
Monte Carlo Simulation on Adiabatic Ensembles and a Genetic Algorithm


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction—Ensembles and Averages
- (1)
- Accept all microstates, weight them with probability and calculate the averages according to Equation (3). This way is, in general, of poor statistical efficiency since most generated microstates have small contributions to the averages. Moreover, it would require the calculation of the partition function, which is not suitable for this particular model.
- (2)
- Accept a new microstate (n) from an old one (o) with probability and calculate the averages by an arithmetic mean. This is the seminal idea of Metropolis et al. [1] that provides an importance sampling because the accepted microstates are the ones that most contribute to the averages. On the other hand, the quotient of eliminates the partition function. This method, dubbed Metropolis Monte Carlo, is implemented in the model of Section 2.
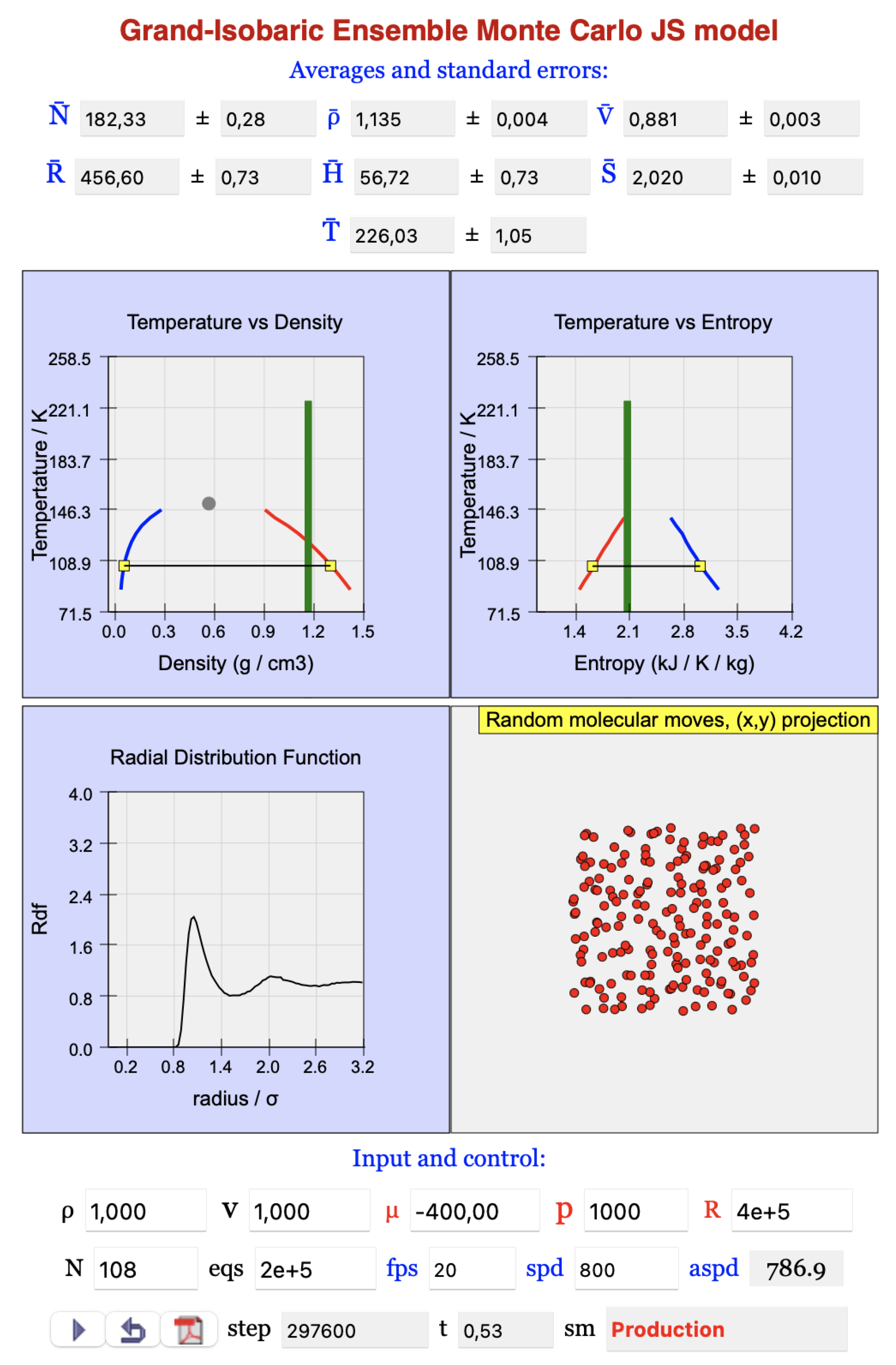
2. Grand-Isobaric Ensemble Model
2.1. Introduction
2.2. Program and Algorithm
- -
- 1% random changes of the volume;
- -
- 33% displacements of particles selected at random;
- -
- 33% insertions of new particles into locations selected at random;
- -
- 33% removals of particles selected at random.
3. Microcanonical Ensemble Model
3.1. Introduction
3.2. Program and Algorithm
- (a)
- Initially, 1000 molecules are set at level 10. Therefore, the total energy of the system is 1000 * 10 u.e.
- (b)
- Two different molecules are successively chosen at random. Then, one of the molecules jumps to the next higher level, increasing the energy by 1 u.e.; the other molecule jumps to the next lower level, decreasing the energy by the same amount. Thus, the total energy of the system is kept constant.
- (c)
- The running entropy is calculated by successive increments of 2000 jumps, and its average displayed every 50 jumps.
- (d)
- Calculation of (Dessaux et al. [38]), for Boltzmann distribution and entropy maximum at equilibrium (red lines in the displays) evaluated at initialization.
3.3. Comments
3.4. Density of States—Boltzmann and Gibbs Entropies
- (i)
- Following Lustig’s method, derive expressions for the thermodynamic properties and apply them.
- (ii)
- From Equation (13), the probability of a microstate is
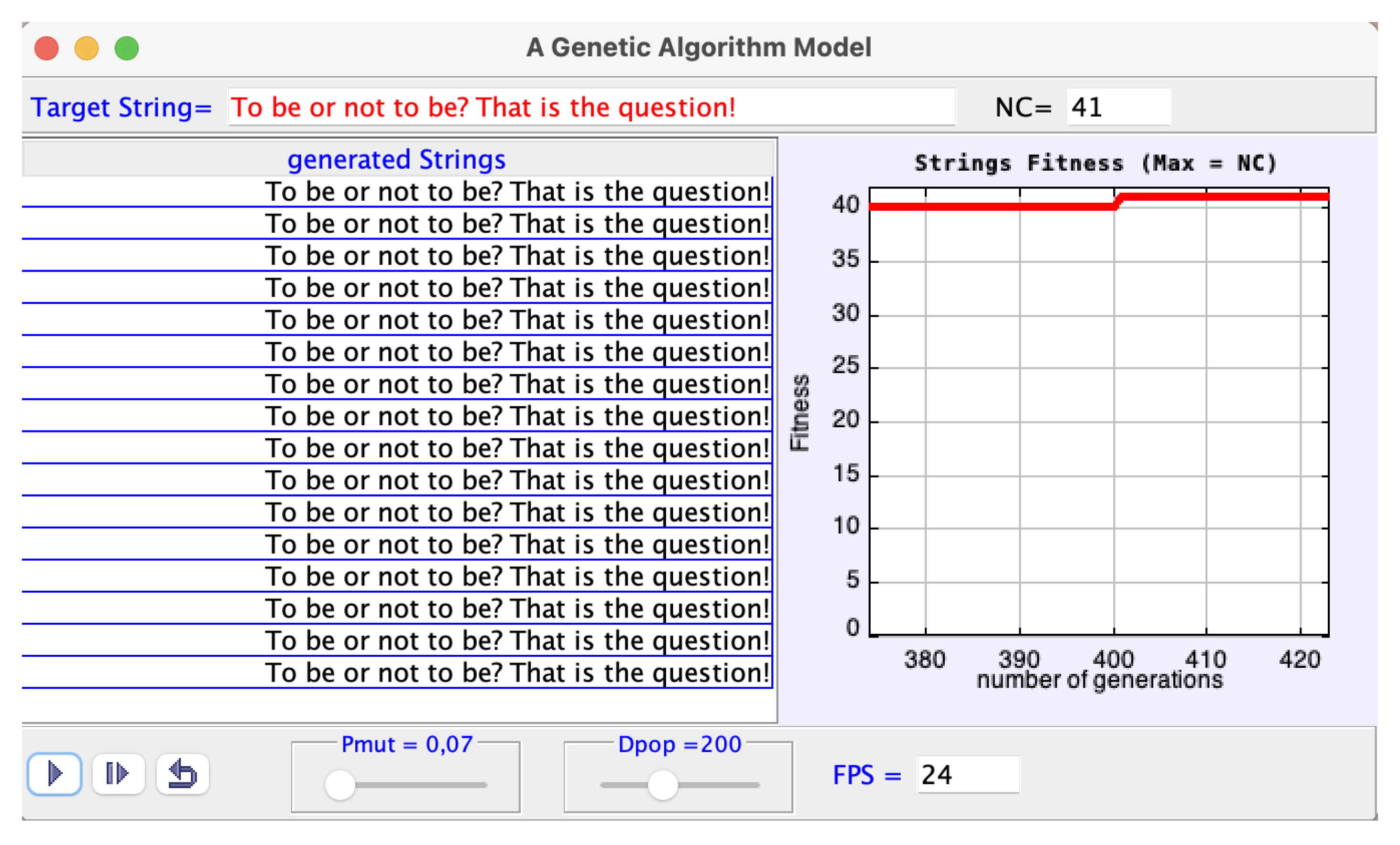
4. A Genetic Algorithm Model
4.1. Introduction
4.2. Program, Algorithm, Guide and Suggested Activities
- (1)
- Set up a random population of strings, with dimension Dpop = 200, each string with 41 random characters.
- (2)
- Determine the fitness of each random string; that is, calculate of the number of characters that eventually coincide with the ones of the target string in their values and respective positions.
- (3)
- Select the string with greatest fitness (“the parent”) and display.
- (4)
- Mutate parent’s characters with probability Pmut = 0.07 to generate a new population of dimension 200.
- (5)
- Go to (3) until the target string is eventually reproduced.
- -
- When the simulation is paused, the user can change the target string, Pmut in the interval [0.0, 1.0] and Dpop in the interval [1, 500] by the respective sliders. As for the FPS field (the number of frames per second), it controls the speed of the displays. It accepts values [1, 24], and 100, and it is editable when the run is paused.
- -
- After editing a data field (target string or/and FPS), the enter key must be pressed; otherwise, the field is not actualized. The same applies after moving the sliders.
- -
- The mouse over a label or button displays a tooltip.
- -
- The program processes only printable characters with ASCII DEC codes from 32 to 126 (https://www.ascii-code.com, accessed on 1 May 2025).
- -
- For Pmut = 0.0, there are no mutations, so the strings do not change.
- -
- For Pmut = 1.0, the characters of the parent are mutated uniformly at random, i.e., at sheer chance, so the successive strings do not converge to the target.
- -
- The default value Pmut = 0.07 leads to a good convergence to the default target.
- -
- If the target is longer than the default one, for example: Shakespeare: To be or not to be? That is the question! is better reproduced with Dpop = 400 even with Pmut = 0.07.
- -
- The shorter target: tobeornottobe is well reproduced with Pmut = 0.1 and Dpop = 100.
- (1)
- The total number of words in Shakespeare works (https://www.opensourceshakespeare.org/statistics/, accessed on 1 May 2025) is 884,421. The number of English characters per word is about 6 (Wikipedia).Henry Bent another comparison [54]:“At room temperature, for example, the conversion of a single calorie of thermal energy completely into potential energy is a less likely event than the production of Shakespeare’s complete works fifteen quadrillion times in succession without error by a tribe of wild monkeys punching randomly on a set of typewriters”.
- (a)
- Calculate the probability of Bent’s monkey metaphor (the Infinite and Finite Monkeys Theorems, cited in Section 4.1, can help).
- (b)
- Compare with the probability he calculated for the complete conversion of thermal energy to work (see Section 4.1).
- (2)
- Reproduce Hamlet’s sentence: ME THINKS IT IS LIKE A WEASEL with different values of Pmut, Dpop and FPS, looking at the fitness and number of generations.
- (3)
- Reproduce the target: CH3-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-NH2 with different values of Pmut, Dpop and FPS, looking at the fitness and number of generations.
- (4)
- Install Java 6.02 modeling tool from Easy Java Simulations (https://www.um.es/fem/Ejs/, accessed on 1 May 2025) in a computer under Unix, Linux or Windows. Run the GeneticAlgortihmModel.jar and follow the instructions in the respective Intro Page to load the program source, automatically named GeneticAlgorithmModel.ejs, into the modeling tool. Then, the code can be inspected and eventually modified. To include crossover, see, e.g, Cartwright [16,58]. In any case, search for the Guides at Easy Java Simulations (https://www.compadre.org/osp/?, accessed on 1 May 2025). This is essential reading, especially by users who have never used it. The modeling tool and respective paths should be configured after its installation.
4.3. Comments
- (1)
- The algorithm steps in Section 4.2. show that the successive populations are generated by mutations, with probability Pmut, from a “parent” i.e., the string with the greatest fitness to the target. If Pmut < 1, the target should be reproduced sooner or later depending on the values of Pmut and Dpop. This is just a simple example of the cumulative selection process which, despite having random mutations, is not at sheer chance because Pmut < 1.
- (2)
- If Pmut = 1, the process is at sheer chance, and the strings chosen by the user are not reproduced, which is in accordance with the improbability of the monkey business and the complete conversion of thermal energy into work.
- (3)
- This Monte Carlo simulation, unlike the grand-isobaric and microcanonical models, does not deal with molecules and ensemble averages. As such, it is not a molecular simulation but rather just an example of an optimization method based on evolutionary principles; however, it is related to physical and biological properties.
- (4)
- The purpose of the simulations is defined at start by the program user (target, Pmut and Dpop). But what about the purpose of natural abiogenesis and biogenesis? Is there no purpose whatsoever, or there is a previous design? These questions are the root of extensive debates (Dawkins [55], Dembski et al. [63], Scientific American [64]); however, it is not in the ambit of this article.
- (5)
- Nevertheless, there is a point about the Second Law of Thermodynamics. Some creationists claim that the 2nd Law contradicts the evolution of species, because the order of the living structures relative to the supposed disorder of their ancestors decreases the entropy of the universe. The creationists’ argument is untrue (Styer [65]). Indeed, it does not account for the role of the environment. In the context of natural evolution, genetic algorithms have also been criticized (Dembski et al. [63]).
- (6)
- Kondepudi et al. [66] revealed that some dissipative structures exhibit organism-like behavior in electrically and chemically driven systems. The highly complex behavior of these systems shows the time evolution to states of higher entropy production. Taking these systems as an example, they present some concepts that give an understanding of biological organisms and their evolution.
- (7)
- Čápek et al. [67], regarding the challenges of the Second Law of Thermodynamics, cite twenty-one formulations of the Law, some not invoking the concept of entropy.
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Alder, B.J.; Wainwright, T.E. Studies in Molecular Dynamics, I. General Method. J. Chem. Phys. 1959, 31, 459–466. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids, 2nd ed.; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Sadus, R.J. Molecular Simulation of Fluids. Theory, Algorithms, Object-Orientation and Parallel Computing, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2024. [Google Scholar]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation. From Algorithms to Applications, 3rd ed.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar]
- Gould, H.; Tobochnik, J.; Christian, W. An Introduction to Computer Simulation Methods, 3rd ed.; Addison-Wesley: Boston, MA, USA, 2016; Available online: https://www.compadre.org/osp/items/detail.cfm?ID=7375 (accessed on 1 May 2025).
- Schoeder, D.V. Interactive molecular dynamics. Am. J. Phys. 2015, 83, 210–218. [Google Scholar] [CrossRef]
- Hoover, W.G.; Hoover, C.G. Time Reversibility, Computer Simulation, Algorithms, Chaos, 2nd ed.; World Scientific: Singapore, 2012. [Google Scholar]
- Kondepudi, D.; Prigogine, I. Modern Thermodynamics. From Heat Engines to Dissipative Structures, 2nd ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Prezhdo, O.V. Advancing Physical Chemistry with Machine Learning. J. Phys. Chem. Lett. 2020, 11, 9656–9658. [Google Scholar] [CrossRef]
- Cartwright, H. Using Artificial Intelligence in Chemistry and Biology. A Practical Guide; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Morcos, F. Information Theory in Molecular Evolution: From Models to Structures and Dynamics. Entropy 2021, 23, 482. [Google Scholar] [CrossRef]
- Ramalho, J.P.; Costa Cabral, B.J.; Silva Fernandes, F.M.S. Improved propagators for the path integral study of quantum systems. J. Chem. Phys. 1993, 98, 3300. [Google Scholar] [CrossRef]
- Klein, M.J. The Physics of J. Willard Gibbs in his Time. Phys. Today 1990, 43, 40–48. [Google Scholar] [CrossRef]
- Chandler, D. Introduction to Modern Statistical Mechanics; Oxford University Press: Oxford, UK, 1987. [Google Scholar]
- Baldovin, M.; Marino, R.; Vulpiani, A. Ergodic observables in non-ergodic systems: The example of the harmonic chain. Phys. A 2023, 630, 129273. [Google Scholar] [CrossRef]
- Kelly, J.J. Semiclassical Statistical Mechanics. 2002. Available online: https://www.physics.umd.edu/courses/Phys603/kelly/Notes/Semiclassical.pdf (accessed on 1 May 2025).
- Desgranges, C.; Delhommelle, J. The central role of entropy in adiabatic ensembles and its application to phase transitions in the grand-isobaric adiabatic ensemble. J. Chem. Phys. 2020, 153, 094114. [Google Scholar] [CrossRef]
- Pearson, E.M.; Halicioglu, T.; Tiller, W.A. Laplace-transform technique for deriving thermodynamic equations from classical microcanonical ensemble. Phys. Rev. A 1985, 32, 3030–3039. [Google Scholar] [CrossRef]
- Graben, H.W.; Ray, J.R. Eight physical systems of thermodynamics, statistical mechanics and computer simulations. Mol. Phys. 1993, 80, 1183–1193. [Google Scholar] [CrossRef]
- Graben, H.W.; Ray, J.R. Unified treatment of adiabatic ensembles. Phys. Rev. A 1991, 43, 4100–4103. [Google Scholar] [CrossRef] [PubMed]
- Site, L.D.; Krekeler, C.; Whittaker, J.; Agarwal, A.; Klein, R.; Höfling, F. Molecular Dynamics of Open Systems: Construction of a Mean-Field Particle Reservoir. Adv. Theory Simul. 2019, 2, 1900014. [Google Scholar] [CrossRef]
- Çagin, T.; Pettitt, M. Grand Molecular Dynamics: A Method for Open Systems. Mol. Simul. 1991, 6, 5–26. [Google Scholar] [CrossRef]
- Desgranges, C.; Delhommelle, J. Evaluation of the grand-canonical partition function using expanded Wang-Landau simulations. I. Thermodynamic properties in the bulk and at the liquid-vapor phase boundary. J. Chem. Phys. 2012, 136, 184107. [Google Scholar] [CrossRef]
- Ströker, P.; Meier, K. Classical statistical mechanics in the (μ,V, L) and (μ, p, R) ensembles. Phys. Rev. E 2023, 107, 064112. [Google Scholar] [CrossRef]
- Lustig, R. Microcanonical Monte Carlo simulation of thermodynamic properties. J. Chem. Phys. 1998, 109, 8816. [Google Scholar] [CrossRef]
- Thermodynamics and Statistical Mechanics of Small Systems. Special Issue Edited by Puglisi, A., Sarracino, A., Vulpani, A., Entropy Journal. 2017–2018. Available online: https://www.mdpi.com/journal/entropy/special_issues/small_systems (accessed on 1 May 2025).
- Rodrigues, P.C.R.; Silva Fernandes, F.M.S. Phase Transitions, coexistence and crystal growth dynamics in ionic nanoclusters: Theory and Simulation. J. Mol. Struct. Theochem 2010, 946, 94–106, Note: In this referred to paper the links to computer animations were deleted. Animations just for KCl nanoclusters. Available online: https://webpages.ciencias.ulisboa.pt/~fmfernandes/clusters_IYC2011/index.htm (accessed on 1 May 2025). [CrossRef]
- Callen, H.B. Thermodynamics and an Introduction to Thermostatistics, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 1985. [Google Scholar]
- Ray, J.R.; Wolf, R.J. Monte Carlo simulations at constant chemical potential and pressure. J. Chem. Phys. 1993, 98, 2263. [Google Scholar] [CrossRef]
- Desgranges, C.; Delhommelle, J. Entropy determination for mixtures in the adiabatic grand-isobaric ensemble. J. Chem. Phys. 2022, 156, 084113. [Google Scholar] [CrossRef]
- Desgranges, C.; Delhommelle, J. Detrmination of mixture properties via a combined Expanded Wang-Landau simulations—Machine Learning approach. Chem. Phys. Lett. 2019, 715, 1–6. [Google Scholar] [CrossRef]
- Wilding, N.B. Computer simulation of fluid phase transitions. Am. J. Phys. 2001, 69, 1147. [Google Scholar] [CrossRef]
- Errington, J.R. Direct calculation of liquid-vapor phase equilibria from transition matrix Monte Carlo simulation. J. Chem. Phys. 2003, 118, 9915. [Google Scholar] [CrossRef]
- Silva Fernandes, F.M.S.; Fartaria, R.P.S. Gibbs ensemble Monte Carlo. Am. J. Phys. 2015, 83, 809. [Google Scholar] [CrossRef]
- Heyes, D.M. The Lennard-Jones Fluid in the Liquid-Vapour Critical Region. Comput. Methods Sci. Technol. 2015, 21, 169–179. [Google Scholar] [CrossRef]
- Schettler, P. A computer problem in statistical thermodynamics. J. Chem. Educ. 1974, 51, 250. [Google Scholar] [CrossRef]
- Dessaux, O.; Goudmand, P.; Langrand, F. Thermodynamique Statistique Chimique, 2nd ed.; Dunod Université: Bordas, Paris, 1982. [Google Scholar]
- Moore, T.A.; Schroeder, D.V. A different approach to introducing statistical mechanics. Am. J. Phys. 1997, 65, 26–36. [Google Scholar] [CrossRef]
- Timberlake, T. The Statistical Interpretation of Entropy: An Activity. Phys. Teach. 2010, 48, 516–519. [Google Scholar] [CrossRef]
- Salagaram, T.; Chetty, N. Enhancing the understandin of entropy through computation. Am. J. Phys. 2011, 79, 1127–1132. [Google Scholar] [CrossRef]
- Baierlein, R. Entropy and the second Law: A pedagogical alternative. Am. J. Phys. 1994, 62, 15–26. [Google Scholar] [CrossRef]
- Ben-Naim, A. Entropy: Order or Information. J. Chem. Educ. 2011, 88, 594–596. [Google Scholar] [CrossRef]
- Lieb, E.H.; Yngvason, J. A Fresh Look at Entropy and the Second Law of Thermodynamics. Phys. Today 2000, 53, 32–37. [Google Scholar] [CrossRef]
- Wolfram, S. Computational Foundations for the Second Law of Thermodynamics. 3 February 2023. Available online: https://writings.stephenwolfram.com/2023/02/computational-foundations-for-the-second-law-of-thermodynamics/ (accessed on 1 May 2025).
- Silva Fernandes, F.M.S.; Ramalho, J.P.P. Hypervolumes in microcanonical Monte Carlo. Comput. Phys. Comm. 1995, 90, 73–80. [Google Scholar] [CrossRef]
- Frenkel, D.; Warren, B. Gibbs, Boltzmann, and negative temperatures. Am. J. Phys. 2015, 83, 163. [Google Scholar] [CrossRef]
- Swendsen, R.H. Thermodynamics of finite systems: A key issues review. Rep. Prog. Phys. 2018, 81, 072001. [Google Scholar] [CrossRef]
- Shirts, R.B. A comparison of Boltzmann and Gibbs definitions of microcanonical entropy of small syatems. AIP Adv. 2021, 11, 125023. [Google Scholar] [CrossRef]
- Rajan, A.G. Resolving the Debate between Boltzmann and Gibbs Entropy: Relative Energy Window Eliminates Thermodynamic Inconsistencies and Allows Negative Absolute Temperatures. J. Phys. Chem. Lett. 2024, 15, 9263–9271. [Google Scholar] [CrossRef]
- Ray, J.R. Microcanonical ensemble Monte Carlo method. Phys. Rev. A 1991, 44, 4061–4064. [Google Scholar] [CrossRef]
- Infinite Monkey Theorem. Posted by Wikipedia. Available online: https://en.wikipedia.org/wiki/Infinite_monkey_theorem (accessed on 1 May 2025).
- Woodcock, S.; Falleta, J. A numerial evalutaion of the Finite Monkeys Theorem. Frankl. Open 2024, 9, 100171. [Google Scholar] [CrossRef]
- Bent, H.A. The Second Law. An Introduction to Classical and Statistical Thermodynamics; Oxford University Press: Oxford, UK, 1965. [Google Scholar]
- Dawkins, R. The Blind Watchmaker, 2nd ed.; W.W. Norton & Company: New York, NY, USA, 1996. [Google Scholar]
- Freeman, J.A. Simulating Neural Networks with Mathematica; Addison Wesley: Boston, MA, USA, 1994. [Google Scholar]
- Phyton Version of Weasel. Posted by GitHub. Available online: https://github.com/explosion/weasel (accessed on 1 May 2025).
- Cartwright, H.G. Applications of Artificial Intelligence in Chemistry. In Oxford Chemistry Primers; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Zupan, J.; Gasteiger, J. Neural Networks in Chemistry and Drug Design, 2nd ed.; Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Desgranges, C.; Delhommelle, J. Crystal nucleation along an entropic pathway: Teaching liquids how to transition. Phys. Rev E 2018, 98, 063307. [Google Scholar] [CrossRef]
- Latino, D.A.R.S.; Fartaria, R.P.S.; Freitas, F.F.M.; Aires-de-Sousa, J.; Silva Fernandes, F.M.S. Mapping Potential Energy Surfaces by Neural Netwoks: The ethanol/Au(111) interface. J. Electroanal. Chem. 2008, 624, 109–120. [Google Scholar] [CrossRef]
- Shwalbe-Koda, D.; Hamel, S.; Babak, S.; Lordi, V. Information theory unifies machine learning, uncertainty quantification, and material thermodynamics. arXiv 2024, arXiv:2404.12367v1. [Google Scholar]
- Dembski, W.A.; Ewert, W. The Design Inference: Eliminating Chance Through Small Probabilities, 2nd ed.; Discovery Institute Press: Seattle, WA, USA, 2023. [Google Scholar]
- The Debate over Creationism vs. Evolution; Scientific American Educational Publishing: New York, NY, USA; The Rosen Publishing Group: New York, NY, USA, 2024.
- Styer, D.F. Entropy and Evolution. Am. J. Phys. 2008, 76, 1031–1033. [Google Scholar] [CrossRef]
- Kondipudi, D.K.; De Bari, B.; Dixon, J.A. Dissipative Structures, Organisms and Evolution. Entropy 2020, 22, 1305. [Google Scholar] [CrossRef]
- Cápek, V.; Sheehan, D.P. Chalenges of the Second Law of Thermodynamics. Theory and Experiment; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva Fernandes, F.M.S. Monte Carlo Simulation on Adiabatic Ensembles and a Genetic Algorithm. Entropy 2025, 27, 565. https://doi.org/10.3390/e27060565
Silva Fernandes FMS. Monte Carlo Simulation on Adiabatic Ensembles and a Genetic Algorithm. Entropy. 2025; 27(6):565. https://doi.org/10.3390/e27060565
Chicago/Turabian StyleSilva Fernandes, Fernando M. S. 2025. "Monte Carlo Simulation on Adiabatic Ensembles and a Genetic Algorithm" Entropy 27, no. 6: 565. https://doi.org/10.3390/e27060565
APA StyleSilva Fernandes, F. M. S. (2025). Monte Carlo Simulation on Adiabatic Ensembles and a Genetic Algorithm. Entropy, 27(6), 565. https://doi.org/10.3390/e27060565