On Enthalpy–Entropy Compensation Characterizing Processes in Aqueous Solution



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
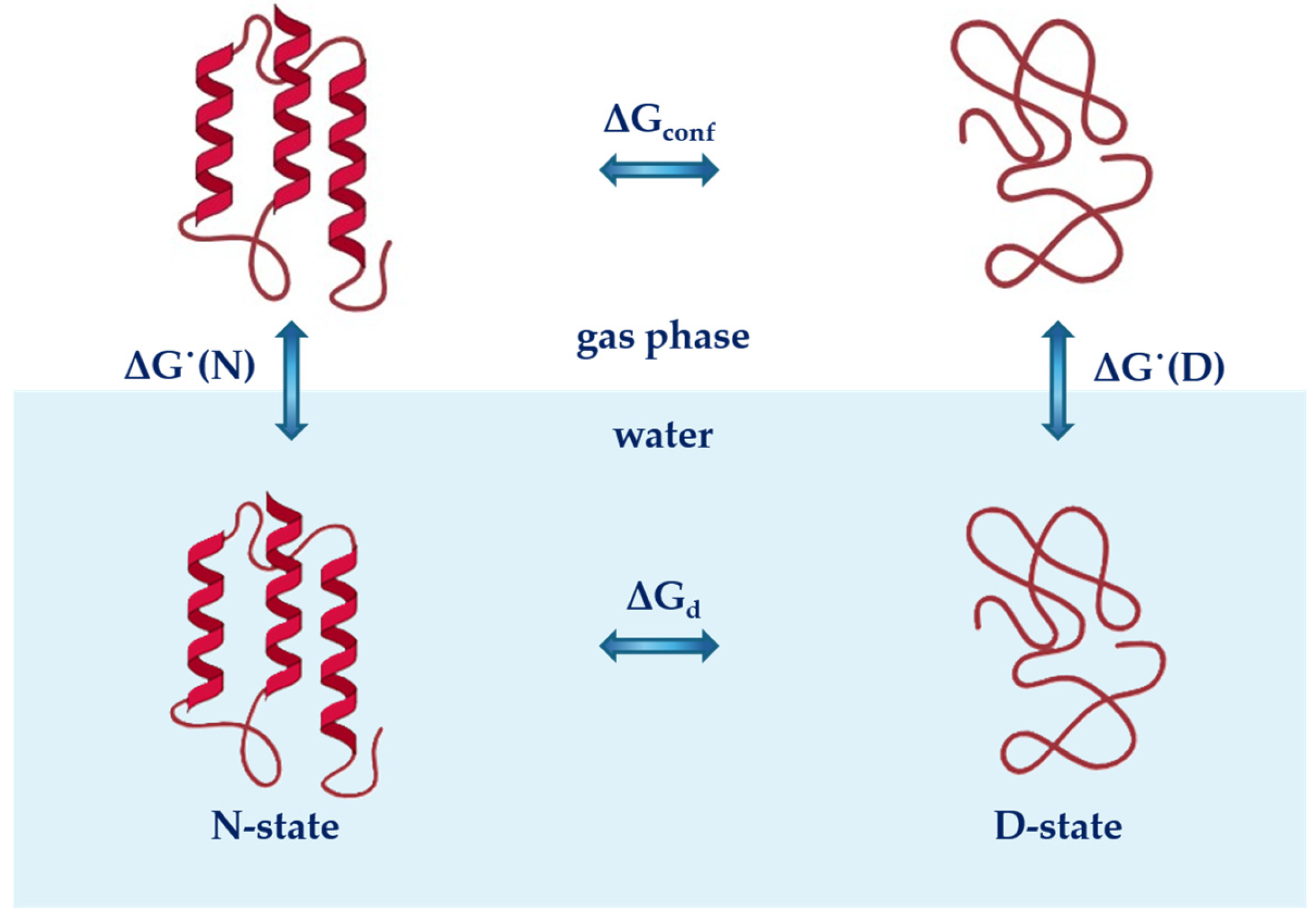
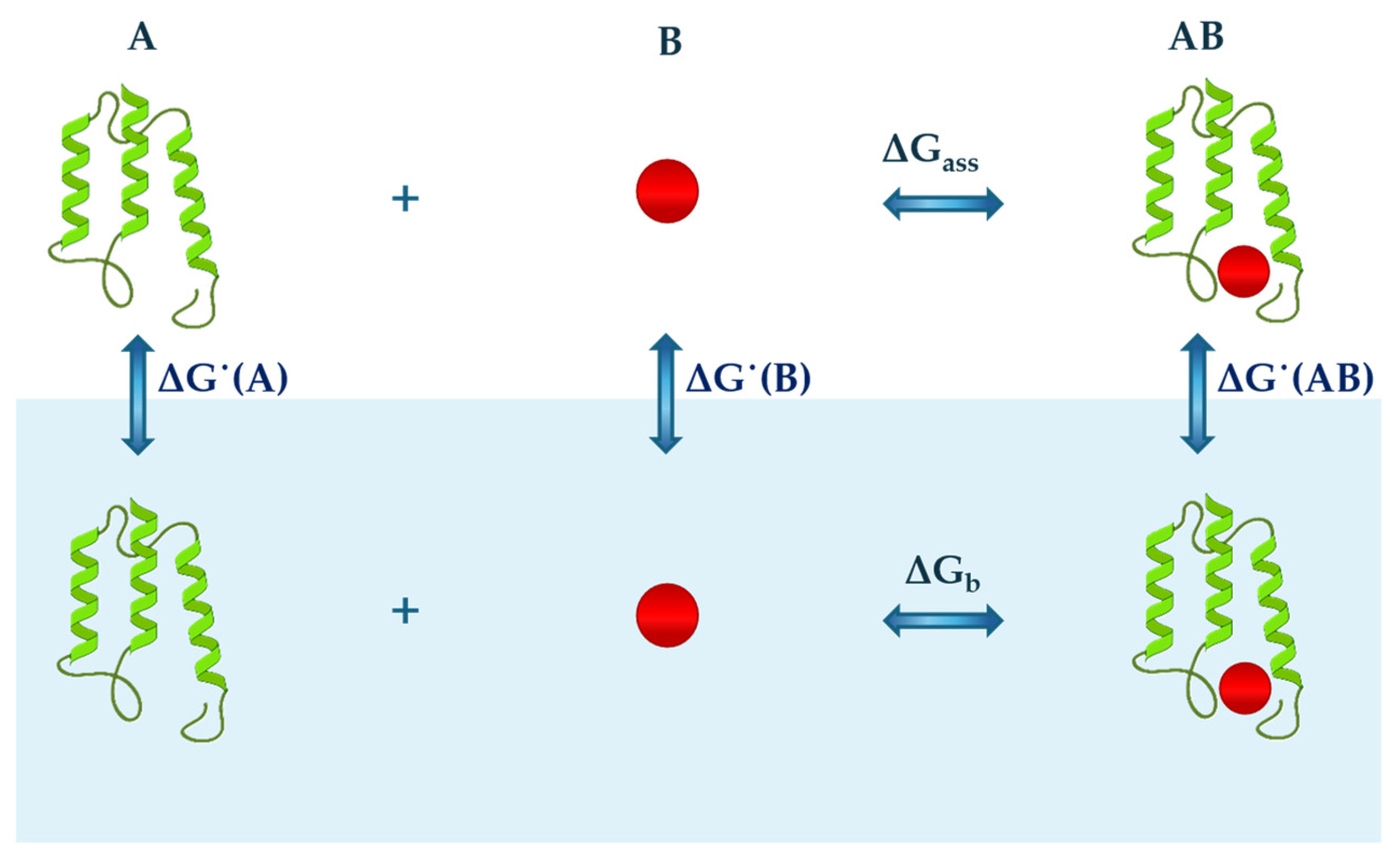
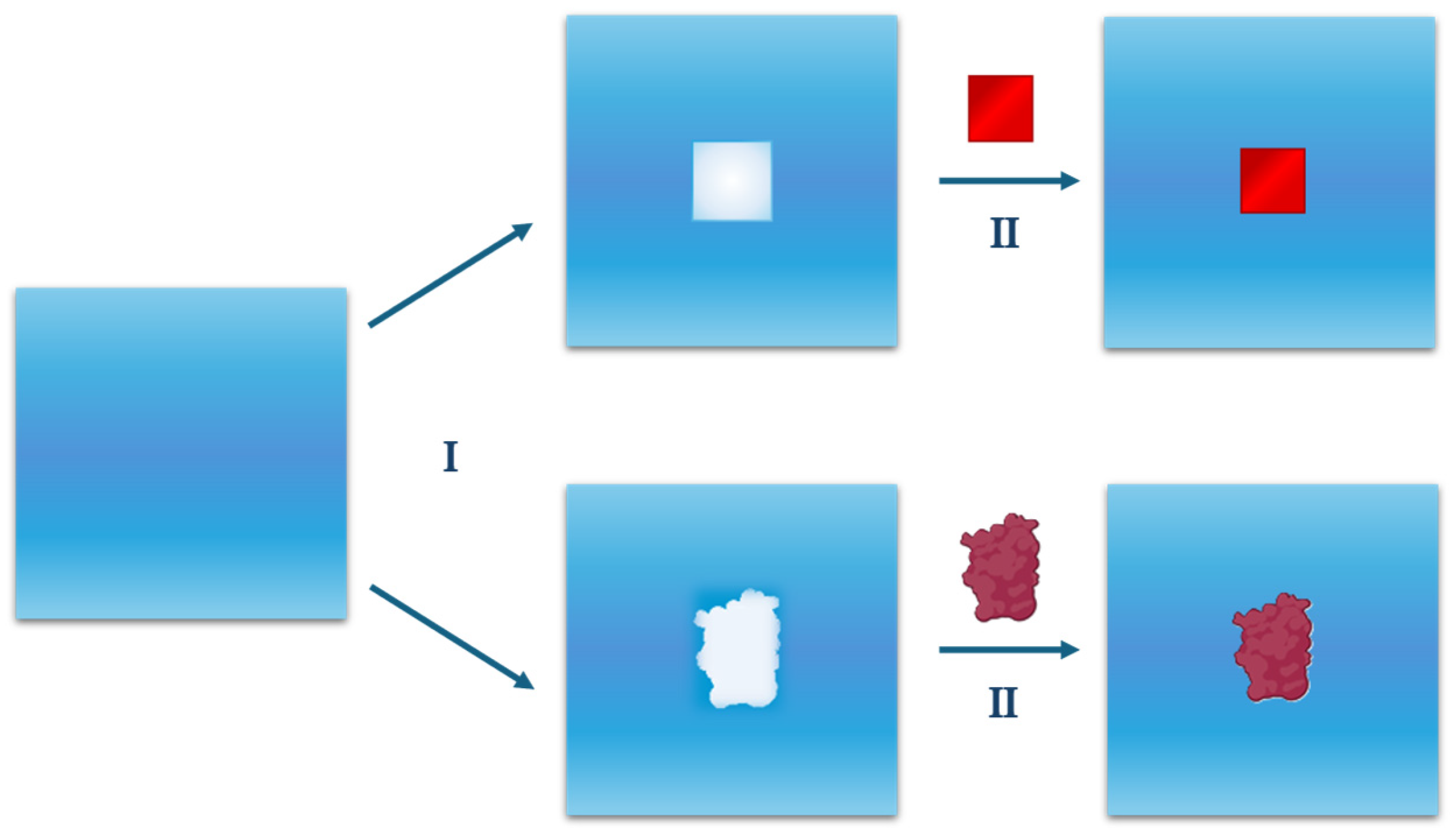
2. Theoretical Considerations
2.1. Cavity Creation
2.2. Activating the Attractive Solute–Water Interactions
3. Structural Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| HO | Harmonic oscillator |
| NPT | Isothermal–isobaric canonical ensemble |
References
- Exner, O. The Enthalpy-Entropy Relationship. In Progress in Physical Organic Chemistry; Streitwieser, A., Taft, R.W., Eds.; Wiley: Hoboken, NJ, USA, 1973; Volume 10, pp. 411–482. ISBN 978-0-471-83356-7. [Google Scholar]
- Krug, R.R.; Hunter, W.G.; Grieger, R.A. Statistical Interpretation of Enthalpy-Entropy Compensation. Nature 1976, 261, 566–567. [Google Scholar] [CrossRef]
- Exner, O. How to Get Wrong Results from Good Experimental Data: A Survey of Incorrect Applications of Regression. J. Phys. Org. Chem. 1997, 10, 797–813. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Enthalpy—Entropy Compensation: A Phantom Phenomenon. J. Biosci. 2002, 27, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Qian, H. An Asymptotic Comparative Analysis of the Thermodynamics of Non-Covalent Association. J. Math. Biol. 2006, 52, 277–289. [Google Scholar] [CrossRef]
- Griessen, R.; Dam, B. Simple Accurate Verification of Enthalpy-Entropy Compensation and Isoequilibrium Relationship. ChemPhysChem 2021, 22, 1774–1784. [Google Scholar] [CrossRef]
- Lumry, R.; Rajender, S. Enthalpy-Entropy Compensation Phenomena in Water Solutions of Proteins and Small Molecules: A Ubiquitous Property of Water. Biopolymers 1970, 9, 1125–1227. [Google Scholar] [CrossRef]
- Liu, L.; Guo, Q.X. Isokinetic Relationship, Isoequilibrium Relationship, and Enthalpy-Entropy Compensation. Chem. Rev. 2001, 101, 673–696. [Google Scholar] [CrossRef]
- Cooper, A.; Johnson, C.M.; Lakey, J.H.; Nöllmann, M. Heat Does Not Come in Different Colours: Entropy-Enthalpy Compensation, Free Energy Windows, Quantum Confinement, Pressure Perturbation Calorimetry, Solvation and the Multiple Causes of Heat Capacity Effects in Biomolecular Interactions. Biophys. Chem. 2001, 93, 215–230. [Google Scholar] [CrossRef]
- Chodera, J.D.; Mobley, D.L. Entropy-Enthalpy Compensation: Role and Ramifications in Biomolecular Ligand Recognition and Design. Annu. Rev. Biophys. 2013, 42, 121–142. [Google Scholar] [CrossRef]
- Movileanu, L.; Schiff, E.A. Entropy–Enthalpy Compensation of Biomolecular Systems in Aqueous Phase: A Dry Perspective. Monatshefte Chem. 2013, 144, 59–65. [Google Scholar] [CrossRef]
- Dragan, A.I.; Read, C.M.; Crane-Robinson, C. Enthalpy–Entropy Compensation: The Role of Solvation. Eur. Biophys. J. 2017, 46, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Makhatadze, G.I.; Privalov, P.L. Energetics of Protein Structure. Adv. Protein Chem. 1995, 47, 307–425. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.D.; Murphy, K.P. Protein Structure and the Energetics of Protein Stability. Chem. Rev. 1997, 97, 1251–1268. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Robertson, A.D. Some Thermodynamic Implications for the Thermostability of Proteins. Protein Sci. 2001, 10, 1187–1194. [Google Scholar] [CrossRef]
- Sawle, L.; Ghosh, K. How Do Thermophilic Proteins and Proteomes Withstand High Temperature? Biophys. J. 2011, 101, 217–227. [Google Scholar] [CrossRef]
- Silverstein, T.P. The Hydrophobic Effect: Is Water Afraid, or Just Not That Interested? ChemTexts 2020, 6, 26. [Google Scholar] [CrossRef]
- Privalov, P.L. Thermodynamic Problems of Protein Structure. Annu. Rev. Biophys. Biophys. Chem. 1989, 18, 47–69. [Google Scholar] [CrossRef]
- Pica, A.; Graziano, G. Shedding Light on the Extra Thermal Stability of Thermophilic Proteins. Biopolymers 2016, 105, 856–863. [Google Scholar] [CrossRef]
- Eftink, M.R.; Anusiem, A.C.; Biltonen, R.L. Enthalpy-Entropy Compensation and Heat Capacity Changes for Protein-Ligand Interactions: General Thermodynamic Models and Data for the Binding of Nucleotides to Ribonuclease A. Biochemistry 1983, 22, 3884–3896. [Google Scholar] [CrossRef]
- Kuroki, R.; Nitta, K.; Yutani, K. Thermodynamic Changes in the Binding of Ca2+ to a Mutant Human Lysozyme (D86/92). Enthalpy-Entropy Compensation Observed upon Ca2+ Binding to Proteins. J. Biol. Chem. 1992, 267, 24297–24301. [Google Scholar] [CrossRef]
- Gilli, P.; Ferretti, V.; Gilli, G.; Borea, P.A. Enthalpy-Entropy Compensation in Drug-Receptor Binding. J. Phys. Chem. 1994, 98, 1515–1518. [Google Scholar] [CrossRef]
- Talhout, R.; Villa, A.; Mark, A.E.; Engberts, J.B.F.N. Understanding Binding Affinity: A Combined Isothermal Titration Calorimetry/Molecular Dynamics Study of the Binding of a Series of Hydrophobically Modified Benzamidinium Chloride Inhibitors to Trypsin. J. Am. Chem. Soc. 2003, 125, 10570–10579. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, V.M.; Bohall, B.R.; Semetey, V.; Whitesides, G.M. The Paradoxical Thermodynamic Basis for the Interaction of Ethylene Glycol, Glycine, and Sarcosine Chains with Bovine Carbonic Anhydrase II: An Unexpected Manifestation of Enthalpy/Entropy Compensation. J. Am. Chem. Soc. 2006, 128, 5802–5812. [Google Scholar] [CrossRef] [PubMed]
- Lafont, V.; Armstrong, A.A.; Ohtaka, H.; Kiso, Y.; Mario Amzel, L.; Freire, E. Compensating Enthalpic and Entropic Changes Hinder Binding Affinity Optimization. Chem. Biol. Drug Des. 2007, 69, 413–422. [Google Scholar] [CrossRef]
- Ward, J.M.; Gorenstein, N.M.; Tian, J.; Martin, S.F.; Post, C.B. Constraining Binding Hot Spots: NMR and Molecular Dynamics Simulations Provide a Structural Explanation for Enthalpy−Entropy Compensation in SH2−Ligand Binding. J. Am. Chem. Soc. 2010, 132, 11058–11070. [Google Scholar] [CrossRef]
- Olsson, T.S.G.; Ladbury, J.E.; Pitt, W.R.; Williams, M.A. Extent of Enthalpy-Entropy Compensation in Protein-Ligand Interactions. Protein Sci. 2011, 20, 1607–1618. [Google Scholar] [CrossRef]
- Ahmad, M.; Helms, V.; Lengauer, T.; Kalinina, O.V. Enthalpy–Entropy Compensation upon Molecular Conformational Changes. J. Chem. Theory Comput. 2015, 11, 1410–1418. [Google Scholar] [CrossRef]
- Fox, J.M.; Zhao, M.; Fink, M.J.; Kang, K.; Whitesides, G.M. The Molecular Origin of Enthalpy/Entropy Compensation in Biomolecular Recognition. Annu. Rev. Biophys. 2018, 47, 223–250. [Google Scholar] [CrossRef]
- Jiménez, J.S.; Benítez, M.J. Gibbs Free Energy and Enthalpy–Entropy Compensation in Protein–Ligand Interactions. Biophysica 2024, 4, 298–309. [Google Scholar] [CrossRef]
- Ben-Naim, A. Hydrophobic Interaction and Structural Changes in the Solvent. Biopolymers 1975, 14, 1337–1355. [Google Scholar] [CrossRef]
- Lumry, R.; Battistel, E.; Jolicoeur, C. Geometric Relaxation in Water. Its Role in Hydrophobic Hydration. Faraday Symp. Chem. Soc. 1982, 17, 93–108. [Google Scholar] [CrossRef]
- Yu, H.-A.; Karplus, M. A Thermodynamic Analysis of Solvation. J. Chem. Phys. 1988, 89, 2366–2379. [Google Scholar] [CrossRef]
- Grunwald, E.; Steel, C. Solvent Reorganization and Thermodynamic Enthalpy-Entropy Compensation. J. Am. Chem. Soc. 1995, 117, 5687–5692. [Google Scholar] [CrossRef]
- Searle, M.S.; Westwell, M.S.; Williams, D.H. Application of a Generalised Enthalpy-Entropy Relationship to Binding Co-Operativity and Weak Associations in Solution. J. Chem. Soc. Perkin Trans. 2 1995, 141–151. [Google Scholar] [CrossRef]
- Dunitz, J.D. Win Some, Lose Some: Enthalpy-Entropy Compensation in Weak Intermolecular Interactions. Chem. Biol. 1995, 2, 709–712. [Google Scholar] [CrossRef]
- Qian, H.; Hopfield, J.J. Entropy-enthalpy Compensation: Perturbation and Relaxation in Thermodynamic Systems. J. Chem. Phys. 1996, 105, 9292–9298. [Google Scholar] [CrossRef]
- Qian, H. Entropy-Enthalpy Compensation: Conformational Fluctuation and Induced-Fit. J. Chem. Phys. 1998, 109, 10015–10017. [Google Scholar] [CrossRef]
- Gallicchio, E.; Kubo, M.M.; Levy, R.M. Entropy−Enthalpy Compensation in Solvation and Ligand Binding Revisited. J. Am. Chem. Soc. 1998, 120, 4526–4527. [Google Scholar] [CrossRef]
- Sharp, K. Entropy-Enthalpy Compensation: Fact or Artifact? Protein Sci. 2001, 10, 661–667. [Google Scholar] [CrossRef]
- Starikov, E.B.; Nordén, B. Enthalpy-Entropy Compensation: A Phantom or Something Useful? J. Phys. Chem. B 2007, 111, 14431–14435. [Google Scholar] [CrossRef]
- Douglas, J.F.; Dudowicz, J.; Freed, K.F. Crowding Induced Self-Assembly and Enthalpy-Entropy Compensation. Phys. Rev. Lett. 2009, 103, 135701. [Google Scholar] [CrossRef] [PubMed]
- Freed, K.F.; Freed, K.F. Entropy-Enthalpy Compensation in Chemical Reactions and Adsorption: An Exactly Solvable Model. J. Phys. Chem. B 2011, 115, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Starikov, E.B.; Nordén, B. Entropy–Enthalpy Compensation as a Fundamental Concept and Analysis Tool for Systematical Experimental Data. Chem. Phys. Lett. 2012, 538, 118–120. [Google Scholar] [CrossRef]
- Ryde, U. A Fundamental View of Enthalpy–Entropy Compensation. Med. Chem. Commun. 2014, 5, 1324–1336. [Google Scholar] [CrossRef]
- Lee, B. Solvent Reorganization Contribution to the Transfer Thermodynamics of Small Nonpolar Molecules. Biopolymers 1991, 31, 993–1008. [Google Scholar] [CrossRef]
- Lee, B. Enthalpy-entropy compensation in the thermodynamics of hydrophobicity. Biophys. Chem. 1994, 51, 271–278. [Google Scholar] [CrossRef]
- Lee, B.; Graziano, G. A Two-State Model of Hydrophobic Hydration That Produces Compensating Enthalpy and Entropy Changes. J. Am. Chem. Soc. 1996, 118, 5163–5168. [Google Scholar] [CrossRef]
- Graziano, G.; Lee, B. Hydration of Aromatic Hydrocarbons. J. Phys. Chem. B 2001, 105, 10367–10372. [Google Scholar] [CrossRef]
- Graziano, G. Case study of enthalpy-entropy non-compensation. J. Chem. Phys. 2004, 120, 4467–4471. [Google Scholar] [CrossRef]
- Graziano, G. Benzene Solubility in Water: A Reassessment. Chem. Phys. Lett. 2006, 429, 114–118. [Google Scholar] [CrossRef]
- Graziano, G. On the Molecular Origin of Cold Denaturation of Globular Proteins. Phys. Chem. Chem. Phys. 2010, 12, 14245–14252. [Google Scholar] [CrossRef] [PubMed]
- Graziano, G. On the Mechanism of Cold Denaturation. Phys. Chem. Chem. Phys. 2014, 16, 21755–21767. [Google Scholar] [CrossRef] [PubMed]
- Graziano, G. Contrasting the Hydration Thermodynamics of Methane and Methanol. Phys. Chem. Chem. Phys. 2019, 21, 21418–21430. [Google Scholar] [CrossRef] [PubMed]
- Ben-Naim, A. Solvation Thermodynamics; Plenum Press: New York, NY, USA, 1987. [Google Scholar]
- Widom, B. Some Topics in the Theory of Fluids. J. Chem. Phys. 1963, 39, 2808–2812. [Google Scholar] [CrossRef]
- Widom, B. Potential-Distribution Theory and the Statistical Mechanics of Fluids. J. Phys. Chem. 1982, 86, 869–872. [Google Scholar] [CrossRef]
- Hansen, J.P.; McDonald, I.R. Theory of Simple Liquids, 3rd ed.; Academic Press: New York, NY, USA, 2005. [Google Scholar]
- Lee, B. A Procedure for Calculating Thermodynamic Functions of Cavity Formation from the Pure Solvent Simulation Data. J. Chem. Phys. 1985, 83, 2421–2425. [Google Scholar] [CrossRef]
- Hummer, G.; Garde, S.; García, A.E.; Paulaitis, M.E.; Pratt, L.R. Hydrophobic Effects on a Molecular Scale. J. Phys. Chem. B 1998, 102, 10469–10482. [Google Scholar] [CrossRef]
- Pratt, L.R.; Pohorille, A. Theory of Hydrophobicity: Transient Cavities in Molecular Liquids. Proc. Natl. Acad. Sci. USA 1992, 89, 2995–2999. [Google Scholar] [CrossRef]
- Madan, B.; Lee, B. Role of Hydrogen Bonds in Hydrophobicity: The Free Energy of Cavity Formation in Water Models with and without the Hydrogen Bonds. Biophys. Chem. 1994, 51, 279–289. [Google Scholar] [CrossRef]
- Ashbaugh, H.S.; Pratt, L.R. Contrasting Nonaqueous against Aqueous Solvation on the Basis of Scaled-Particle Theory. J. Phys. Chem. B 2007, 111, 9330–9336. [Google Scholar] [CrossRef]
- Patel, A.J.; Varilly, P.; Chandler, D.; Garde, S. Quantifying Density Fluctuations in Volumes of All Shapes and Sizes Using Indirect Umbrella Sampling. J. Stat. Phys. 2011, 145, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Sosso, G.C.; Caravati, S.; Rotskoff, G.; Vaikuntanathan, S.; Hassanali, A. On the Role of Nonspherical Cavities in Short Length-Scale Density Fluctuations in Water. J. Phys. Chem. A 2017, 121, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; Bromberg, S. Molecular Driving Forces: Statistical Thermodynamics in Chemistry and Biology, 1st ed.; Garland Science: New York, NY, USA, 2003; ISBN 978-0-8153-2051-7. [Google Scholar]
- Reiss, H. Scaled Particle Methods in the Statistical Thermodynamics of Fluids. In Advances in Chemical Physics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1965; pp. 1–84. ISBN 978-0-470-14355-1. [Google Scholar]
- Pierotti, R.A. Aqueous solutions of nonpolar gases. J. Phys. Chem. 1965, 69, 281–288. [Google Scholar] [CrossRef]
- Frank, H.S.; Evans, M.W. Free Volume and Entropy in Condensed Systems. III. Entropy in Binary Liquid Mixtures; Partial Molal Entropy in Dilute Solutions; Structure and Thermodynamics in Aqueous Electrolytes. J. Chem. Phys. 1945, 13, 507–532. [Google Scholar] [CrossRef]
- Graziano, G. Comment on “Water’s Structure around Hydrophobic Solutes and the Iceberg Model”. J. Phys. Chem. B 2014, 118, 2598–2599. [Google Scholar] [CrossRef]
- Pratt, L.R.; Chandler, D. Theory of the Hydrophobic Effect. J. Chem. Phys. 1977, 67, 3683–3704. [Google Scholar] [CrossRef]
- Juurinen, I.; Pylkkänen, T.; Sahle, C.J.; Simonelli, L.; Hämäläinen, K.; Huotari, S.; Hakala, M. Effect of the Hydrophobic Alcohol Chain Length on the Hydrogen-Bond Network of Water. J. Phys. Chem. B 2014, 118, 8750–8755. [Google Scholar] [CrossRef]
- Fidler, J.; Rodger, P.M. Solvation Structure around Aqueous Alcohols. J. Phys. Chem. B 1999, 103, 7695–7703. [Google Scholar] [CrossRef]
- Guillot, B.; Guissani, Y. A Computer Simulation Study of the Temperature Dependence of the Hydrophobic Hydration. J. Chem. Phys. 1993, 99, 8075–8094. [Google Scholar] [CrossRef]
- Cooper, A. Heat Capacity of Hydrogen-Bonded Networks: An Alternative View of Protein Folding Thermodynamics. Biophys. Chem. 2000, 85, 25–39. [Google Scholar] [CrossRef]
- Cooper, A. Heat Capacity Effects in Protein Folding and Ligand Binding: A Re-Evaluation of the Role of Water in Biomolecular Thermodynamics. Biophys. Chem. 2005, 115, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.R.; Sharp, K.A. A New Angle on Heat Capacity Changes in Hydrophobic Solvation. J. Am. Chem. Soc. 2003, 125, 9853–9860. [Google Scholar] [CrossRef]
- Prabhu, N.V.; Sharp, K.A. Heat Capacity in Proteins. Annu. Rev. Phys. Chem. 2005, 56, 521–548. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, 3rd ed.; Cornell University Press: Ithaca, NY, USA, 1960; p. 468. ISBN 978-0-8014-0333-0. [Google Scholar]
- McQuarrie, D.A. Statistical Mechanics; Harper & Row: New York, NY, USA, 1976. [Google Scholar]
- Graziano, G. On the Temperature Dependence of Hydration Thermodynamics for Noble Gases. Phys. Chem. Chem. Phys. 1999, 1, 1877–1886. [Google Scholar] [CrossRef]
- Graziano, G. Hydration Thermodynamics of Aliphatic Alcohols. Phys. Chem. Chem. Phys. 1999, 1, 3567–3576. [Google Scholar] [CrossRef]
- Muller, N. Search for a Realistic View of Hydrophobic Effects. Acc. Chem. Res. 1990, 23, 23–28. [Google Scholar] [CrossRef]
- Graziano, G.; Lee, B. On the Intactness of Hydrogen Bonds around Nonpolar Solutes Dissolved in Water. J. Phys. Chem. B 2005, 109, 8103–8107. [Google Scholar] [CrossRef]
- Graziano, G. Structural Order in the Hydration Shell of Nonpolar Groups versus That in Bulk Water. ChemPhysChem 2024, 25, e202400102. [Google Scholar] [CrossRef]
- Ford, D.M. Enthalpy−Entropy Compensation Is Not a General Feature of Weak Association. J. Am. Chem. Soc. 2005, 127, 16167–16170. [Google Scholar] [CrossRef]
- Walrafen, G.E. Raman Spectrum of Water: Transverse and Longitudinal Acoustic Modes below. Apprxeq. 300 cm−1 and Optic Modes above. Apprxeq. 300 cm−1. J. Phys. Chem. 1990, 94, 2237–2239. [Google Scholar] [CrossRef]
- Heyden, M.; Sun, J.; Funkner, S.; Mathias, G.; Forbert, H.; Havenith, M.; Marx, D. Dissecting the THz Spectrum of Liquid Water from First Principles via Correlations in Time and Space. Proc. Natl. Acad. Sci. USA 2010, 107, 12068–12073. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.E.; Sorensen, C.M. Raman Spectroscopic Study of Dilute HOD in Liquid H2O in the Temperature Range −31.5 to 160 °C. J. Chem. Phys. 1990, 93, 6954–6961. [Google Scholar] [CrossRef]
- Silverstein, K.A.T.; Haymet, A.D.J.; Dill, K.A. The Strength of Hydrogen Bonds in Liquid Water and Around Nonpolar Solutes. J. Am. Chem. Soc. 2000, 122, 8037–8041. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancini, F.; Graziano, G. On Enthalpy–Entropy Compensation Characterizing Processes in Aqueous Solution. Entropy 2025, 27, 716. https://doi.org/10.3390/e27070716
Mancini F, Graziano G. On Enthalpy–Entropy Compensation Characterizing Processes in Aqueous Solution. Entropy. 2025; 27(7):716. https://doi.org/10.3390/e27070716
Chicago/Turabian StyleMancini, Fiorella, and Giuseppe Graziano. 2025. "On Enthalpy–Entropy Compensation Characterizing Processes in Aqueous Solution" Entropy 27, no. 7: 716. https://doi.org/10.3390/e27070716
APA StyleMancini, F., & Graziano, G. (2025). On Enthalpy–Entropy Compensation Characterizing Processes in Aqueous Solution. Entropy, 27(7), 716. https://doi.org/10.3390/e27070716