1. Introduction
The concept of a local equilibrium in non-equilibrium thermodynamics comes up in analyses of transport processes such as heat and mass transport. The term “local equilibrium” is not well defined [
1]. Moreover, depending on whether a perturbation of an equilibrium state is temporal or spacial, the concept of local equilibrium in the perturbed state may have different meanings. Our use of the term is related to so-called linear non-equilibrium thermodynamics, where one assumes a local equilibrium, meaning that the Gibbs–Duhem equation is assumed valid with local values of the variables. The assumption is closely related to assuming that the local non-equilibrium entropy can be approximated by its equilibrium value in the entropy balance, which is the basis for finding the entropy production and coupled flux–force relations [
2]. Whereas entropy is well understood and modeled for thermodynamic systems at equilibrium, the situation for systems out of equilibrium is less clear [
3]. Usually, the assumption is introduced as an approximation and justified with successful consistency tests as “circumstantial evidence”. This is not satisfactory, and the extremely large forces and fluxes used in computer simulations that are necessary for acceptable signal-to-noise ratios in the results raise a need for a quantitative examination of local equilibrium approximation (LEA).
An alternative approach is to
assume the opposite, viz. a local non-equilibrium, derive the corresponding theory whenever possible, and determine the deviation from the local equilibrium. A powerful criterion for this deviation is the difference in equilibrium and non-equilibrium entropy. Several theories for non-equilibrium entropy have been proposed for heat transport and diffusion, see, e.g., [
4,
5], and references therein. Cimmelli et al. [
4] have given a very thorough discussion of four different theories and compared their pros and cons. They discuss the theories’ relation to kinetic theory, valid for low-density gases and conclude that in general, continuum theories that are compatible with kinetic theory are less universal, but more predictable than other theories.
In the present work, we pursue developing a theory for transport processes that do not depend on LEA. The theory is based on the Boltzmann equation and developed for diffusion in a binary gas mixture. It is then used to analyze results from molecular dynamics (MD) simulations of binary diffusion in a Lennard–Jones/spline (LJs) system [
6]. We use an MD method introduced by Holian for diffusion simulations, which will be described in detail in
Section 3. (Information about this method was communicated by B. L. Holian to W. G. Hoover in 1973, see Hoover and Ashurst [
7] and Allen and Tildesley, p. 370 [
8]. A very similar method has been used by Dong and Cao to compute self-diffusion coefficients [
9]).
The two components are physically identical but they are identified as two types, “1” and “2” or “red” and “blue”. Therefore, the system’s properties are those of a one-component system, which simplifies the analysis and enables comparison with self-diffusion data computed with other methods. This is one of the simplest cases one can imagine for a study of a transport process. Our intention is to force the diffusion process as much as the system allows in an attempt to provoke a deviation from the local equilibrium. The results are compared with what we obtain from Fick’s law and with data from equilibrium simulations (mean square displacement).
According to Darken’s second equation [
10], the mutual diffusion coefficient
D in a binary mixture relates to the self-diffusion coefficients
and
, as
where
is the mole fraction of component 1 (
) and
is the thermodynamic factor. In our ideal case,
and
, so that the diffusion coefficient we compute by generating a diffusive flux in the two-component system is simply the self-diffusion coefficient. This enables a comparison between the binary diffusion coefficient, generated with extreme fluxes and forces, and self-diffusion coefficients generated at global equilibrium.
3. MD Simulations
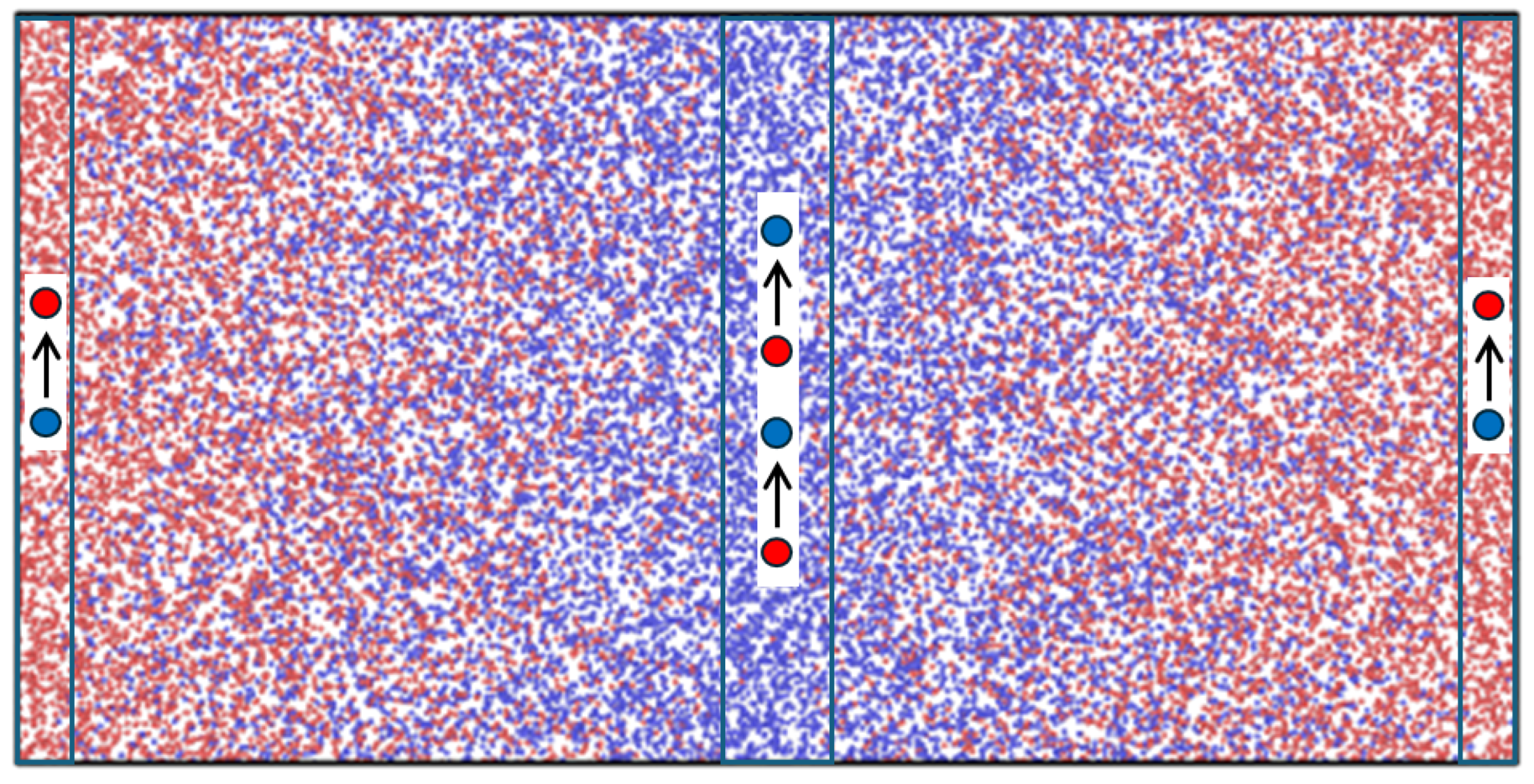
Non-equilibrium molecular dynamics (NEMD) simulations were carried out to provide data for use with the theory. The layout of the MD system is shown in
Figure 1.
The system consisted of
Lennard–Jones/spline (LJs) particles [
6] in a rectangular cuboid with aspect ratio
. The LJs potential is a smoothly truncated Lennard–Jones potential with cut-off distance
. The MD box had periodic boundary conditions in all three directions. The box was divided into 64 layers of equal thickness in the
x-direction for recording local data of temperature, density, composition, etc. The particles were of two types, 1 (“red”) and 2 (“blue”), with exactly the same mass and potential parameters [
8].
Concentration gradients and diffusive mass fluxes were generated with an in-house NEMD code using the MEX algorithm [
1]. The MEX algorithm works so that a randomly picked particle of type 2 is changed to type 1 in one of the control volumes at the box ends (see
Figure 1) while a particle of type 1 is changed to type 2 in the central volume at the same time. The overall concentration was thus kept constant. The identity swapping was tried at regular intervals, which varied between every time step and every 100th time step. The swapping acts as source and sink terms at the boundaries (ends and center of the MD box). At steady state, the mass flux in the bulk fluid is related to the swapping frequency,
,
, where
A is the cross-sectional area of the box. By changing the swapping frequency, the diffusion flux was controlled from zero (equilibrium) to maximum (saturated control volumes, i.e.,
at the box ends and
in the box center). If there were no candidate particles to swap in either control volume, the swapping was skipped and the actual swapping frequency was reduced accordingly. The reported mass fluxes in
Table 1 were computed from
for each layer in the fluid and then averaged over the layers in the bulk. The logged swapping frequency was used for consistency check. Typical swapping frequencies varied from 9 to 22 (in LJ units).The mass current is half the swapping frequency (due to the fact that particles move both left and right from the swapping layers), and the mass flux is the current divided by the cross-sectional area. For the results reported here, the time between each swap was on average between 0.02 and 0.06, which is three orders of magnitude shorter than the characteristic time
. We can therefore expect that the non-continuous particle swapping at the boundaries leads to a mass flux that is smooth in time (which was also observed).
The simulations started from an FCC lattice configuration with
. To prevent heating of the system due to the entropy production, the system temperature was controlled by thermostating each of the 64 layers to the same set temperature and constant density was assured by rapid equilibration of the pressure in the fluid. The overall density was controlled by the total number of particles in the fixed box volume. The thermostat was a simple velocity scaling and shifting subject to set the local momentum to zero [
1]. The scaling factor was the same in
x-,
y-, and
z-directions, but the shift was direction specific. The thermostat was activated every 20 time steps.
One may argue that enforcing a constant temperature through the system has an impact on properties derived from temperature gradients such as Soret and Dufour effects. This should be considered for systems where the components do not have identical properties, but for the present system, such coupled effects vanish.
Six cases were generated with the density and temperature as listed in
Table 1. All values of physical properties reported here are in Lennard–Jones units. The mean free path,
, was estimated from elementary kinetic theory as
and compared with the half box length,
. Case 1 is a very dilute (ideal) gas which is not well suited for MD simulations due to the fact that the particles are mostly in free flight; the mean free path is of the same order as
(the distance between the swapping layers) in this case. Nevertheless, this low-density case was included here because of the kinetic theory basis for the analysis. Case 6 is close to the LJs system’s dew point (at
for
). When the mean free path is much larger than the range of the potential, particles may overlap at the end of a time step. To avoid strong accelerations if this happens, the particles were given a reflecting “shield” at
.
A total of 4,000,000 time steps of length in Lennard–Jones units were used to produce data for analysis. The final 2,000,000 time steps were used to represent steady state with .
4. Results and Discussion
We shall now analyze the binary diffusion data using the theory developed in
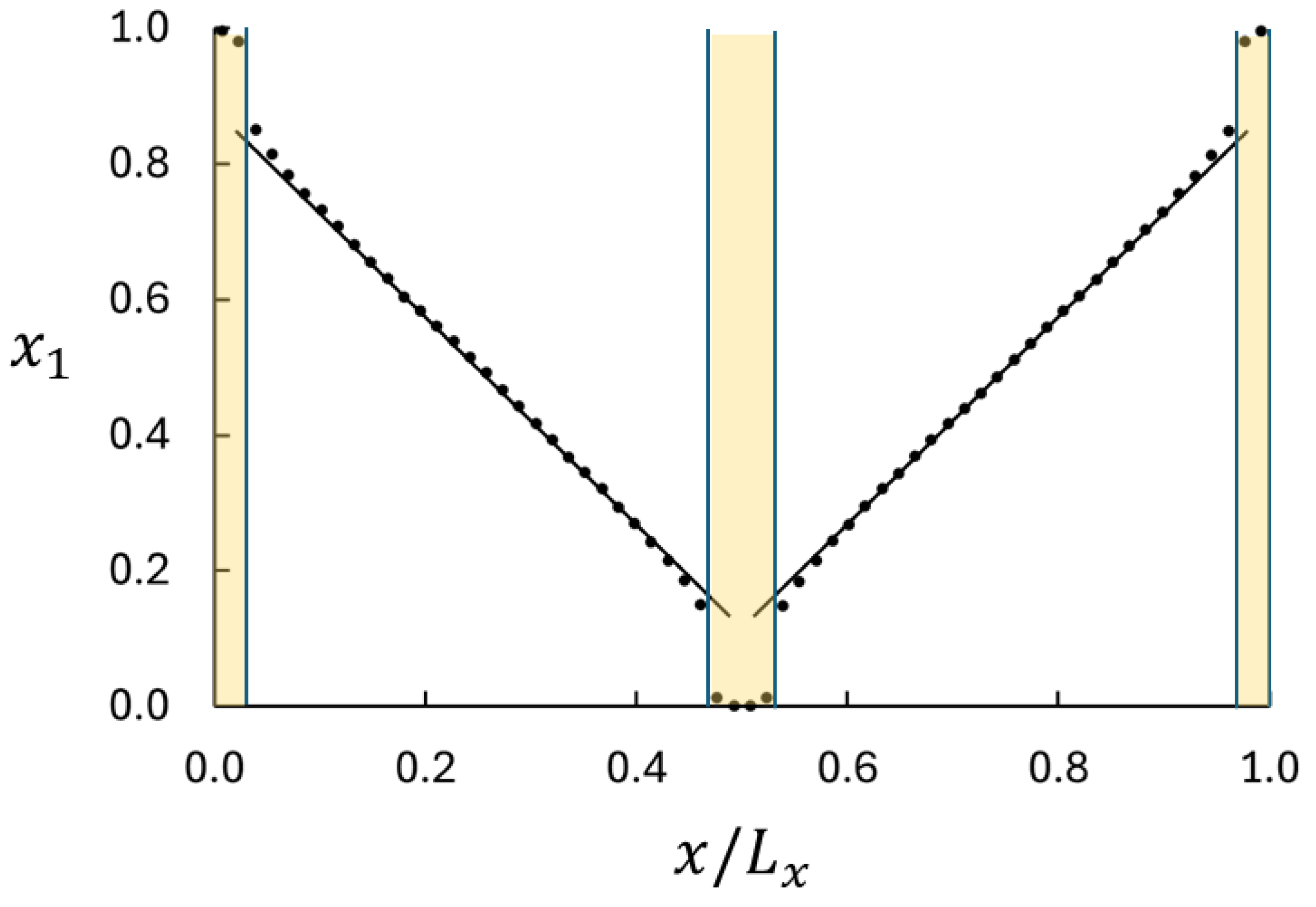
Section 2.2. Central in this analysis is the curvature of the mole fraction profile and the deviation from the local equilibrium. A typical mole fraction profile is shown in
Figure 2. The following are three important features: (i) the profile appears to be linear in the central part of the bulk regions, (ii) the profile is slightly curved near the ends of the bulk regions, and (iii) there are significant jumps in
between the swapping volumes and the bulk. We shall focus on the profile in the bulk near the boundaries where the profile is slightly curved. Equation (
29) shows that for
and
D to be constant in the bulk region, the fraction
must follow from
where
and
. In other words, there is a direct relation between the nonlinearity in the mole fraction profile and
. Hence, we use the curvature in the mole fraction profile to analyze the deviation from the local equilibrium.
The jumps in
are consequences of the large imposed mass flux and the resolution of the data acquisition in the
x-direction. Similar jumps are seen for temperature profiles in simulations of heat transport [
15,
16]. A weaker imposed mass flux would bring the system closer to equilibrium and show smaller jumps, but the objective here is to bring the system as far from equilibrium as possible. Clearly, the maximum difference in composition, and therefore the maximum mass flux, is obtained when the swapping layers are saturated with one of the components.
A central question in this work is to what extent LEA is valid. Equation (
28) gives a direct measure of the deviation from the equilibrium chemical potential. We see that
is positive. From Equation (
29), we see that Fick’s law will always underestimate the value of
D. The difference can be quantified with the second term in the square bracket in Equation (
29). If the fraction
is small compared to 1, the local equilibrium is a good approximation, reducing Equation (
29) to Fick’s law. Otherwise, the approximation is poor. Exactly what we mean by an acceptable approximation is a matter of choice, but the important point is that whatever choice is made, it can be quantified and communicated. Equation (
28) shows that the deviation from equilibrium depends on
with the minimum value at
. The minimum value of
is then
with
.
Lack of local equilibrium will depend on density, temperature, and the mass flux.
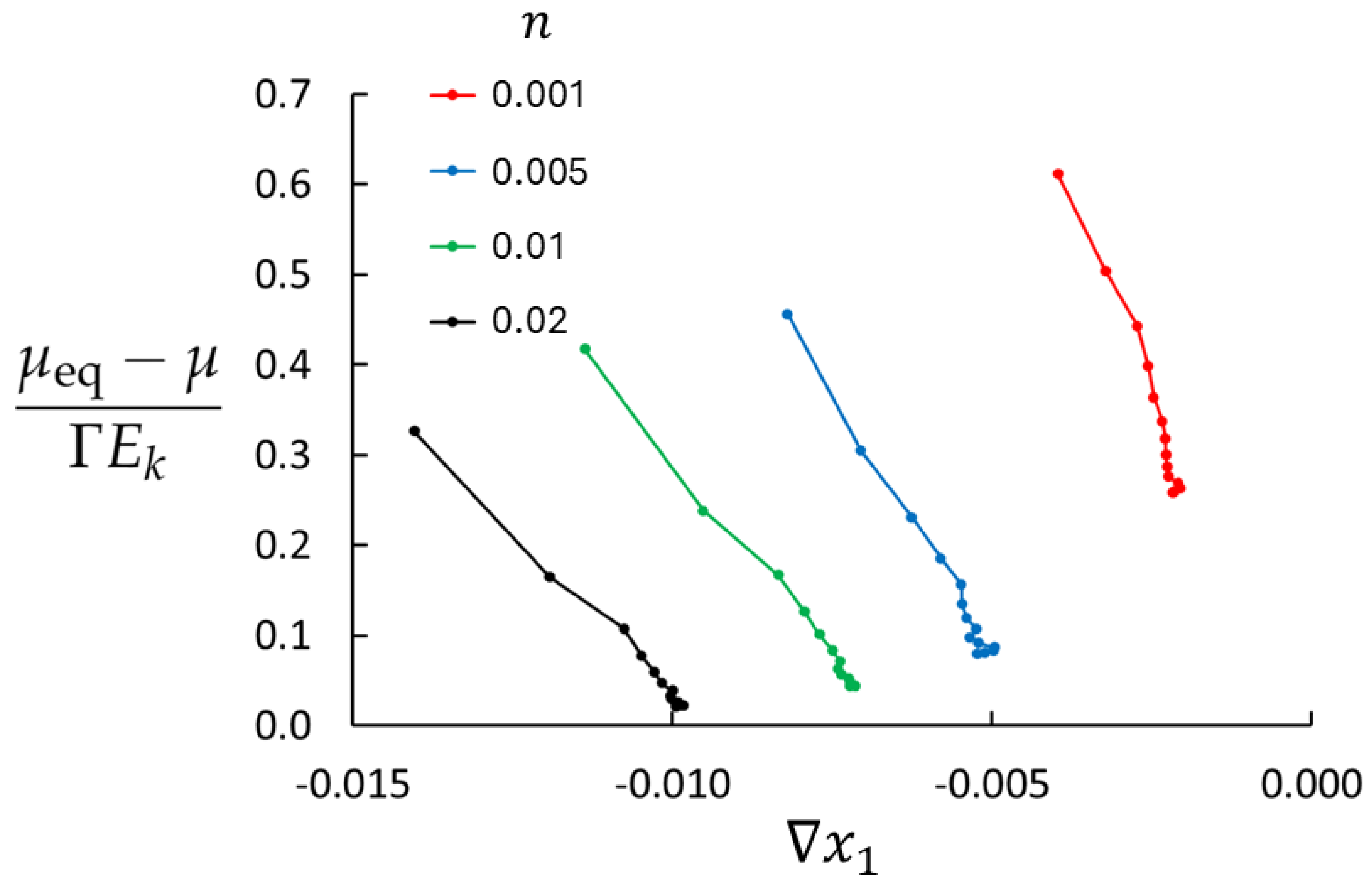
Figure 3 shows the deviations from the local equilibrium for four densities used in this study at
and maximum imposed mass flux (saturated control volumes). The right endpoint of each graph represents the minimum value of
, found in the central part of the bulk region where the system is closest to equilibrium. The maximum value of
is the left endpoint (near the swapping layers) where the deviation from equilibrium is largest. LEA appears to be very poor for the lowest density, at best approximately 25% (in the central part of the bulk) and worse towards the swapping layers. In this region, the diffusive velocity of the rare component is large,
, and the right-hand side of Equation (
28) becomes large. Furthermore, for
, molecular collisions are rare and mass transport is to a large extent ballistic. The situation is much better for higher densities. For
, the deviation from equilibrium is approximately 2 % in the central part of the bulk, rising to about 30 % near the swapping layers.
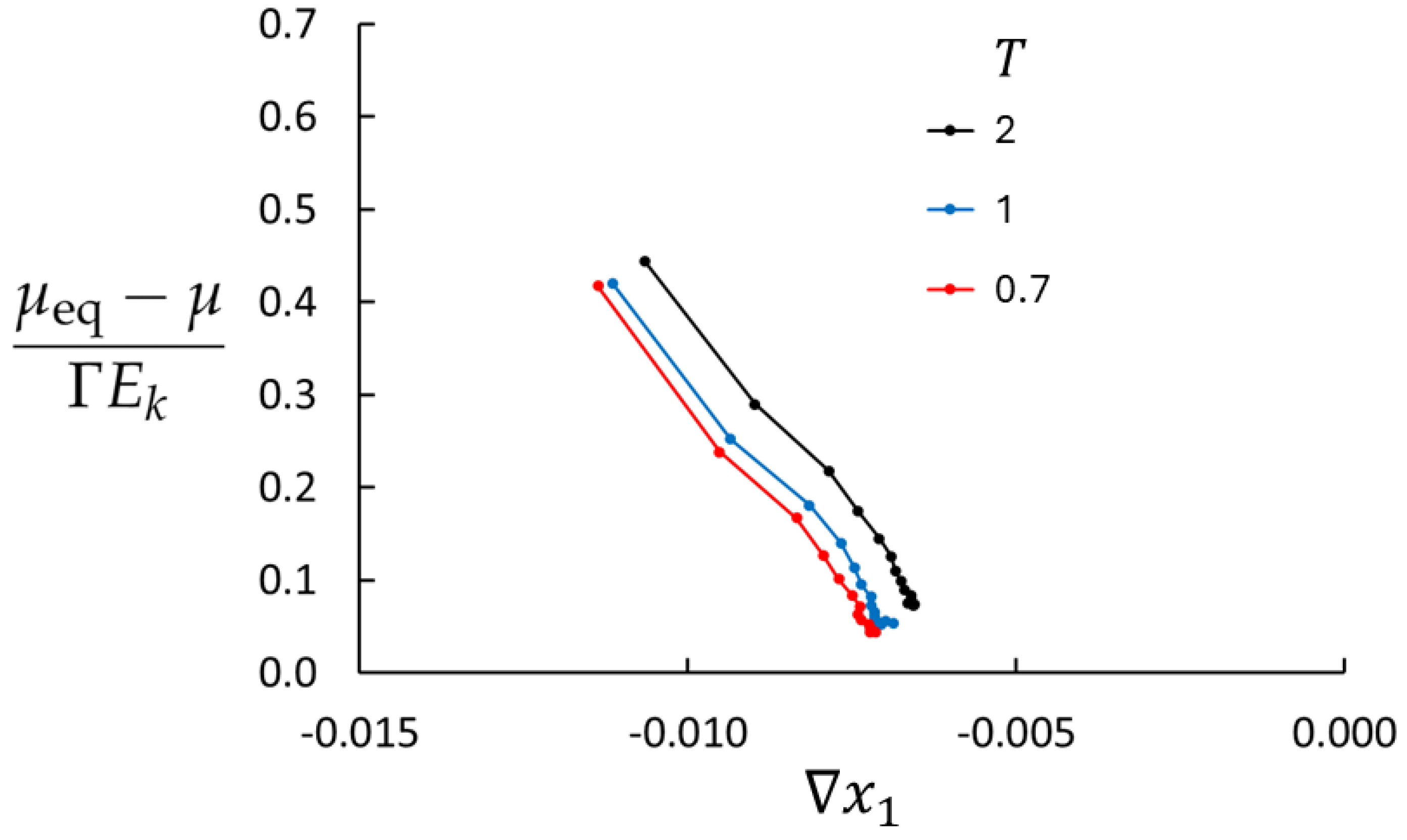
Figure 4 shows the deviations from the local equilibrium for three temperatures (
, and
),
, and maximum mass flux. LEA is less than 4% off in the central part of the bulk for the highest temperature and better for the lower temperatures.
LEA will clearly be better for reduced mass flux. According to Equations (
28) and (
29), the
is proportional to
. We simulated Case 4 for six additional imposed fluxes. The results are shown in
Figure 5. In addition to the reduced value of
with reduced flux, it is worth noting that the range spanned by
is also reduced with reduced flux, meaning that the mole fraction profile is less curved.
The parameter
in Equation (
28), which is implicit in Equation (
29), was determined as follows. In principle,
D may be a function of
and
. However, the system is an ideal mixture of particles with identical physical properties at uniform density and temperature. The diffusion coefficient in Equation (
29) must therefore be independent of
and
. The value of
was adjusted to satisfy this condition. An example of the relation between
and
D is shown in
Figure 6. A value of
that is too large (small) gives too much (little) emphasis on the curvature in
, especially near the ends of the mole fraction range (cf.
Figure 2). By fitting a linear function to
D vs.
,
was determined as the value giving zero slope (=2.4 for Case 4 shown in
Figure 6). We note that the value of
D for the largest values of
, where the
-profile is linear, is not very sensitive to the value of
. This procedure gave almost the same value for
for the six cases studied here (
Table 2).
We note that the condition on D applies to the special case of identical components, in a more general case one must expect that D depends on the composition.
Numerical values for the diffusion coefficient obtained from Equation (
29) for the six cases are shown in
Table 2. The coefficient obtained from Fick’s law is consistently smaller than
D and the agreement between
and
D is better for higher densities with fixed temperature and for higher temperature with fixed density. Both trends can be understood in terms of the system’s ability to dissipate the effect of the perturbation.
As mentioned in the Introduction, the main purpose of this work is to develop a tool for analysis of LEA in non-equilibrium thermodynamics. We considered a very special case, mutual diffusion of an ideal system in gas phase, and used kinetic theory as the basis. As it stands, the method has limited value for higher densities, which requires an improved version of the kinetic theory. Another limitation is that we approximated
,
, and
with ideal-gas values. These properties must be determined from a more realistic equation of state, which is available for the LJs model [
17].
The theory was developed for binary diffusion at low density in general and simplified for a system with identical components. Application to binary systems with different components is therefore possible. For instance, analysis of mutual diffusion in a binary mixture with two different components will be possible with the same tool, although it will require more information to evaluate the terms
and
in Equations (
9) and (
10). The conclusions with respect to LEA may or may not be the same as we draw in this work, but the point is that the theory presented here can be used for the analysis. The results presented in
Table 2 suggest that increased density will improve LEA due to more frequent molecular collisions.
Another interesting application is heat transport, where the same considerations of the combination of ballistic and diffusive transport mechanisms apply.
Equation (
30) gives us a tool to compute the difference between the non-equilibrium and equilibrium entropy. We found for
:
where we used the Sackur–Tetrode ideal gas entropy,
, as representing
. Using Case 2 as an example and assuming that the LJs model represents argon with the potential parameters,
K and
nm, we find that the molar equilibrium entropy is 125 J mol
−1 K
−1 and the difference between the molar non-equilibrium and equilibrium entropies is 0.5 J mol
−1 K
−1 for
. The relative deviation is
for
and 0.01 for
(the extreme value in the bulk,
cf Figure 2). These results are additional demonstrations of the excellence of LEA in this diffusion case.
The importance of the kinetic energy of diffusion [
14] can be illustrated by the ratio between the kinetic energy of diffusion and the thermal energy,
Again using Case 2 as an example, we found that the ratio is 0.04 and 0.46 for
and
, respectively. This result shows that even if the kinetic energy of diffusion is large compared with the thermal energy, the deviation from the local equilibrium is small and LEA is good.
A more detailed analysis of the velocities should include the velocity distributions. A local Maxwellian distribution would indicate a local equilibrium. An analysis of the velocity part of the Boltzmann
H-function can be used to quantify the deviation from equilibrium [
1]. The velocity distributions can also be used for a discussion of the local temperature and the role of the kinetic energy of diffusion in computing the local temperature, which we have not considered here. Based on the thermodynamic results we have, which show that the system is close to equilibrium, we expect that the velocity distribution for each component will be Gaussian in the
y- and
z-directions, a shifted Gaussian in the
x-direction, and Gaussians with zero mean for the combined distributions of the two components in all three directions. The variance of the distributions will give us the local kinetic temperature.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}