
Elastic Entropic Forces in Polymer Deformation



Abstract
:
1. Introduction
2. Methods: Accounting for Entropic Elastic Forces in Polymer Deformation Processes
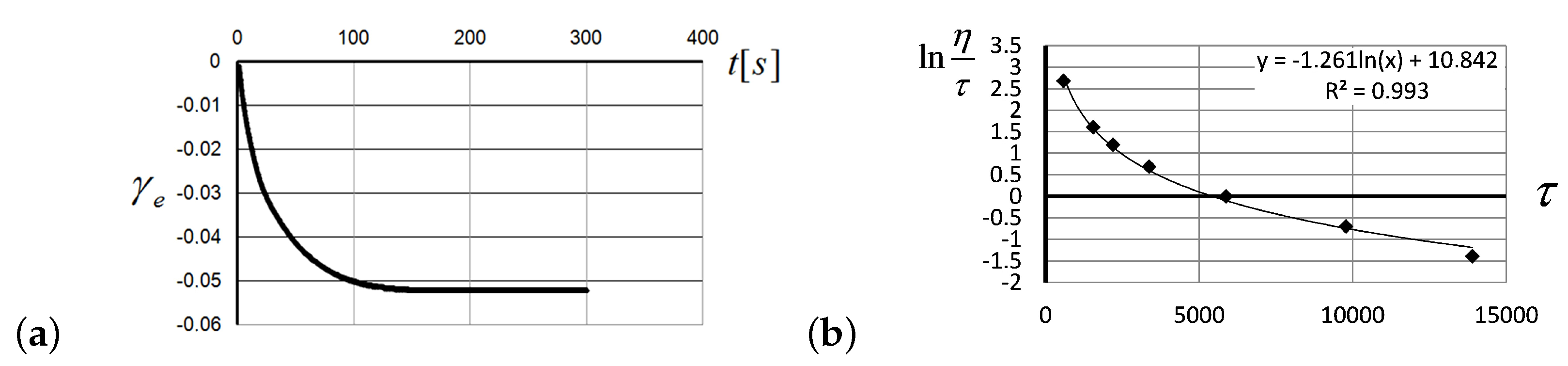
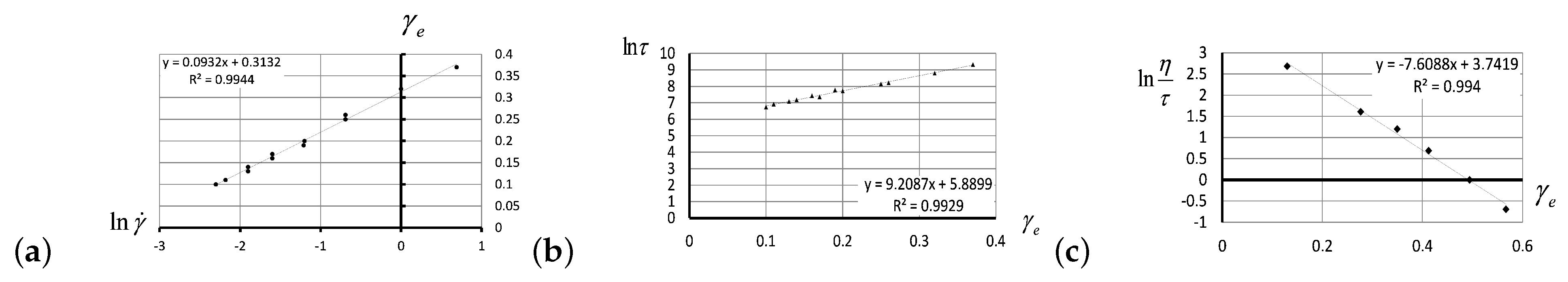
2.1. Viscosity Anomalies in Polymer Flows
2.2. Dependence of Elastomer Tensile Curves on Elastic Entropic Forces
- (1)
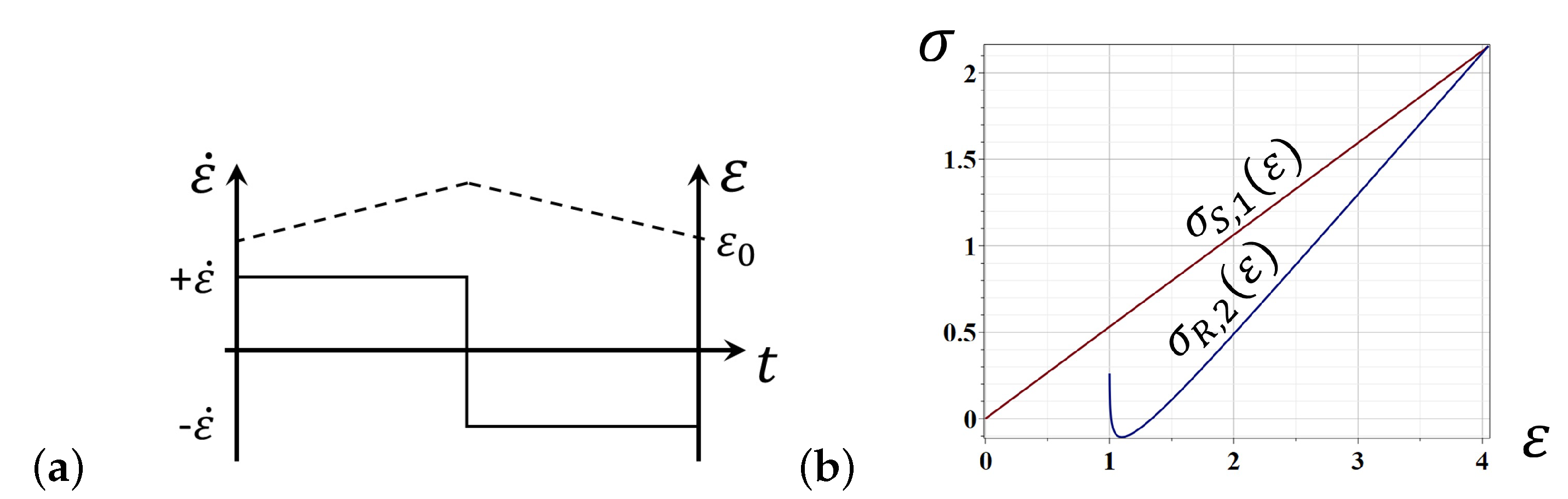
- With increasing deformation over the stretching phase, the EEF reinforces the resistance of molecular network against the tensile stress fostering an increase of the jump activation energy in Eyring’s formula (3) by an amount where is a viscous volume coefficient related to the stretching deformation [5,14,30], viz.,where is the pre-exponential factor calculated by absolute reaction rate theory, and the index S pertains to the stretching phase of deformation.
- (2)
- In a retraction phase of deformation, the EEF decreases the jump activation energy by an amount , as the action of EEF coincides with the retraction direction of the specimen deformation, and, therefore,where the corresponding pre-exponential factor and viscous volume coefficient are indexed by being related to the retraction phase of deformation.
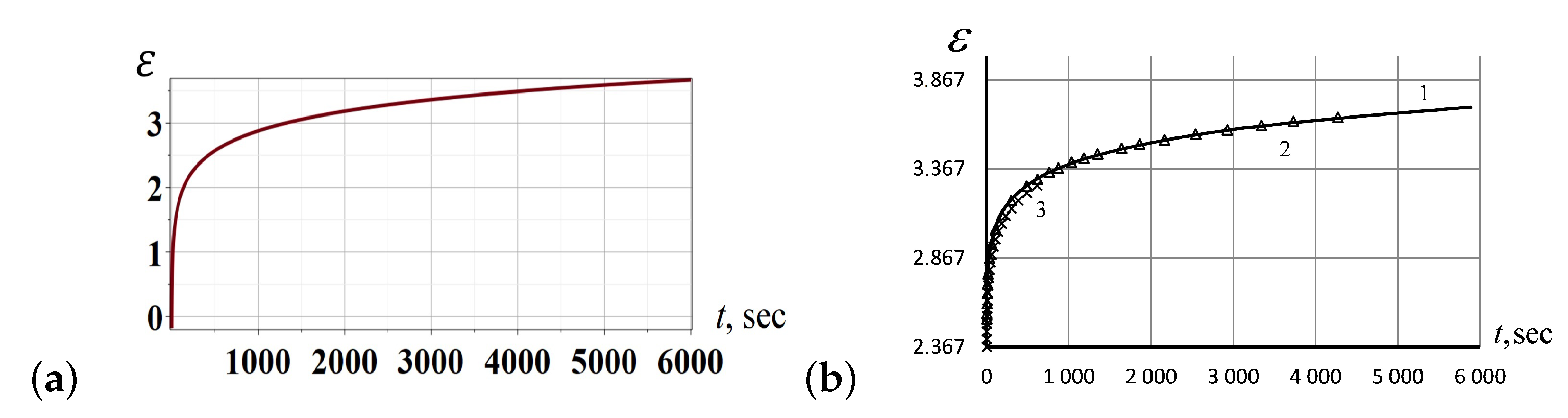
2.3. Entropic Elastic Forces for Creeping Prediction
3. Results: Experimental Verification of Entropic Elastic Forces
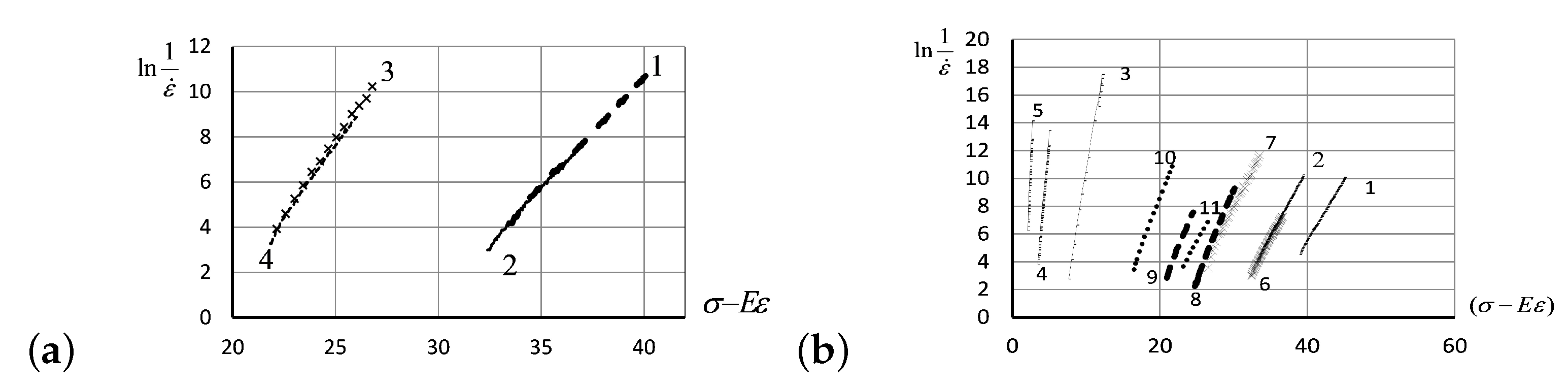
3.1. Experimental Verification of the Entropic Nature of Viscosity Anomaly
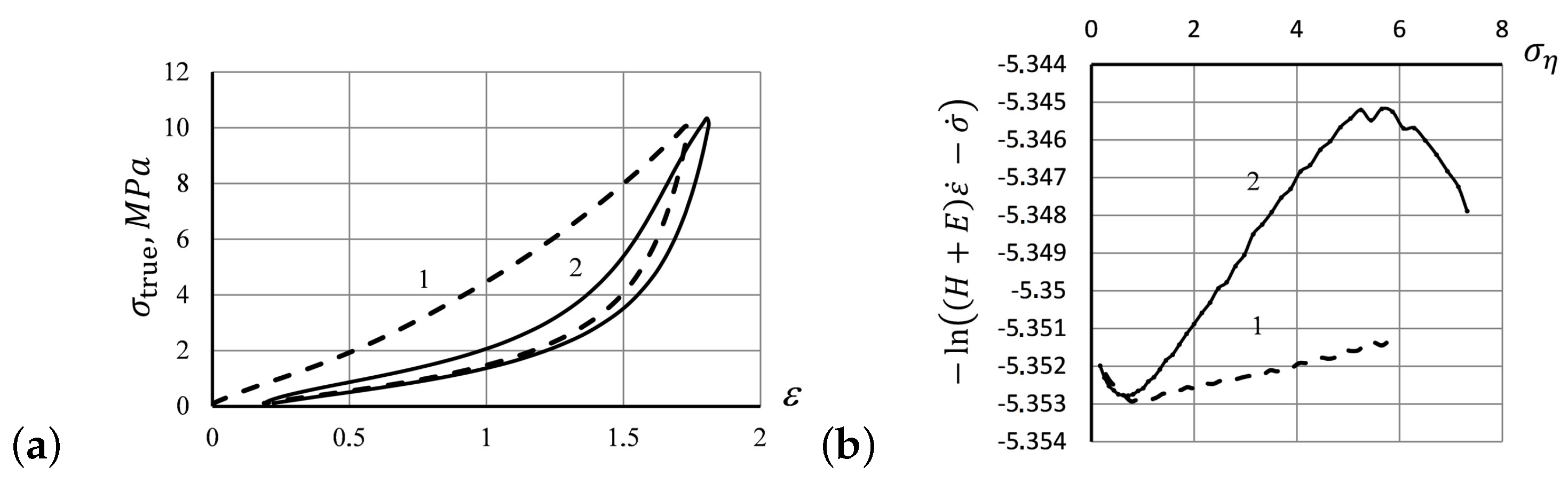
3.2. Experimental Verification of the Effect of Elastic Entropic Forces on Elastomer Tensile Curves
3.3. Experimental Study of Creeping Behavior in Silicon Rubber
4. Discussion
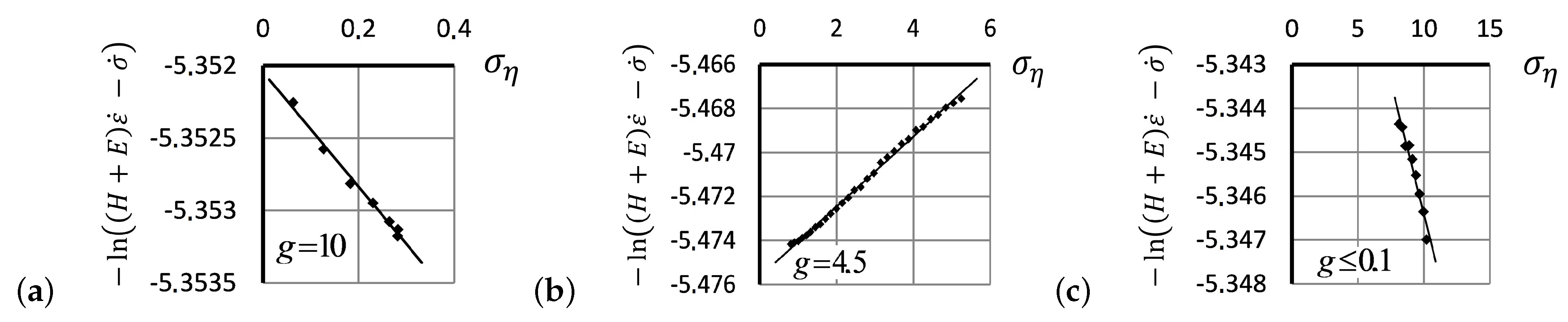
- The experimentally recorded values of the Flory correction coefficient g depend on neither temperature nor stretching rate. We, therefore, assume that the value of g may characterize the tendency of polymers to maintain a stable structure in mechanical deformation.
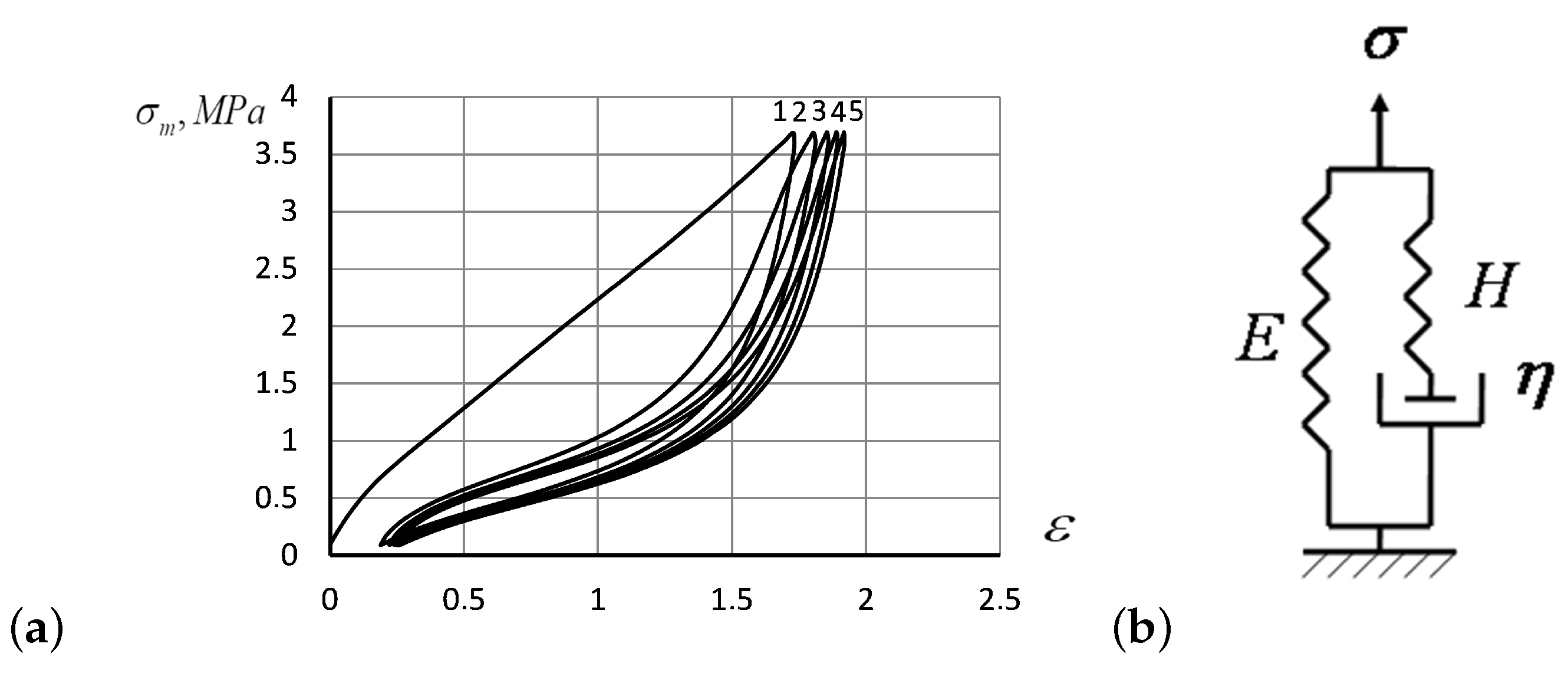
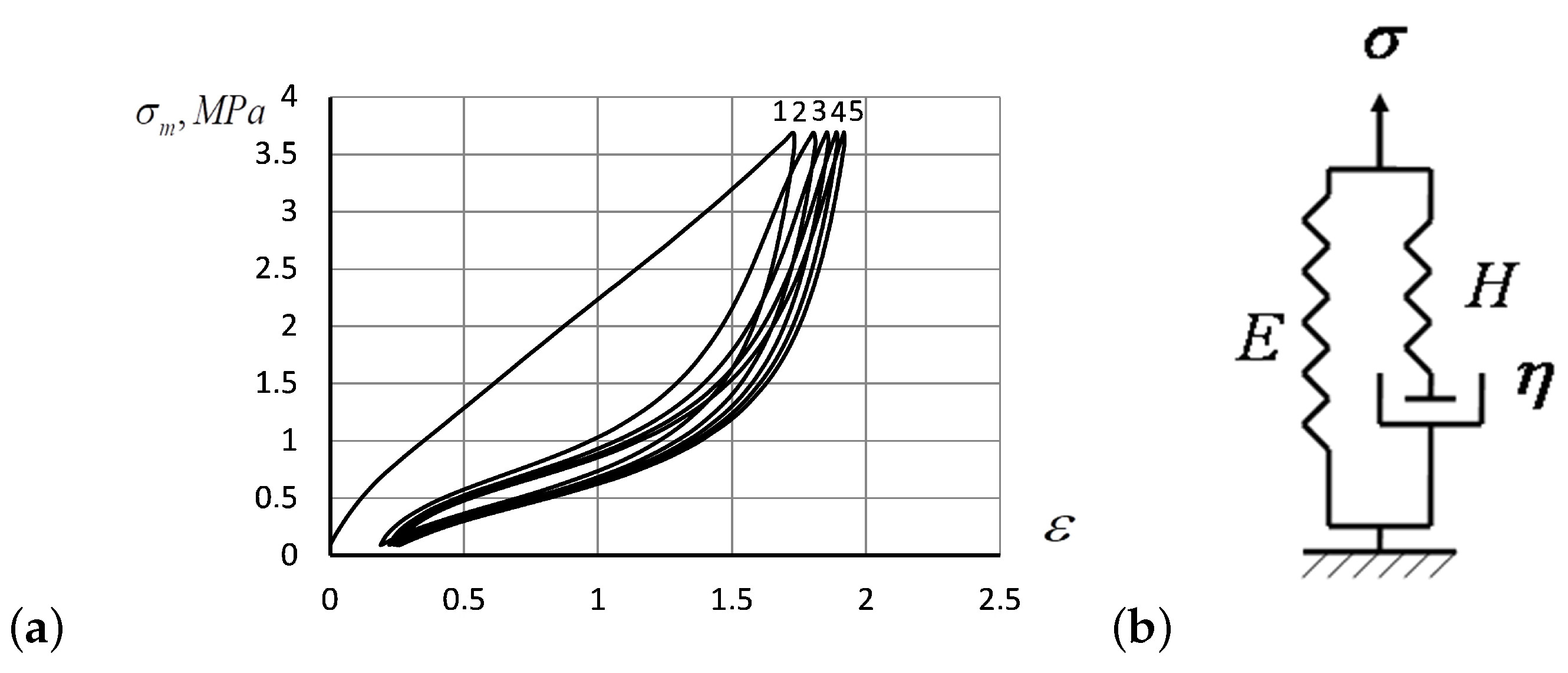
- Flory’s corrections measured for the repeated hysteresis loops were close to each other. Only the first hysteresis round seems to differ substantially from the others, as also predicted by the analytic solutions of the model Equations (18)–(20). The prominent distinction between the first stretching of the specimen and the subsequent rounds indicates an irreversible change that occurs in the polymer structure due to the rupture of weak structural constituents, after which the system acquires a more deformation resistible structure, as manifested by the Mullins effect. The specimen acquires a slight residual deformation, which changes a little during subsequent hysteresis cycles (see Figure 1a)
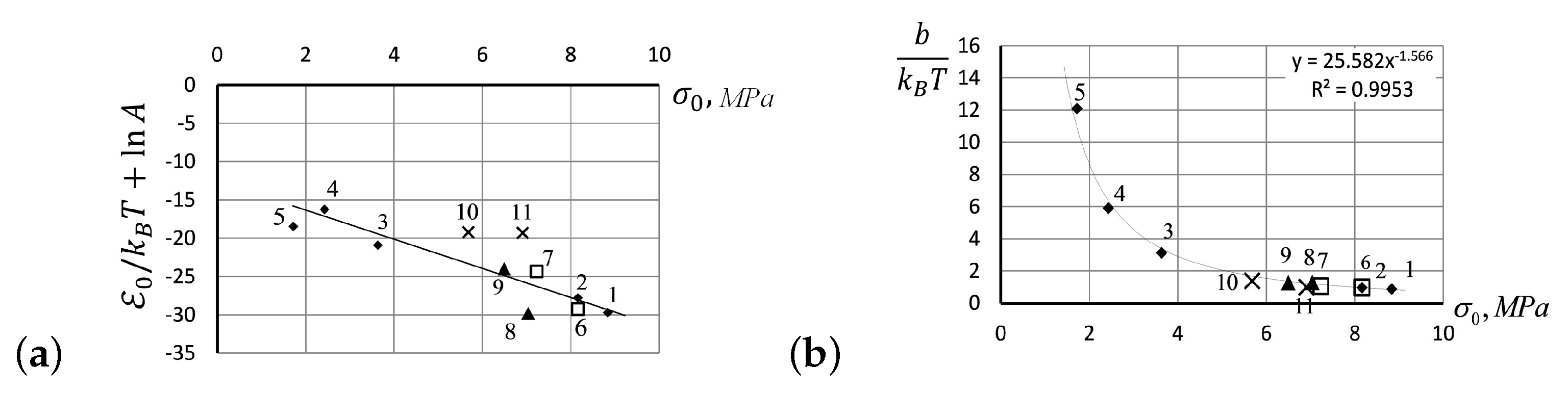
- Up to five stable segments can be identified visually on the experimental hysteresis curves. In particular, there are three regions of increasing deformation and two regions of reversible deformation. The measured values of Flory corrections exhibited sufficient reproducibility for all tested samples. For the tested PMVA specimen, the recorded Flory correction factor was 5–7 units.
- A quantitative description of elastomer deformation can be obtained, using the basic equations of the statistical theory of rubber elasticity and Eyring’s equation modified to take into account the entropic nature of deformation in polymers. For the stretching phase of rubber deformation, the elastic forces increase the activation energy, while they decrease during the retraction deformation.
- The small segments at the beginning and at the end of tensile curves (denoted as the initial and final segments of hysteresis cycles) show a stress growth slowdown, which may be associated with a decrease in the activation energy. There was a significant change in the values of Flory corrections in these segments at the same time. The final section of the return hysteresis curve has a particularly sharp increase in the Flory correction factor. This can be interpreted as a result of a strong increase in the number of physical cross-links at the final stage of the elastomer chain folding process.
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EEF | Elastic Entropic Force |
| IEP | Irreversible Entropy Production |
| KVDE | Kelvin–Voigt Differential Equation |
| KVM | Kelvin–Voigt Model |
| MFI | Melt Flow Index |
| MKU | Molecular Kinetic Units |
| PMVS | Polymethylvinylsiloxane (rubber) |
| RUL | Remaining Useful Life |
| SLS | Standard Linear Solid |
Appendix A. Entropic Force of Elasticity
References
- Frenkel, J. Über die Wärmebewegung in festen und fluessigen Köerpern. Zeits. Phys. 1926, 36, 652–669. [Google Scholar] [CrossRef]
- Eyring, H. Viscosity, Plasticity, and Diffusion as Examples of Absolute Reaction Rates. J. Chem. Phys. 1936, 4, 283–291. [Google Scholar] [CrossRef]
- Glasstone, S.; Laidler, K.J.; Eyring, H. The Theory of Rate Processes; Springer Science & Business Media: New York, NY, USA; London, UK, 1941. [Google Scholar]
- Lei, Q.; Hou, Y.; Lin, R. Correlation of viscosities of pure liquids in a wide temperature range. Fluid Phase Equilibr 1997, 140, 221–231. [Google Scholar]
- Kartsovnik, V.I. Changes of Activation Energy during Deformation of Rubber. J. Macromol. Sci. Part B Phys. 2011, 5, 75–88. [Google Scholar] [CrossRef]
- He, M.; Zhu, C.; Liu, X. Estimating the viscosity of ionic liquid at high pressure using Eyring’s absolute rate theory. Fluid Phase Equilibria 2018, 458, 170–176. [Google Scholar] [CrossRef]
- Kartsovnik, V.I. Prediction of the Creep of Elastomers Taking into Account the Forces of Entropic Elasticity of Macromolecules (prediction of Creep of Elastomers). J. Macromol. Sci. Part B Phys. 2018, 57, 447–464. [Google Scholar] [CrossRef]
- Honeycombe, R.W.K. Plastic Deformation of Metals; Edward Arnold: London, UK, 1968. [Google Scholar]
- Vinogradow, G.W.; Malkin, A.Y. The Rheology of Polymers; Chemistry: Moscow, Russia, 1977. (In Russian) [Google Scholar]
- Bauchy, M.; Guillot, B.; Micoulaut, M.; Sator, N. Viscosity and viscosity anomalies of model silicates and magmas: A numerical investigation. Chem. Geol. 2013, 346, 47–56. [Google Scholar] [CrossRef]
- Rudin, A. The Elements of Polymer Science and Engineering; Academic Press: Cambridge, MA, USA, 1982; pp. 209–211. [Google Scholar]
- Shende, T.; Niasar, V.J.; Babaei, M. An empirical equation for shear viscosity of shear thickening fluids. J. Mol. Liq. 2021, 325, 115220. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, J.; Zeng, H. Chapter 8—Polymer Processing and Rheology. In Polymer Science and Nanotechnology; Narain, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 149–178. [Google Scholar]
- Kartsovnik, V.I.; Worlitsch, R.; Hermann, F.; Tsoglin, Y. Calculation of the Viscosity of Polymer Melts Based on Measurements of the Recovered Rubber-like Deformation. J. Macromol. Sci. Part B Phys. 2016, 55, 149–157. [Google Scholar] [CrossRef]
- Kartsovnik, V.I.; Pelekh, V.V. On the mechanism of the flow of polymers. arXiv 2007, arXiv:0707.0789. [Google Scholar]
- Kröger, M. NEMD Computer Simulation of Polymer Melt Rheology. Appl. Rheol. 1995, 5, 66–71. [Google Scholar] [CrossRef]
- Wagner, M.H. The effect of dynamic tube dilation on chain stretch in nonlinear polymer melt rheology. J. Non-Newton. Fluid Mech. 2011, 166, 915–924. [Google Scholar] [CrossRef]
- Tsouka, S.; Dimakopoulos, Y.; Mavrantzas, V.G.; Tsamopoulos, J. Stress-gradient induced migration of polymers in corrugated channels. J. Rheol. 2014, 58, 911–947. [Google Scholar] [CrossRef]
- Treloar, L.R.G. The Physics of Rubber Elasticity; Clarendon Press: Oxford, UK, 1975. [Google Scholar]
- Buche, M.R.; Silberstein, M.N. Statistical mechanical constitutive theory of polymer networks: The inextricable links between distribution, behavior, and ensemble. Phys. Rev. E 2020, 102, 012501. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, G.V.; Malkin, A.Y.; Shumsky, V.F. High Elasticity, Normal and Shear Stresses on Shear Deformation of Low-molecular-weight Polyisobutylene. Rheol. Acta 1970, 9, 155–163. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R. Polymer Physics; OUP Oxford: Oxford, UK, 2003. [Google Scholar]
- Graessley, W.W. Polymeric Liquids and Networks: Structure and Properties; Taylor and Francis: Abingdon, UK, 2004. [Google Scholar]
- Morozinis, A.K.; Tzoumanekas, C.; Anogiannakis, S.D.; Theodorou, D.N. Atomistic simulations of cavitation in a model polyethylene network. Polym. Sci. Ser. C 2013, 55, 212–218. [Google Scholar] [CrossRef]
- Tobolsky, A.V. Properties and Structure of Polymers; John Wiley & Sons: New York, NY, USA; London, UK, 1960. [Google Scholar]
- Malkin, A.Y. High Elasticity and Viscoelasticity of Melts and Solutions of Polymers on Shear Flow. Mekh. Polim. 1975, 1, 173–187. (In Russian) [Google Scholar]
- Heinrich, G.; Straub, E.; Helmis, G. Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 1988; Volume 88, pp. 33–87. [Google Scholar]
- Hanson, D.E.; Martin, R.L. Quantum chemistry and molecular dynamics studies of the entropic elasticity of localized molecular kinks in polyisoprene chains. J. Chem. Phys. 2010, 133, 084903. [Google Scholar] [CrossRef] [PubMed]
- Hanson, D.E.; Martin, R.L. How far can a rubber molecule stretch before breaking? ab initio study of tensile elasticity and failure in single-molecule polyisoprene and polybutadiene. J. Chem. Phys. 2009, 130, 064903. [Google Scholar] [CrossRef]
- Kartsovnik, V.I. Relationship between the Deformation Processes Occurring in Rubbers and Their Molecular Structure. Structure of Rubbers under Strain. J. Macromol. Sci. Part B Phys. 2021, 61, 324–343. [Google Scholar] [CrossRef]
- Meyer, H.K.; von Susich, G. Die elastischen Eigenschaften der organischen Hochpolymeren und ihre kinetische Deutung. Kolloid-Zeitschrift 1932, 59, 208–216. [Google Scholar] [CrossRef]
- Kuhn, W. Beziehungen zwischen Molekülgröße, statistischer Molekülgestalt und elastischen Eigenschaften hochpolymerer Stoffe. Kolloid-Zeitschrift 1936, 76, 258–271. [Google Scholar] [CrossRef]
- Guth, E.; Mark, H. Zur innermolekularen, Statistik, insbesondere bei Kettenmolekülen. Monatshefte für Chemie und verwandte Teile anderer Wissenschaften 1934, 65, 93–121. [Google Scholar] [CrossRef]
- James, H.M.; Guth, E. Theory of the Elastic Properties of Rubber. J. Chem. Phys. 1943, 11, 455. [Google Scholar] [CrossRef]
- Eyring, H. The Resultant Electric Moment of Complex Molecules. Phys. Rev. 1932, 39, 746. [Google Scholar] [CrossRef]
- Wall, F.T. Statistical Thermodynamics of Rubber I. Rubb. Chem. Technol. 1942, 15, 468–472. [Google Scholar] [CrossRef]
- Wall, F.T. Statistical Thermodynamics of Rubber II. J. Chem. Phys. 1942, 10, 485–488. [Google Scholar] [CrossRef]
- Treloar, L.R.G. The elasticity of a network of long-chain molecules. I. Trans. Faraday Soc. 1943, 39, 36–41. [Google Scholar] [CrossRef]
- Treloar, L.R.G. The elasticity of a network of long-chain molecules. II. Rubb. Chem. Technol. 1944, 17, 296–302. [Google Scholar] [CrossRef]
- Flory, P.J.; Rehner, J.J. Statistical Mechanics of Cross-Linked Polymer Networks I. Rubber like Elasticity. J. Chem. Phys. 1943, 11, 512. [Google Scholar] [CrossRef]
- Flory, P.L. Principles of Polymer Chemistry; Ithaca: New York, NY, USA, 1953. [Google Scholar]
- Fleer, G.J.; Cohen Stuart, M.A.; Scheutjens, J.M.H.M.; Cosgrove, T.; Vincent, B. Polymers at Intefaces; Chapman & Hill: Cambridge, UK, 1993. [Google Scholar]
- Askadskii, A.A. The Deformation of Polymers; Khimya: Moskow, Russia, 1973. [Google Scholar]
- Diani, J.; Fayolle, B.; Gilormini, P. A Review on the Mullins Effect. Eur. Polym. J. 2009, 45, 601–612. [Google Scholar] [CrossRef]
- Golberg, I.I. Mechanical Behavior of Polymers (the Mathematical Description); Khimiya (Chemistry): Moscow, Russia, 1970. [Google Scholar]
- Parisi, D.; Coppola, S.; Righi, S.; Gagliardi, G.; Grasso, F.S.; Bacchelli, F. Alternative Use of the Sentmanat Extensional Rheometer to Investigate the Rheological Behavior of Industrial Rubbers at very Large Deformations. Rubber Chem. Technol. 2022, 95, 241–276. [Google Scholar] [CrossRef]
- Landi, G.T.; Paternostro, M. Irreversible entropy production: From classical to quantum. Rev. Mod. Phys. 2021, 93, 035008. [Google Scholar] [CrossRef]
- Kostina, A.; Plekhov, O. The Entropy of an Armco Iron under Irreversible Deformation. Entropy 2015, 17, 264–276. [Google Scholar] [CrossRef]
- Goold, J.M.; Huber, A.; Riera, L.d.R.; Skrzypczyk, P. The role of quantum information in thermodynamics—A topical review. J. Phys. Math. Theor. 2016, 49, 143001. [Google Scholar] [CrossRef]
- Vinjanampathy, S.; Anders, J. Quantum thermodynamics. Contemp. Phys. 2016, 57, 545. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hysteresis Cycle | Ascending Strain Interval | g | Determination, | Tensile Slope, |
|---|---|---|---|---|
| 1 | [1.8–20%] | 24 | ||
| 1 | [21–170%] | |||
| 2 | [21–56%] | 10 | ||
| 2 | [65–164%] | |||
| 2 | [166–179%] |
| Hysteresis Cycle | Ascending Strain Interval | g | Determination, | Tensile Slope, |
|---|---|---|---|---|
| 1 | [173–103%] | 7 | ||
| 1 | [65–25%] | 70 | ||
| 2 | [178–81%] | |||
| 2 | [70–29%] | 81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kartsovnik, V.I.; Volchenkov, D. Elastic Entropic Forces in Polymer Deformation. Entropy 2022, 24, 1260. https://doi.org/10.3390/e24091260
Kartsovnik VI, Volchenkov D. Elastic Entropic Forces in Polymer Deformation. Entropy. 2022; 24(9):1260. https://doi.org/10.3390/e24091260
Chicago/Turabian StyleKartsovnik, Vladimir I., and Dimitri Volchenkov. 2022. "Elastic Entropic Forces in Polymer Deformation" Entropy 24, no. 9: 1260. https://doi.org/10.3390/e24091260
APA StyleKartsovnik, V. I., & Volchenkov, D. (2022). Elastic Entropic Forces in Polymer Deformation. Entropy, 24(9), 1260. https://doi.org/10.3390/e24091260