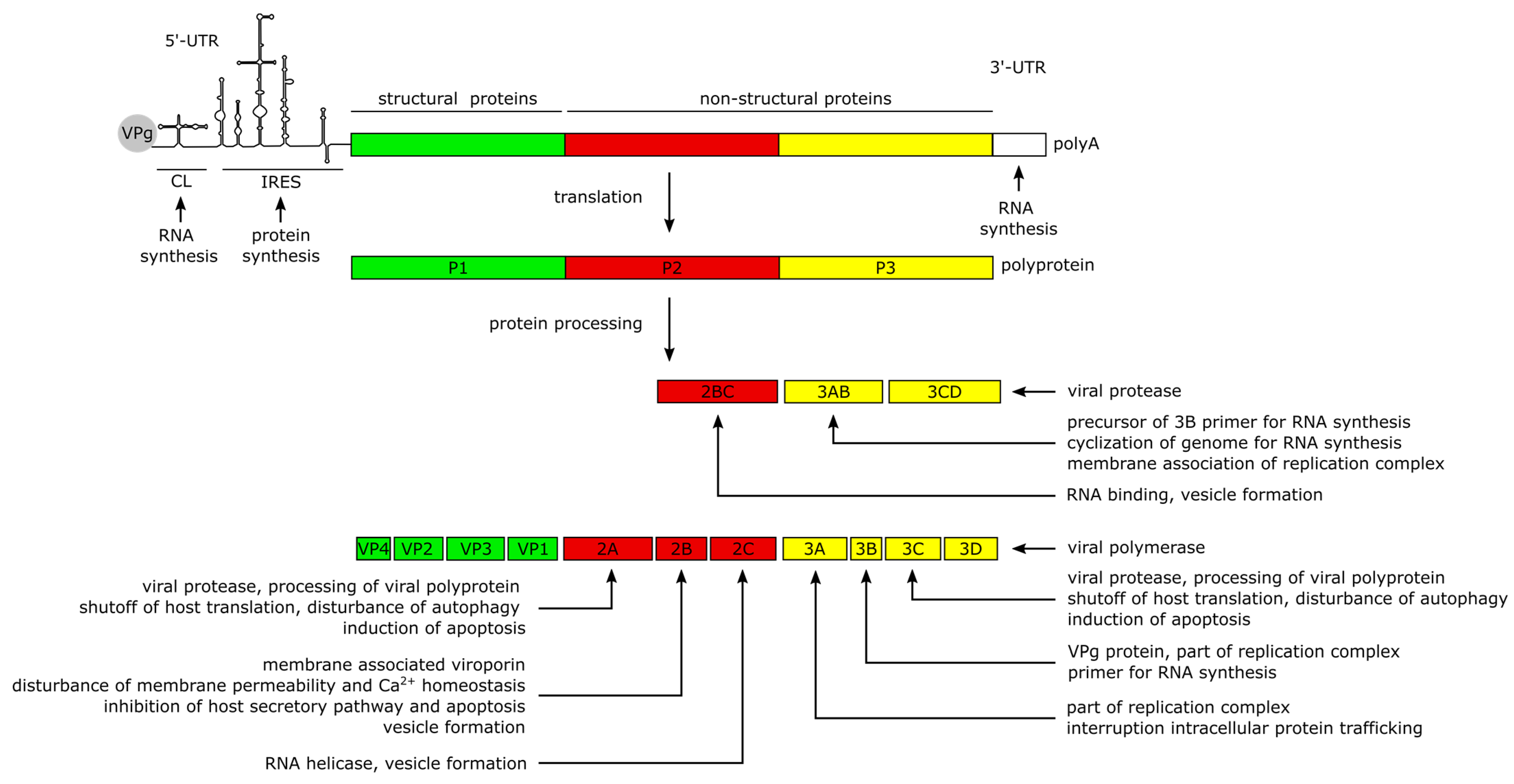
2. CVB3 Structure, Genome and Protein Functions
3. CVB3 Infections in Humans and in Experimentally Infected Mice
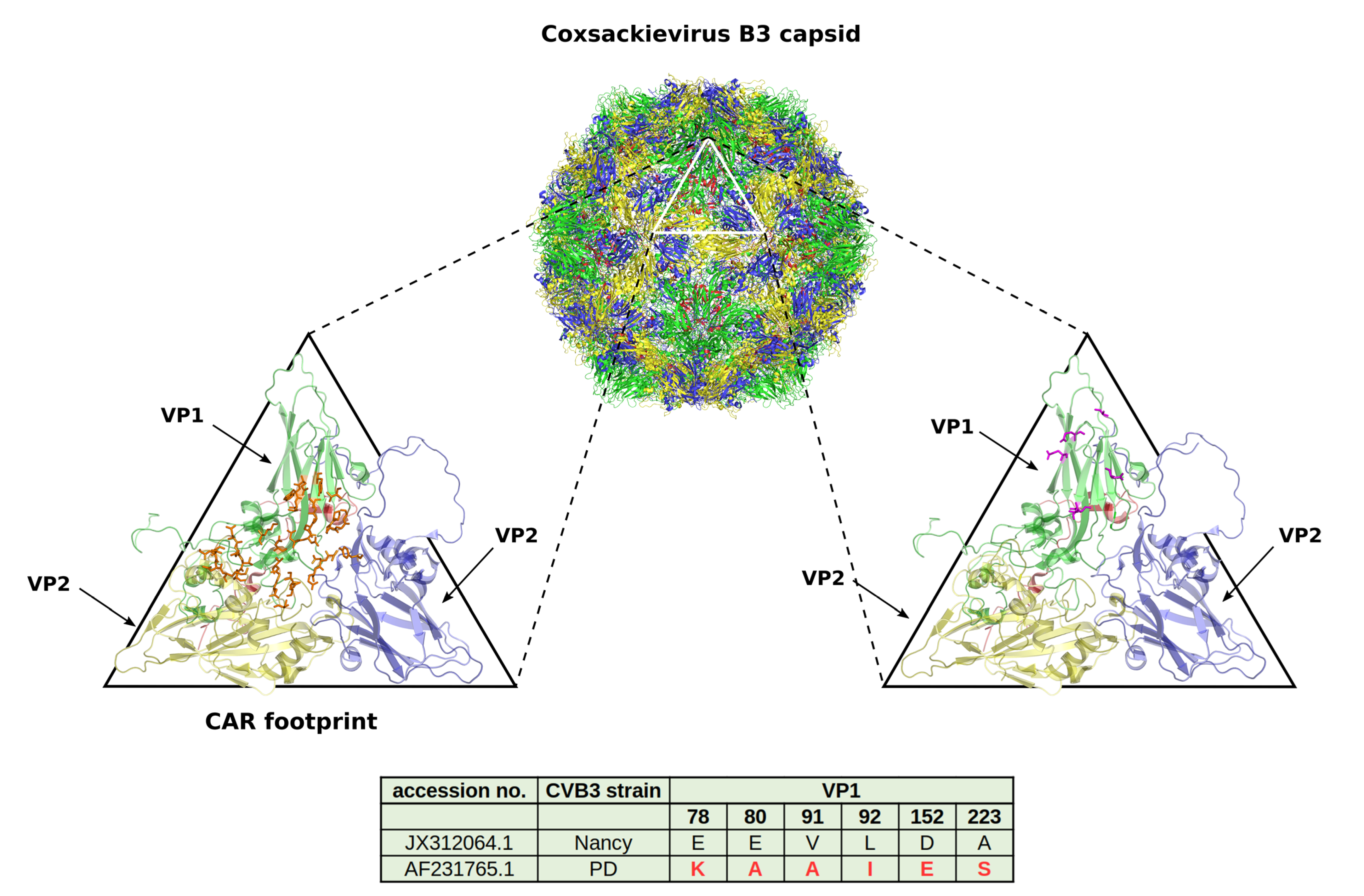
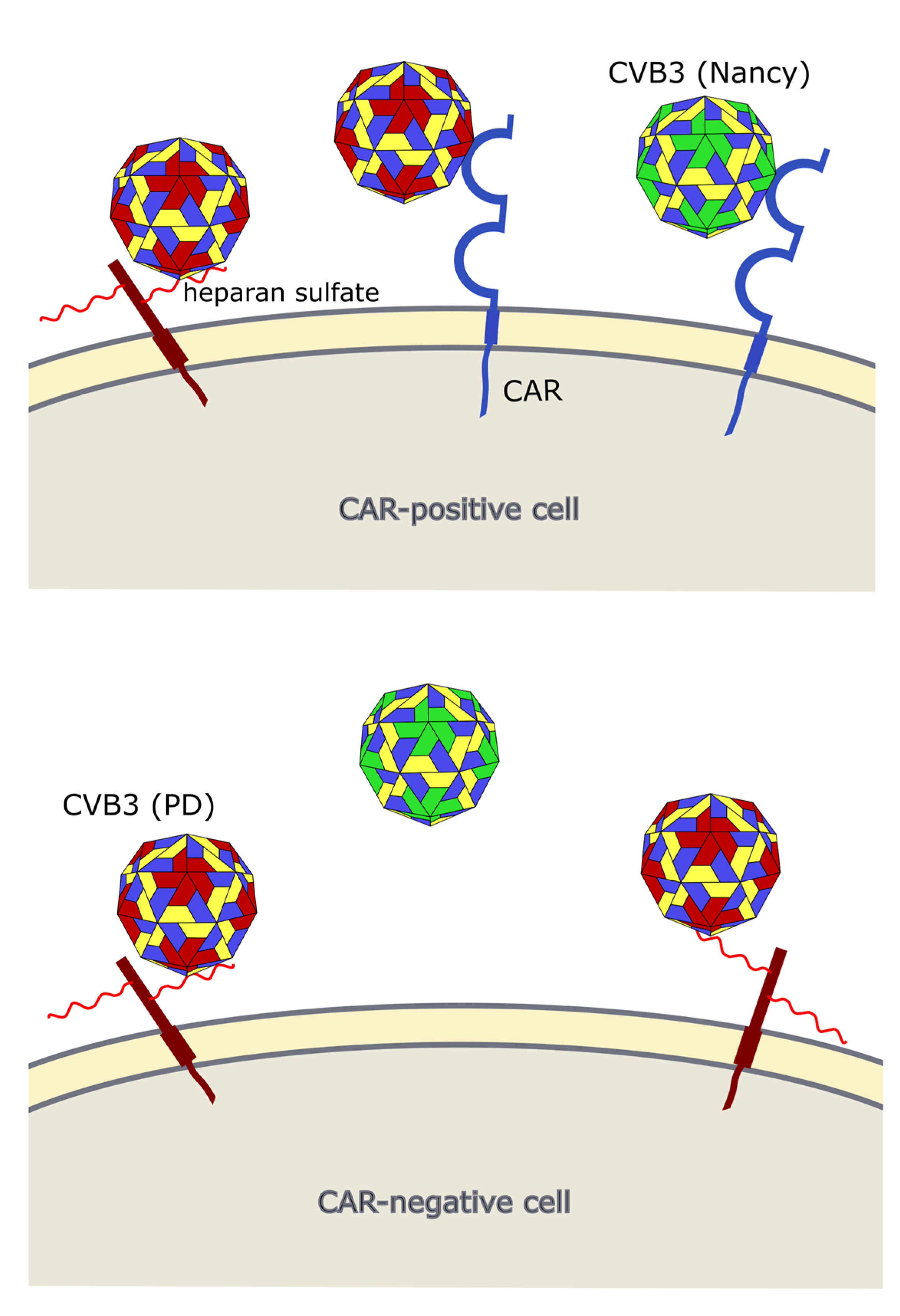
4. CVB3 Receptors and Its Importance for CVB3 Targeting of Cancer
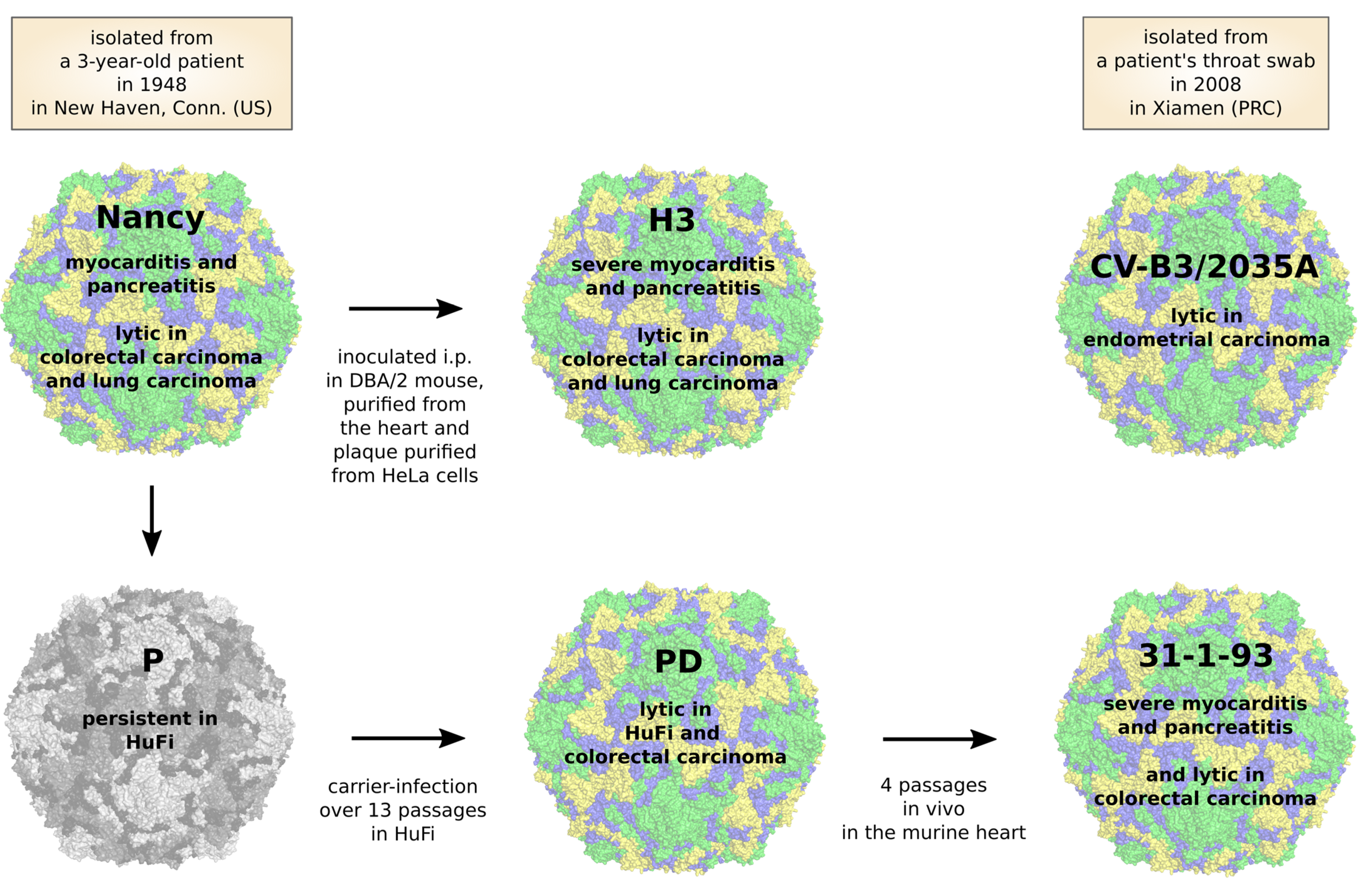
5. CVB3 Strains, Their Oncolytic Activity and Treatment-Related Side Effects
6. Influence of Oncolytic CVB3 on the Tumor Microenvironment
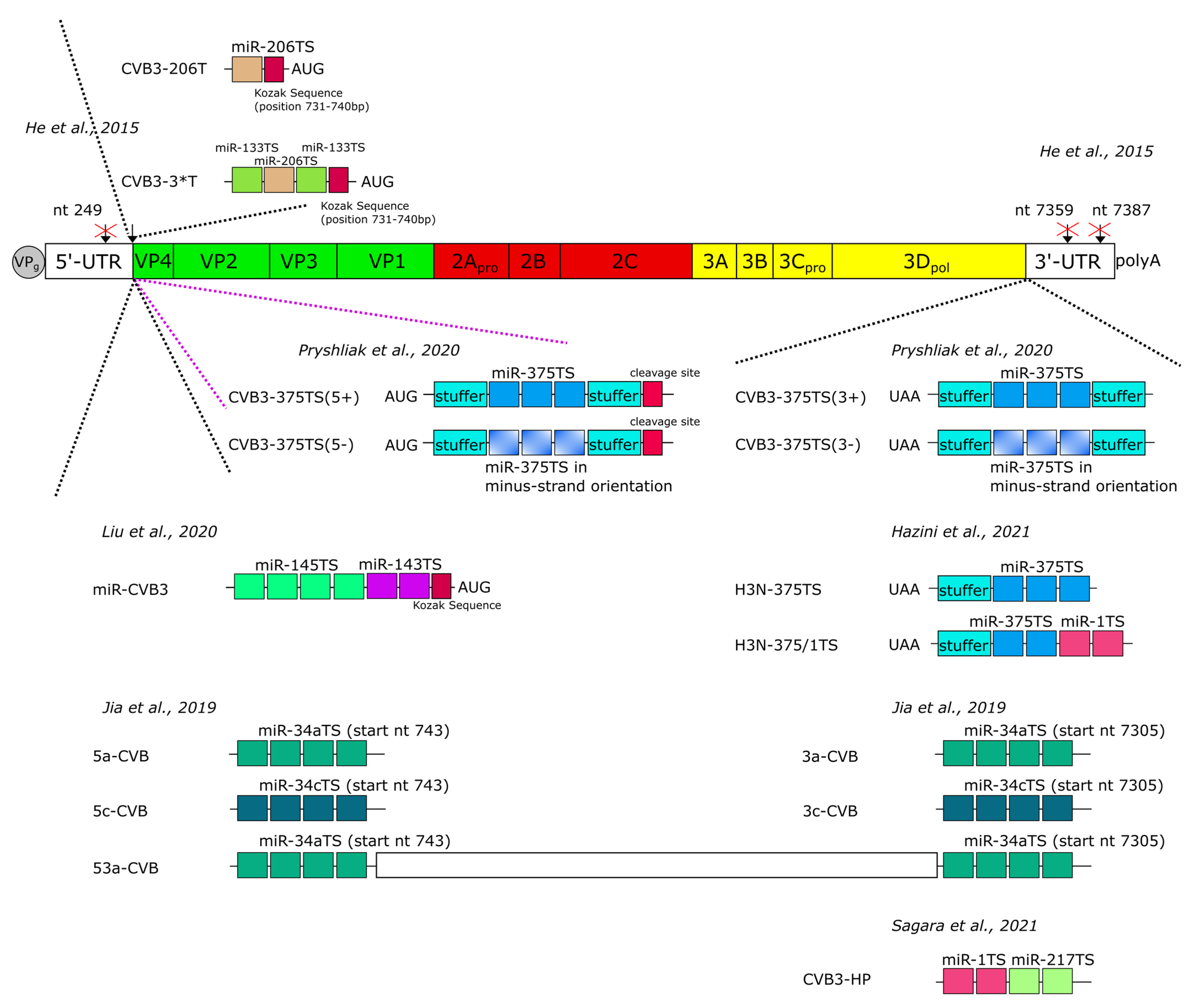
7. Improvement of the Safety of Oncolytic CVB3 by MicroRNA-Mediated Regulation of Virus Replication
8. Directed Virus Evolution as a Strategy to Increase Anti-Tumor Efficiency of Oncolytic CVB3
9. Genetic Engineering of CVB3 to Enhance Its Anti-Tumor Efficiency
10. Oncolytic Coxsackievirus B3 Versus Coxsackievirus A21
11. Conclusions/Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef]
- Anderson, B.D.; Nakamura, T.; Russell, S.J.; Peng, K.-W. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004, 64, 4919–4926. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Huang, J.; Wang, L.; Jia, D.; Yang, J.; Dillon, D.A.; Zurakowski, D.; Mao, H.; Moses, M.A.; Auguste, D.T. ICAM-1 as a molecular target for triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 14710–14715. [Google Scholar] [CrossRef]
- Moradi, A.; Zhand, S.; Hosseini, S.M.; Tabarraei, A.; Saeidi, M. Analysis of poliovirus receptor, CD155 expression in different human colorectal cancer cell lines: Implications for poliovirus virotherapy. J. Cancer Res. Ther. 2018, 15, 61–67. [Google Scholar] [CrossRef]
- Au, G.G.; Beagley, L.G.; Haley, E.S.; Barry, R.D.; Shafren, D.R. Oncolysis of malignant human melanoma tumors by Coxsackieviruses A13, A15 and A18. Virol. J. 2011, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Achard, C.; Boisgerault, N.; Delaunay, T.; Roulois, D.; Nedellec, S.; Royer, P.-J.; Pain, M.; Combredet, C.; Mesel-Lemoine, M.; Cellerin, L.; et al. Sensitivity of human pleural mesothelioma to oncolytic measles virus depends on defects of the type I interferon response. Oncotarget 2015, 6, 44892–44904. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Althof, N.; Harkins, S.; Kemball, C.C.; Flynn, C.T.; Alirezaei, M.; Whitton, J.L. In vivo ablation of type I interferon receptor from cardiomyocytes delays coxsackieviral clearance and accelerates myocardial disease. J. Virol. 2014, 88, 5087–5099. [Google Scholar] [CrossRef] [PubMed]
- Moerdyk-Schauwecker, M.; Shah, N.R.; Murphy, A.M.; Hastie, E.; Mukherjee, P.; Grdzelishvili, V.Z. Resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus: Role of type I interferon signaling. Virology 2013, 436, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Zarate, D.; Liu, Y.-P.; Suksanpaisan, L.; Russell, S.J.; Peng, K.-W. Overcoming cancer cell resistance to VSV oncolysis with JAK1/2 inhibitors. Cancer Gene Ther. 2013, 20, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.D.; Mezhir, J.J.; Bickenbach, K.; Veerapong, J.; Charron, J.; Posner, M.C.; Roizman, B.; Weichselbaum, R.R. Activated MEK suppresses activation of PKR and enables efficient replication and in vivo oncolysis by Δγ134.5 mutants of herpes simplex virus 1. J. Virol. 2006, 80, 1110–1120. [Google Scholar] [CrossRef]
- Poppers, J.; Mulvey, M.; Khoo, D.; Mohr, I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 2000, 74, 11215–11221. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef] [PubMed]
- Toda, M.; Rabkin, S.D.; Kojima, H.; Martuza, R.L. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum. Gene Ther. 1999, 10, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic parvoviruses: From basic virology to clinical applications. Virol. J. 2015, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Fang, H. Clinical trials with oncolytic adenovirus in China. Curr. Cancer Drug Targets 2007, 7, 141–148. [Google Scholar] [CrossRef]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef]
- Suskind, R.G.; Huebner, R.J.; Rowe, W.P.; Love, R. Viral agents oncolytic for human tumors in heterologous host. oncolytic effect of coxsackie B viruses. Proc. Soc. Exp. Biol. Med. 1957, 94, 309–318. [Google Scholar] [CrossRef]
- Miyamoto, S.; Inoue, H.; Nakamura, T.; Yamada, M.; Sakamoto, C.; Urata, Y.; Okazaki, T.; Marumoto, T.; Takahashi, A.; Takayama, K.; et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012, 72, 2609–2621. [Google Scholar] [CrossRef]
- Miyamoto, S.; Sagara, M.; Kohara, H.; Tani, K. Oncolytic coxsackievirus therapy as an immunostimulator. Rinsho Ketsueki 2017, 58, 977–982. [Google Scholar] [CrossRef]
- Garmaroudi, F.S.; Marchant, D.; Hendry, R.; Luo, H.; Yang, D.; Ye, X.; Shi, J.; McManus, B.M. Coxsackievirus B3 replication and pathogenesis. Future Microbiol. 2015, 10, 629–653. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Ramsingh, A.I.; Collins, D.N. A point mutation in the VP4 coding sequence of coxsackievirus B4 influences virulence. J. Virol. 1995, 69, 7278–7281. [Google Scholar] [CrossRef] [PubMed]
- Ansardi, D.C.; Porter, D.C.; Morrow, C.D. Myristylation of poliovirus capsid precursor P1 is required for assembly of subviral particles. J. Virol. 1992, 66, 4556–4563. [Google Scholar] [CrossRef] [PubMed]
- Organtini, L.J.; Makhov, A.M.; Conway, J.F.; Hafenstein, S.; Carson, S.D. Kinetic and structural analysis of coxsackievirus B3 receptor interactions and formation of the A-particle. J. Virol. 2014, 88, 5755–5765. [Google Scholar] [CrossRef]
- Muckelbauer, J.K.; Kremer, M.; Minor, I.; Diana, G.; Dutko, F.J.; Groarke, J.; Pevear, D.C.; Rossmann, M.G. The structure of coxsackievirus B3 at 3.5 å resolution. Structure 1995, 3, 653–667. [Google Scholar] [CrossRef]
- Oliveira, M.A.; Zhao, R.; Lee, W.-M.; Kremer, M.J.; Minor, I.; Rueckert, R.R.; Diana, G.D.; Pevear, D.C.; Dutko, F.J.; A McKinlay, M.; et al. The structure of human rhinovirus 16. Structure 1993, 1, 51–68. [Google Scholar] [CrossRef]
- Milestone, A.M.; Petrella, J.E.; Sanchez, M.D.; Mahmud, M.; Whitbeck, J.C.; Bergelson, J.M. Interaction with coxsackievirus and adenovirus receptor, but not with decay-accelerating factor (DAF), induces A-particle formation in a DAF-binding coxsackievirus B3 isolate. J. Virol. 2005, 79, 655–660. [Google Scholar] [CrossRef]
- Bergelson, J.M.; Krithivas, A.; Celi, L.; Droguett, G.; Horwitz, M.S.; Wickham, T.; Crowell, R.L.; Finberg, R.W. The murine CAR homolog is a receptor for coxsackie B viruses and adenoviruses. J. Virol. 1998, 72, 415–419. [Google Scholar] [CrossRef]
- Tomko, R.P.; Xu, R.; Philipson, L. HCAR and MCAR: The human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. USA 1997, 94, 3352–3356. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.G. Viral cell recognition and entry. Protein Sci. 1994, 3, 1712–1725. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.G.; Arnold, E.; Erickson, J.W.; Frankenberger, E.A.; Griffith, J.P.; Hecht, H.J.; Johnson, J.E.; Kamer, G.; Luo, M.; Mosser, A.G.; et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nat. Cell Biol. 1985, 317, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Hogle, J.M.; Chow, M.; Filman, D.J. Three-dimensional structure of poliovirus at 2.9 A resolution. Science 1985, 229, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Hafenstein, S.; Bowman, V.D.; Chipman, P.R.; Kelly, C.M.B.; Lin, F.; Medof, M.E.; Rossmann, M.G. Interaction of decay-accelerating factor with coxsackievirus B3. J. Virol. 2007, 81, 12927–12935. [Google Scholar] [CrossRef] [PubMed]
- Yoder, J.D.; Cifuente, J.O.; Pan, J.; Bergelson, J.M.; Hafenstein, S. The crystal structure of a coxsackievirus B3-RD variant and a refined 9-angstrom cryo-electron microscopy reconstruction of the virus complexed with decay-accelerating factor (DAF) Provide a new footprint of DAF on the virus surface. J. Virol. 2012, 86, 12571–12581. [Google Scholar] [CrossRef]
- Andino, R.; Rieckhof, G.E.; Baltimore, D. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell 1990, 63, 369–380. [Google Scholar] [CrossRef]
- Pelletier, J.; Sonenberg, N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nat. Cell Biol. 1988, 334, 320–325. [Google Scholar] [CrossRef]
- Zell, R.; Sidigi, K.; Bucci, E.; Stelzner, A.; Görlach, M. Determinants of the recognition of enteroviral cloverleaf RNA by cox-sackievirus B3 proteinase 3C. RNA 2002, 8, 188–201. [Google Scholar] [CrossRef]
- Bailey, J.M.; Tapprich, W.E. Structure of the 5′ nontranslated region of the coxsackievirus B3 genome: Chemical modification and comparative sequence analysis. J. Virol. 2006, 81, 650–668. [Google Scholar] [CrossRef]
- Lin, J.-Y.; Chen, T.-C.; Weng, K.-F.; Chang, S.-C.; Chen, L.-L.; Shih, S.-R. Viral and host proteins involved in picornavirus life cycle. J. Biomed. Sci. 2009, 16, 103. [Google Scholar] [CrossRef]
- Peischard, S.; Ho, H.T.; Theiss, C.; Strutz-Seebohm, N.; Seebohm, G. A kidnapping story: How coxsackievirus B3 and its host cell interact. Cell. Physiol. Biochem. 2019, 53, 121–140. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Harris, K.S.; Alexander, L.; Wimmer, E. Interaction between the 5′-terminal cloverleaf and 3AB/3CDpro of po-liovirus is essential for RNA replication. J. Virol. 1995, 69, 3658–3667. [Google Scholar] [CrossRef]
- Andino, R.; Rieckhof, G.; Achacoso, P.; Baltimore, D. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 1993, 12, 3587–3598. [Google Scholar] [CrossRef]
- Melchers, W.J.; Hoenderop, J.G.; Slot, H.J.B.; Pleij, C.W.; Pilipenko, E.V.; I Agol, V.; Galama, J.M. Kissing of the two predominant hairpin loops in the coxsackie B virus 3’ untranslated region is the essential structural feature of the origin of replication required for negative-strand RNA synthesis. J. Virol. 1997, 71, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Chau, D.H.W.; Yuan, J.; Zhang, H.; Cheung, P.; Lim, T.; Liu, Z.; Sall, A.; Yang, D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 2006, 12, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Kerekatte, V.; Keiper, B.D.; Badorff, C.; Cai, A.; Knowlton, K.U.; Rhoads, R.E. Cleavage of poly(A)-binding protein by cox-sackievirus 2A protease in vitro and in vivo: Another mechanism for host protein synthesis shutoff? J. Virol. 1999, 73, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Kahvejian, A.; Svitkin, Y.V.; Sukarieh, R.; M’Boutchou, M.N.; Sonenberg, N. Mammalian poly(A)-binding protein is a eu-karyotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 2005, 19, 104–113. [Google Scholar] [CrossRef]
- Hanson, P.J.; Ye, X.; Qiu, Y.; Zhang, H.M.; Hemida, M.G.; Wang, F.; Lim, T.; Gu, A.; Cho, B.; Kim, H.; et al. Cleavage of DAP5 by coxsackievirus B3 2A protease facilitates viral replication and enhances apoptosis by altering translation of IRES-containing genes. Cell Death Differ. 2015, 23, 828–840. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef]
- Lim, B.-K.; Peter, A.K.; Xiong, D.; Narezkina, A.; Yung, A.; Dalton, N.D.; Hwang, K.-K.; Yajima, T.; Chen, J.; Knowlton, K.U. Inhibition of Coxsackievirus-associated dystrophin cleavage prevents cardiomyopathy. J. Clin. Investig. 2013, 123, 5146–5151. [Google Scholar] [CrossRef] [PubMed]
- de Jong, A.S.; Wessels, E.; Dijkman, H.B.P.M.; Galama, J.M.D.; Melchers, W.J.G.; Willems, P.H.G.M.; van Kuppeveld, F.J.M. Determinants for membrane association and permeabilization of the coxsackievirus 2B protein and the identification of the golgi complex as the target organelle. J. Biol. Chem. 2003, 278, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- de Jong, A.S.; Visch, H.-J.; de Mattia, F.; van Dommelen, M.M.; Swarts, H.G.; Luyten, T.; Callewaert, G.; Melchers, W.J.; Willems, P.H.; van Kuppeveld, F.J. The coxsackievirus 2B protein increases efflux of ions from the endoplasmic reticulum and golgi, thereby inhibiting protein trafficking through the golgi. J. Biol. Chem. 2006, 281, 14144–14150. [Google Scholar] [CrossRef] [PubMed]
- De Jong, A.S.; Schrama, I.W.J.; Willems, P.H.G.M.; Galama, J.M.D.; Melchers, W.J.G.; Van Kuppeveld, F.J.M. Multimerization reactions of coxsackievirus proteins 2B, 2C and 2BC: A mammalian two-hybrid analysis. J. Gen. Virol. 2002, 83, 783–793. [Google Scholar] [CrossRef]
- Van Kuppeveld, F.J.; Hoenderop, J.G.; Smeets, R.L.; Willems, P.H.; Dijkman, H.B.; Galama, J.M.; Melchers, W.J. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. 1997, 16, 3519–3532. [Google Scholar] [CrossRef]
- Buenz, E.J.; Howe, C.L. Picornaviruses and cell death. Trends Microbiol. 2006, 14, 28–36. [Google Scholar] [CrossRef]
- Campanella, M.; de Jong, A.S.; Lanke, K.W.H.; Melchers, W.J.G.; Willems, P.H.G.M.; Pinton, P.; Rizzuto, R.; van Kuppeveld, F.J.M. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J. Biol. Chem. 2004, 279, 18440–18450. [Google Scholar] [CrossRef]
- Van Kuppeveld, F.J.; Galama, J.M.; Zoll, J.; Hurk, P.J.V.D.; Melchers, W.J. Coxsackie B3 virus protein 2B contains cationic amphipathic helix that is required for viral RNA replication. J. Virol. 1996, 70, 3876–3886. [Google Scholar] [CrossRef]
- Xia, H.; Wang, P.; Wang, G.C.; Yang, J.; Sun, X.; Wu, W.; Qiu, Y.; Shu, T.; Zhao, X.; Yin, L.; et al. Human enterovirus nonstructural protein 2CATPase functions as both an RNA helicase and ATP-independent RNA chaperone. PLoS Pathog. 2015, 11, e1005067. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, C.; Yang, R.; Bai, P.; Zhang, X.-Y.; Kong, J.; Yin, L.; Qiu, Y.; Zhou, X. Antiviral peptides targeting the helicase activity of enterovirus nonstructural protein 2C. J. Virol. 2021. [Google Scholar] [CrossRef]
- Yun, S.-H.; Shin, H.-H.; Ju, E.-S.; Lee, Y.-J.; Lim, B.-K.; Jeon, E.-S. Inhibition of RNA helicase activity prevents coxsackievirus B3-induced myocarditis in human iPS cardiomyocytes. Int. J. Mol. Sci. 2020, 21, 3041. [Google Scholar] [CrossRef]
- Paul, A.V.; van Boom, J.H.; Filippov, D.; Wimmer, E. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature 1998, 393, 280–284. [Google Scholar] [CrossRef]
- Molla, A.; Harris, K.S.; Paul, A.V.; Shin, S.H.; Mugavero, J.; Wimmer, E. Stimulation of poliovirus proteinase 3Cpro-related proteolysis by the genome-linked protein VPg and its precursor 3AB. J. Biol. Chem. 1994, 269, 27015–27020. [Google Scholar] [CrossRef]
- Harris, K.S.; Xiang, W.; Alexander, L.; Lane, W.S.; Paul, A.V.; Wimmer, E. Interaction of poliovirus polypeptide 3CDpro with the 5′ and 3′ termini of the poliovirus genome. Identification of viral and cellular cofactors needed for efficient binding. J. Biol. Chem. 1994, 269, 27004–27014. [Google Scholar] [CrossRef]
- Bowles, N.E.; Richardson, P.J.; Olsen, E.G.; Archard, L.C. Detection of Coxsackie-B-virus-specific RNA sequences in myocardial biopsy samples from patients with myocarditis and dilated cardiomyopathy. Lancet 1986, 1, 1120–1123. [Google Scholar] [CrossRef]
- Compagnoli Carmona, R.D.; Caetano Machado, B.; Aparecida de Sousa, C.; Vieira, H.R.; Moraes Alves, M.R.; Farias de Souza, K.A.; de Souza Gregório, D.; Costa Vilanova, B.; Sampaio Tavares Timenetsky, M.D. Distribution of species enterovirus B in patients with central nervous system infections in São Paulo State, Brazil. J. Med. Virol. 2020, 92, 3849–3856. [Google Scholar] [CrossRef] [PubMed]
- Foulis, A.K.; Farquharson, M.A.; Cameron, S.O.; McGill, M.; Schonke, H.; Kandolf, R. A search for the presence of the enteroviral capsid protein VP1 in pancreases of patients with type 1 (insulin-dependent) diabetes and pancreases and hearts of infants who died of coxsackieviral myocarditis. Diabetologia 1990, 33, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Tracy, S.; Hofling, K.; Pirruccello, S.; Lane, P.H.; Reyna, S.M.; Gauntt, C.J. Group B coxsackievirus myocarditis and pancreatitis: Connection between viral virulence phenotypes in mice. J. Med. Virol. 2000, 62, 70–81. [Google Scholar] [CrossRef]
- Wang, S.-M.; Liu, C.-C.; Yang, Y.-J.; Yang, H.-B.; Lin, C.-H.; Wang, J.-R. Fatao coxsackievirus B infection in early infancy characterized by fulminant hepatitis. J. Infect. 1998, 37, 270–273. [Google Scholar] [CrossRef]
- Tariq, N.; Kyriakopoulos, C. Group B Coxsackie Virus; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Iwasaki, T.; Monma, N.; Satodate, R.; Kawana, R.; Kurata, T. An immunofluorescent study of generalized Coxsackie virus B3 infection in a newborn infant. Acta Pathol. Jpn. 1985, 35, 741–748. [Google Scholar] [CrossRef]
- Ronellenfitsch, S.; Tabatabai, J.; Bottcher, S.; Diedrich, S.; Frommhold, D.; Schubert-Bast, S.; Poeschl, J.; Schnitzler, P. First report of a Chinese strain of coxsackie B3 virus infection in a newborn in Germany in 2011: A case report. J. Med. Case Rep. 2014, 8, 164. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Wang, S.-M.; Chen, I.-C.; Yu, C.-K.; Liu, C.-C. Hepatic damage caused by coxsackievirus B3 is dependent on age-related tissue tropisms associated with the coxsackievirus-adenovirus receptor. Pathog. Dis. 2013, 68, 52–60. [Google Scholar] [CrossRef]
- Pinkert, S.; Pryshliak, M.; Pappritz, K.; Knoch, K.; Hazini, A.; Dieringer, B.; Schaar, K.; Dong, F.; Hinze, L.; Lin, J.; et al. Development of a new mouse model for coxsackievirus-induced myocarditis by attenuating coxsackievirus B3 virulence in the pancreas. Cardiovasc. Res. 2020, 116, 1756–1766. [Google Scholar] [CrossRef]
- Kallewaard, N.L.; Zhang, L.; Chen, J.W.; Guttenberg, M.; Sanchez, M.D.; Bergelson, J.M. Tissue-specific deletion of the coxsackievirus and adenovirus receptor protects mice from virus-induced pancreatitis and myocarditis. Cell Host Microbe 2009, 6, 91–98. [Google Scholar] [CrossRef]
- Pinkert, S.; Westermann, D.; Wang, X.; Klingel, K.; Dorner, A.; Savvatis, K.; Grossl, T.; Krohn, S.; Tschope, C.; Zeichhardt, H.; et al. Prevention of cardiac dysfunction in acute coxsackievirus B3 cardiomyopathy by inducible expression of a soluble coxsackievirus-adenovirus receptor. Circulation 2009, 120, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, M.; Merkle, I.; Klingel, K.; Hammerschmidt, E.; Zautner, A.E.; Wutzler, P. The viral genetic background determines the outcome of coxsackievirus B3 infection in outbred NMRI mice. J. Med. Virol. 2007, 79, 1334–1342. [Google Scholar] [CrossRef]
- Koenig, A.; Buskiewicz, I.; Huber, S.A. Age-associated changes in estrogen receptor ratios correlate with increased female susceptibility to coxsackievirus B3-induced myocarditis. Front. Immunol. 2017, 8, 1585. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Xu, W.; Guo, Q.; Jiang, Z.; Wang, P.; Yue, Y.; Xiong, S. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ. Res. 2009, 105, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Leipner, C.; Grun, K.; Schneider, I.; Gluck, B.; Sigusch, H.H.; Stelzner, A. Coxsackievirus B3-induced myocarditis: Differences in the immune response of C57BL/6 and Balb/c mice. Med. Microbiol. Immunol. 2004, 193, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Carson, S.D.; Chapman, N.N.; Tracy, S.M. Purification of the putative coxsackievirus B receptor from HeLa cells. Biochem. Biophys. Res. Commun. 1997, 233, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Zapater, E.; Santis, G.; Parsons, M. CAR: A key regulator of adhesion and inflammation. Int. J. Biochem. Cell Biol. 2017, 89, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Mohanty, J.G.; Crowell, R.L.; St John, N.F.; Lublin, D.M.; Finberg, R.W. Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55). J. Virol. 1995, 69, 1903–1906. [Google Scholar] [CrossRef]
- Shafren, D.R.; Bates, R.C.; Agrez, M.V.; Herd, R.L.; Burns, G.F.; Barry, R.D. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J. Virol. 1995, 69, 3873–3877. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chipman, P.R.; Howitt, J.; Bator, C.M.; Whitt, M.A.; Baker, T.S.; Kuhn, R.J.; Anderson, C.W.; Freimuth, P.; Rossmann, M.G. Interaction of coxsackievirus B3 with the full length coxsackievirus-adenovirus receptor. Nat. Genet. 2001, 8, 874–878. [Google Scholar] [CrossRef]
- Fechner, H.; Haack, A.; Wang, H.; Wang, X.; Eizema, K.; Pauschinger, M.; Schoemaker, R.; Veghel, R.; Houtsmuller, A.; Schultheiss, H.P.; et al. Expression of coxsackie adenovirus receptor and alphav-integrin does not correlate with adenovector targeting in vivo indicating anatomical vector barriers. Gene Ther. 1999, 6, 1520–1535. [Google Scholar] [CrossRef]
- Zanone, M.M.; Favaro, E.; Ferioli, E.; Huang, G.C.; Klein, N.J.; Perin, P.C.; Peakman, M.; Conaldi, P.G.; Camussi, G. Human pancreatic islet endothelial cells express coxsackievirus and adenovirus receptor and are activated by coxsackie B virus infection. FASEB J. 2007, 21, 3308–3317. [Google Scholar] [CrossRef]
- Fechner, H.; Noutsias, M.; Tschoepe, C.; Hinze, K.; Wang, X.; Escher, F.; Pauschinger, M.; Dekkers, D.; Vetter, R.; Paul, M.; et al. Induction of coxsackievirus-adenovirus-receptor expression during myocardial tissue formation and remodeling: Identification of a cell-to-cell contact-dependent regulatory mechanism. Circulation 2003, 107, 876–882. [Google Scholar] [CrossRef]
- Reeh, M.; Bockhorn, M.; Görgens, D.; Vieth, M.; Hoffmann, T.; Simon, R.; Izbicki, J.R.; Sauter, G.; Schumacher, U.; Anders, M. Presence of the coxsackievirus and adenovirus receptor (CAR) in human neoplasms: A multitumour array analysis. Br. J. Cancer 2013, 109, 1848–1858. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, W.; Wan, J.; Yang, Y.; Fu, W.; Pan, D.; Cai, L.; Cheng, T.; Huang, X.; Wang, Y. Oncolytic activity of a coxsackievirus B3 strain in human endometrial cancer cell lines. Virol. J. 2018, 15, 65. [Google Scholar] [CrossRef]
- Hazini, A.; Pryshliak, M.; Bruckner, V.; Klingel, K.; Sauter, M.; Pinkert, S.; Kurreck, J.; Fechner, H. Heparan sulfate binding coxsackievirus B3 strain PD: A novel avirulent oncolytic agent against human colorectal carcinoma. Hum. Gene Ther. 2018, 29, 1301–1314. [Google Scholar] [CrossRef]
- Zautner, A.E.; Korner, U.; Henke, A.; Badorff, C.; Schmidtke, M. Heparan sulfates and coxsackievirus-adenovirus receptor: Each one mediates coxsackievirus B3 PD infection. J. Virol. 2003, 77, 10071–10077. [Google Scholar] [CrossRef]
- Zautner, A.E.; Jahn, B.; Hammerschmidt, E.; Wutzler, P.; Schmidtke, M. N- and 6-O-sulfated heparan sulfates mediate internalization of coxsackievirus B3 variant PD into CHO-K1 cells. J. Virol. 2006, 80, 6629–6636. [Google Scholar] [CrossRef]
- Hatabe, S.; Kimura, H.; Arao, T.; Kato, H.; Hayashi, H.; Nagai, T.; Matsumoto, K.; de Velasco, M.; Fujita, Y.; Yamanouchi, G.; et al. Overexpression of heparan sulfate 6-O-sulfotransferase-2 in colorectal cancer. Mol. Clin. Oncol. 2013, 1, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.L.; Rushton, G.; Jayson, G.C.; Avizienyte, E. Ovarian cancer cell heparan sulfate 6-O-sulfotransferases regulate an angiogenic program induced by heparin-binding epidermal growth factor (EGF)-like growth factor/EGF receptor signaling. J. Biol. Chem. 2014, 289, 10488–10501. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; He, J.; Du, J.; Zhang, R.-X.; Yao, H.-B.; Shao, Q.-S. Overexpression of HS6ST2 is associated with poor prognosis in patients with gastric cancer. Oncol. Lett. 2017, 14, 6191–6197. [Google Scholar] [CrossRef]
- Song, K.; Li, Q.; Peng, Y.-B.; Li, J.; Ding, K.; Chen, L.-J.; Shao, C.-H.; Zhang, L.-J.; Li, P. Silencing of hHS6ST2 inhibits progression of pancreatic cancer through inhibition of Notch signalling. Biochem. J. 2011, 436, 271–282. [Google Scholar] [CrossRef]
- Melnick, J.L. Portraits of viruses: The picornaviruses. Intervirology 1983, 20, 61–100. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, K.U.; Jeon, E.S.; Berkley, N.; Wessely, R.; Huber, S. A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J. Virol. 1996, 70, 7811–7818. [Google Scholar] [CrossRef]
- Kandolf, R.; Hofschneider, P.H. Molecular cloning of the genome of a cardiotropic Coxsackie B3 virus: Full-length reverse-transcribed recombinant cDNA generates infectious virus in mammalian cells. Proc. Natl. Acad. Sci. USA 1985, 82, 4818–4822. [Google Scholar] [CrossRef] [PubMed]
- Tonew, M.; Hartmann, M.; Schmidtke, M.; Stelzner, A. Replication and persistence of coxsackieviruses B3 in human fibroblasts. Zentralbl. Bakteriol. 1995, 282, 92–101. [Google Scholar] [CrossRef]
- Jia, Y.; Miyamoto, S.; Soda, Y.; Takishima, Y.; Sagara, M.; Liao, J.; Hirose, L.; Hijikata, Y.; Miura, Y.; Hara, K.; et al. Extremely low organ toxicity and strong antitumor activity of miR-34-regulated oncolytic coxsackievirus B3. Mol. Ther. Oncolytics 2019, 12, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Liu, H.; de Silva, T.; Xue, Y.; Mohamud, Y.; Ng, C.S.; Qu, J.; Zhang, J.; Jia, W.W.G.; Lockwood, W.W.; et al. Coxsackievirus type B3 is a potent oncolytic virus against KRAS-mutant lung adenocarcinoma. Mol. Ther. Oncolytics 2019, 14, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xue, Y.C.; Deng, H.; Mohamud, Y.; Ng, C.S.; Chu, A.; Lim, C.J.; Lockwood, W.W.; Jia, W.W.G.; Luo, H. MicroRNA modification of coxsackievirus B3 decreases its toxicity, while retaining oncolytic potency against lung cancer. Mol. Ther. Oncolytics 2020, 16, 207–218. [Google Scholar] [CrossRef]
- Schmidtke, M.; Selinka, H.C.; Heim, A.; Jahn, B.; Tonew, M.; Kandolf, R.; Stelzner, A.; Zell, R. Attachment of coxsackievirus B3 variants to various cell lines: Mapping of phenotypic differences to capsid protein VP1. Virology 2000, 275, 77–88. [Google Scholar] [CrossRef]
- Hazini, A.; Dieringer, B.; Pryshliak, M.; Knoch, K.-P.; Heimann, L.; Tolksdorf, B.; Pappritz, K.; El-Shafeey, M.; Solimena, M.; Beling, A.; et al. miR-375- and miR-1-regulated coxsackievirus B3 has no pancreas and heart toxicity but strong antitumor efficiency in colorectal carcinomas. Hum. Gene Ther. 2021. [Google Scholar] [CrossRef]
- Sagara, M.; Miyamoto, S.; Itoh, S.; Soda, Y.; Tani, K. Development of new oncolytic virotherapy targeting breast cancer using coxsackievirus B3. Anticancer. Res. 2021, 41, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Pryshliak, M.; Hazini, A.; Knoch, K.; Dieringer, B.; Tolksdorf, B.; Solimena, M.; Kurreck, J.; Pinkert, S.; Fechner, H. MiR-375-mediated suppression of engineered coxsackievirus B3 in pancreatic cells. FEBS Lett. 2020, 594, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Liu, Z. Novel recombinant coxsackievirus B3 with genetically inserted basic peptide elicits robust antitumor activity against lung cancer. Cancer Med. 2020, 9, 5210–5220. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic viruses and immune checkpoint inhibition: The best of both worlds. Mol. Ther. Oncolytics 2019, 13, 93–106. [Google Scholar] [CrossRef]
- Ma, J.; Ramachandran, M.; Jin, C.; Quijano-Rubio, C.; Martikainen, M.; Yu, D.; Essand, M. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis. 2020, 11, 48. [Google Scholar] [CrossRef]
- Marconi, R.; Strolin, S.; Bossi, G.; Strigari, L. A meta-analysis of the abscopal effect in preclinical models: Is the biologically effective dose a relevant physical trigger? PLoS ONE 2017, 12, e0171559. [Google Scholar] [CrossRef]
- Ilett, E.J.; Prestwich, R.J.; Kottke, T.; Errington, F.; Thompson, J.M.; Harrington, K.J.; Pandha, H.S.; Coffey, M.; Selby, P.J.; Vile, R.G.; et al. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther. 2009, 16, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Bunuales, M.; Garcia-Aragoncillo, E.; Casado, R.; Quetglas, J.I.; Hervas-Stubbs, S.; Bortolanza, S.; Benavides-Vallve, C.; Ortiz-de-Solorzano, C.; Prieto, J.; Hernandez-Alcoceba, R. Evaluation of monocytes as carriers for armed oncolytic adenoviruses in murine and Syrian hamster models of cancer. Hum. Gene Ther. 2012, 23, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Adair, R.A.; Scott, K.J.; Fraser, S.; Errington-Mais, F.; Pandha, H.; Coffey, M.; Selby, P.; Cook, G.P.; Vile, R.; Harrington, K.J.; et al. Cytotoxic and immune-mediated killing of human colorectal cancer by reovirus-loaded blood and liver mononuclear cells. Int. J. Cancer 2013, 132, 2327–2338. [Google Scholar] [CrossRef]
- Kemball, C.C.; Flynn, C.T.; Hosking, M.P.; Botten, J.; Whitton, J.L. Wild-type coxsackievirus infection dramatically alters the abundance, heterogeneity, and immunostimulatory capacity of conventional dendritic cells in vivo. Virology 2012, 429, 74–90. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brown, M.C.; Holl, E.K.; Boczkowski, D.; Dobrikova, E.; Mosaheb, M.; Chandramohan, V.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Müller, L.M.E.; Holmes, M.; Michael, J.L.; Scott, G.B.; West, E.J.; Scott, K.J.; Parrish, C.; Hall, K.; Stäble, S.; Jennings, V.A.; et al. Plasmacytoid dendritic cells orchestrate innate and adaptive anti-tumor immunity induced by oncolytic coxsackievirus A21. J. Immunother. Cancer 2019, 7, 164. [Google Scholar] [CrossRef]
- LaRocca, C.J.; Warner, S.G. Oncolytic viruses and checkpoint inhibitors: Combination therapy in clinical trials. Clin. Transl. Med. 2018, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Poller, W.; Hajjar, R.; Schultheiss, H.P.; Fechner, H. Cardiac-targeted delivery of regulatory RNA molecules and genes for the treatment of heart failure. Cardiovasc. Res. 2010, 86, 353–364. [Google Scholar] [CrossRef]
- Brown, B.D.; Gentner, B.; Cantore, A.; Colleoni, S.; Amendola, M.; Zingale, A.; Baccarini, A.; Lazzari, G.; Galli, C.; Naldini, L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat. Biotechnol. 2007, 25, 1457–1467. [Google Scholar] [CrossRef]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef]
- Liang, Y.; Ridzon, D.; Wong, L.; Chen, C. Characterization of microRNA expression profiles in normal human tissues. BMC Genom. 2007, 8, 166. [Google Scholar] [CrossRef]
- O’Neill, C.P.; Dwyer, R.M. Nanoparticle-based delivery of tumor suppressor microRNA for cancer therapy. Cells 2020, 9, 521. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, J.; Chen, Q.; Cheng, J.; Chen, X.; Mao, Y.; Chen, W.; Liu, C.; Wu, H.; Lv, Y.; et al. MicroRNA‑200a and microRNA‑141 have a synergetic effect on the suppression of epithelial‑mesenchymal transition in liver cancer by targeting STAT4. Oncol. Lett. 2020, 21, 1. [Google Scholar] [CrossRef]
- Kelly, E.J.; Hadac, E.M.; Greiner, S.; Russell, S.J. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 2008, 14, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Leber, M.F.; Baertsch, M.-A.; Anker, S.C.; Henkel, L.; Singh, H.M.; Bossow, S.; Engeland, C.E.; Barkley, R.; Hoyler, B.; Albert, J.; et al. Enhanced control of oncolytic measles virus using microRNA target sites. Mol. Ther. Oncolytics 2018, 9, 30–40. [Google Scholar] [CrossRef]
- Geisler, A.; Fechner, H. MicroRNA-regulated viral vectors for gene therapy. World J. Exp. Med. 2016, 6, 37–54. [Google Scholar] [CrossRef]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.J.; Nace, R.; Barber, G.N.; Russell, S.J. Attenuation of vesicular stomatitis virus encephalitis through microRNA targeting. J. Virol. 2009, 84, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Leber, M.F.; Bossow, S.; Leonard, V.H.; Zaoui, K.; Grossardt, C.; Frenzke, M.; Miest, T.; Sawall, S.; Cattaneo, R.; Von Kalle, C.; et al. MicroRNA-sensitive oncolytic measles viruses for cancer-specific vector tropism. Mol. Ther. 2011, 19, 1097–1106. [Google Scholar] [CrossRef]
- Ruiz, A.J.; Hadac, E.M.; Nace, R.A.; Russell, S.J. MicroRNA-detargeted mengovirus for oncolytic virotherapy. J. Virol. 2016, 90, 4078–4092. [Google Scholar] [CrossRef] [PubMed]
- Shayestehpour, M.; Moghim, S.; Salimi, V.; Jalilvand, S.; Yavarian, J.; Romani, B.; Mokhtari-Azad, T. Targeting human breast cancer cells by an oncolytic adenovirus using microRNA-targeting strategy. Virus Res. 2017, 240, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Cawood, R.; Wong, S.-L.; Di, Y.; Baban, D.F.; Seymour, L.W. MicroRNA controlled adenovirus mediates anti-cancer efficacy without affecting endogenous microRNA activity. PLoS ONE 2011, 6, e16152. [Google Scholar] [CrossRef]
- He, F.; Yao, H.; Wang, J.; Xiao, Z.; Xin, L.; Liu, Z.; Ma, X.; Sun, J.; Jin, Q.; Liu, Z. Coxsackievirus B3 engineered to contain microRNA targets for muscle-specific microRNAs displays attenuated cardiotropic virulence in mice. J. Virol. 2015, 89, 908–916. [Google Scholar] [CrossRef]
- Liu, Z.; Carthy, C.M.; Cheung, P.; Bohunek, L.; Wilson, J.E.; McManus, B.M.; Yang, D. Structural and functional analysis of the 5′ untranslated region of coxsackievirus B3 RNA: In vivo translational and infectivity studies of full-length mutants. Virology 1999, 265, 206–217. [Google Scholar] [CrossRef]
- Elsedawy, N.B.; Nace, R.A.; Russell, S.J.; Schulze, A.J. Oncolytic activity of targeted picornaviruses formulated as synthetic infectious RNA. Mol. Ther. Oncolytics 2020, 17, 484–495. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Hohenadl, C.; Klingel, K.; Mertsching, J.; Hofschneider, P.; Kandolf, R. Strand-specific detection of enteroviral RNA in myocardial tissue by in situ hybridization. Mol. Cell. Probes 1991, 5, 11–20. [Google Scholar] [CrossRef]
- Schubert, S.; Rothe, D.; Werk, D.; Grunert, H.-P.; Zeichhardt, H.; Erdmann, V.A.; Kurreck, J. Strand-specific silencing of a picornavirus by RNA interference: Evidence for the superiority of plus-strand specific siRNAs. Antivir. Res. 2007, 73, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Bilsel, P.A.; Nichol, S.T. Polymerase errors accumulating during natural evolution of the glycoprotein gene of vesicular sto-matitis virus Indiana serotype isolates. J. Virol. 1990, 64, 4873–4883. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.; Spindler, K.; Horodyski, F.; Grabau, E.; Nichol, S.; Vandepol, S. Rapid evolution of RNA genomes. Science 1982, 215, 1577–1585. [Google Scholar] [CrossRef]
- Steinhauer, D.A.; De La Torre, J.C.; Meier, E.; Holland, J.J. Extreme heterogeneity in populations of vesicular stomatitis virus. J. Virol. 1989, 63, 2072–2080. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.J.; de la Torre, J.C.; Clarke, D.K.; Duarte, E. Quantitation of relative fitness and great adaptability of clonal popu-lations of RNA viruses. J. Virol. 1991, 65, 2960–2967. [Google Scholar] [CrossRef]
- Hiyoshi, Y.; Schetter, A.J.; Okayama, H.; Inamura, K.; Anami, K.; Nguyen, G.H.; Horikawa, I.; Hawkes, J.E.; Bowman, E.D.; Leung, S.Y.; et al. Increased MicroRNA-34b and -34c predominantly expressed in stromal tissues is associated with poor prognosis in human colon cancer. PLoS ONE 2015, 10, e0124899. [Google Scholar] [CrossRef]
- Bordería, A.V.; Isakov, O.; Moratorio, G.; Henningsson, R.; Agüera-González, S.; Organtini, L.; Gnädig, N.F.; Blanc, H.; Alcover, A.; Hafenstein, S.; et al. Group selection and contribution of minority variants during virus adaptation determines virus fitness and phenotype. PLOS Pathog. 2015, 11, e1004838. [Google Scholar] [CrossRef]
- Svyatchenko, V.A.; Ternovoy, V.A.; Kiselev, N.N.; Demina, A.V.; Loktev, V.B.; Netesov, S.V.; Chumakov, P.M. Bioselection of coxsackievirus B6 strain variants with altered tropism to human cancer cell lines. Arch. Virol. 2017, 162, 3355–3362. [Google Scholar] [CrossRef]
- Thorne, S.H.; Sampath, P. Arming viruses in multi-mechanistic oncolytic viral therapy: Current research and future developments, with emphasis on poxviruses. Oncolytic Virother. 2013, 3, 1–9. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lim, B.-K.; Shin, J.-O.; Lee, S.-C.; Kim, D.-K.; Choi, N.-J.; Choe, S.-C.; Knowlton, K.U.; Jeon, E.-S. Long-term cardiac gene expression using a coxsackieviral vector. J. Mol. Cell. Cardiol. 2005, 38, 745–751. [Google Scholar] [CrossRef]
- Zeng, J.; Chen, X.X.; Dai, J.P.; Zhao, X.F.; Xin, G.; Su, Y.; Wang, G.F.; Li, R.; Yan, Y.X.; Su, J.H.; et al. An attenuated coxsackievirus B3 vector: A potential tool for viral tracking study and gene delivery. PLoS ONE 2013, 8, e83753. [Google Scholar] [CrossRef] [PubMed]
- Slifka, M.K.; Pagarigan, R.; Mena, I.; Feuer, R.; Whitton, J.L. Using recombinant coxsackievirus B3 to evaluate the induction and protective efficacy of CD8+ T cells during picornavirus infection. J. Virol. 2001, 75, 2377–2387. [Google Scholar] [CrossRef]
- Tong, L.; Lin, L.; Zhao, W.; Wang, B.; Wu, S.; Liu, H.; Zhong, X.; Cui, Y.; Gu, H.; Zhang, F.; et al. Destabilization of coxsackievirus B3 genome integrated with enhanced green fluorescent protein gene. Intervirology 2011, 54, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Kim, K.-S.; Tracy, S.; Jackson, J.; Höfling, K.; Leser, J.S.; Malone, J.; Kolbeck, P. Coxsackievirus expression of the murine secretory protein interleukin-4 induces increased synthesis of immunoglobulin G1 in mice. J. Virol. 2000, 74, 7952–7962. [Google Scholar] [CrossRef]
- Dekel, B.; Yoeli, R.; Shulman, L.; Padeh, S.; Passwell, J. Localized thigh swelling mimicking a neoplastic process: Involvement of coxsackie virus type A21. Acta Paediatr. 2002, 91, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Yi, L.; Song, Y.; Zhang, X.; Liang, L.; Ni, H.; Ke, C.; Wu, J.; Lu, J. A cluster of coxsackievirus A21 associated acute respiratory illness: The evidence of efficient transmission of CVA21. Arch. Virol. 2016, 162, 1057–1059. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Aoki, Y.; Komabayashi, K.; Itagaki, T.; Mizuta, K. Isolation of coxsackievirus A21 from patients with acute respiratory infection in Yamagata, Japan in 2019. Jpn. J. Infect. Dis. 2021, 74, 172–174. [Google Scholar] [CrossRef]
- Bradley, S.; Jakes, A.D.; Harrington, K.; Pandha, H.; Melcher, A.; Errington-Mais, F. Applications of coxsackievirus A21 in oncology. Oncolytic Virother. 2014, 3, 47–55. [Google Scholar] [CrossRef]
- Annels, N.E.; Mansfield, D.; Arif, M.; Ballesteros-Merino, C.; Simpson, G.R.; Denyer, M.; Sandhu, S.S.; Melcher, A.A.; Harrington, K.J.; Davies, B.; et al. Phase I trial of an ICAM-1-targeted immunotherapeutic-coxsackievirus A21 (CVA21) as an oncolytic agent against non muscle-invasive bladder cancer. Clin. Cancer Res. 2019, 25, 5818–5831. [Google Scholar] [CrossRef]
- Shafren, D.R.; Barry, R.D.; Dorahy, D.J.; Thorne, R.F. Cytoplasmic interactions between decay-accelerating factor and intercellular adhesion molecule-1 are not required for coxsackievirus A21 cell infection. J. Gen. Virol. 2000, 81, 889–894. [Google Scholar] [CrossRef]
- Shafren, D.R.; Dorahy, D.J.; Greive, S.J.; Burns, G.F.; Barry, R.D. Mouse cells expressing human intercellular adhesion mole-cule-1 are susceptible to infection by coxsackievirus A21. J. Virol. 1997, 71, 785–789. [Google Scholar] [CrossRef]
- Shafren, D.R.; Dorahy, D.J.; A Ingham, R.; Burns, G.F.; Barry, R.D. Coxsackievirus A21 binds to decay-accelerating factor but requires intercellular adhesion molecule 1 for cell entry. J. Virol. 1997, 71, 4736–4743. [Google Scholar] [CrossRef] [PubMed]
- Shafren, D.R.; Au, G.G.; Nguyen, T.; Newcombe, N.G.; Haley, E.S.; Beagley, L.; Johansson, E.S.; Hersey, P.; Barry, R.D. Systemic therapy of malignant human melanoma tumors by a common cold-producing enterovirus, coxsackievirus A21. Clin. Cancer Res. 2004, 10, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Au, G.G.; Lindberg, A.M.; Barry, R.D.; Shafren, D.R. Oncolysis of vascular malignant human melanoma tumors by Coxsackievirus A21. Int. J. Oncol. 2005, 26, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Vaeteewoottacharn, K.; Kariya, R. Application of highly immunocompromised mice for the establishment of patient-derived xenograft (PDX) models. Cells 2019, 8, 889. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Cells for CVB3 | Cancer Model; Route of Application | CVB3 Variant | MiR-Regulation | Aimed de-Targeting of | Reference | |
|---|---|---|---|---|---|---|
| human breast cancer cells (TNBC cells) | mouse MDA-MB-468 xenograft; intratumoral | not stated | tissue‑specific | miR-1 miR-217 | pancreas, heart | [108] |
| human colorectal carcinoma cells | mouse DLD-1 xenograft; intratumoral | PD | miR-375 miR-1 | pancreas, heart | [107] | |
| human colorectal cancer cells | colorectal cancer cells | attenuated H3 | miR-375 | pancreatic cells | [109] | |
| human lung cancer cells (KRASmut lung adenocarcinoma non‑SCLC, TP53mut/RB1mut SCLC cells) | mouse H526-derived TP53mut/RB1mut SCLC xenograft; intraperitoneal | Nancy | tumor suppressor | miR-145 miR-143 | heart, lung (lung epithelial cells, cardiomyocytes) | [105] |
| lung cancer cells (non‑SCLC H1299, TC-1) | mouse H1299 xenograft and TC‑1 syngeneic lung cancer model; intratumoral | Nancy | miR-34a miR-34c | normal cells | [103] | |
| human lung cancer cells (non‑SCLC) | mouse GLC-82, A549, H460 xenograft; intravenous | attenuated Nancy—modified with basic peptide | - | - | - | [110] |
| human lung cancer cells (KRASmut lung adenocarcinoma non‑SCLC) | mouse H2030 xenograft; intratumoral | Nancy | - | - | - | [104] |
| various cancer cell lines, esp. human non‑SCLC lung cancer cells | mouse A549, H1299, EBC-1 xenograft and TC‑1 syngeneic lung cancer model; intratumoral | Nancy | - | - | - | [20] |
| human EC cells | mouse HEC-1-A, HEC-1-B, Ishikawa xenograft; intratumoral, intravenous | CV-B3/2035A | - | - | - | [91] |
| human colorectal cancer cells | mouse DLD-1 xenograft; intratumoral | PD, Nancy, H3 and 31-1-93 | - | - | - | [92] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geisler, A.; Hazini, A.; Heimann, L.; Kurreck, J.; Fechner, H. Coxsackievirus B3—Its Potential as an Oncolytic Virus. Viruses 2021, 13, 718. https://doi.org/10.3390/v13050718
Geisler A, Hazini A, Heimann L, Kurreck J, Fechner H. Coxsackievirus B3—Its Potential as an Oncolytic Virus. Viruses. 2021; 13(5):718. https://doi.org/10.3390/v13050718
Chicago/Turabian StyleGeisler, Anja, Ahmet Hazini, Lisanne Heimann, Jens Kurreck, and Henry Fechner. 2021. "Coxsackievirus B3—Its Potential as an Oncolytic Virus" Viruses 13, no. 5: 718. https://doi.org/10.3390/v13050718
APA StyleGeisler, A., Hazini, A., Heimann, L., Kurreck, J., & Fechner, H. (2021). Coxsackievirus B3—Its Potential as an Oncolytic Virus. Viruses, 13(5), 718. https://doi.org/10.3390/v13050718