
Application of Coarse-Grained (CG) Models to Explore Conformational Pathway of Large-Scale Protein Machines



Abstract
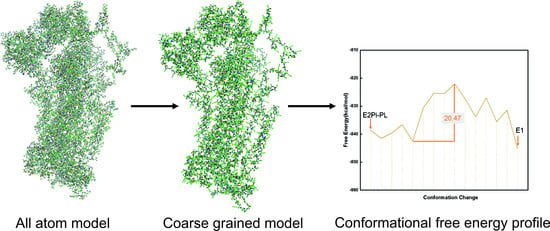
:
1. Introduction
2. Methods
2.1. Coarse-Grained (CG) Model
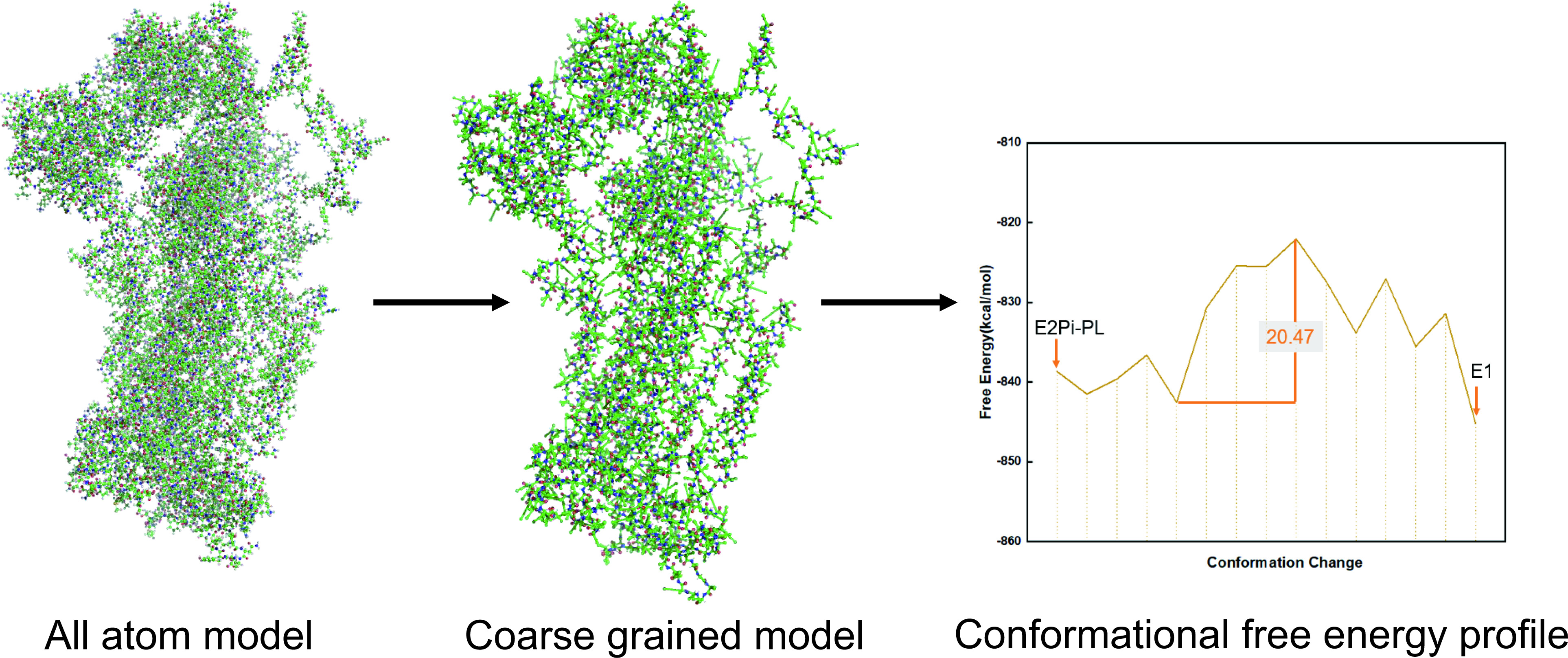
2.2. Workflow Integrated with Coarse-Grained (CG) Model
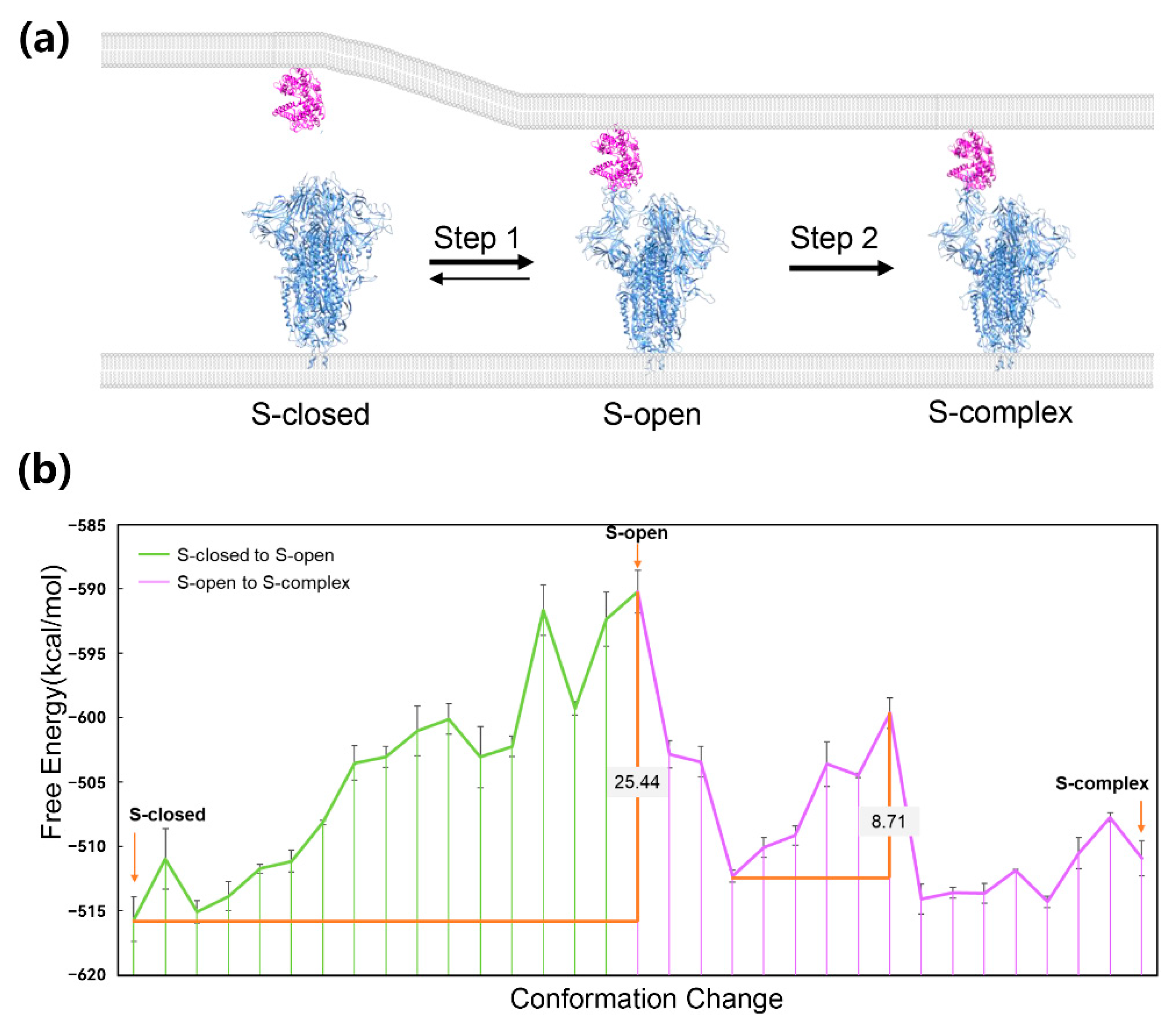
3. Application to Protein–Protein Interaction System
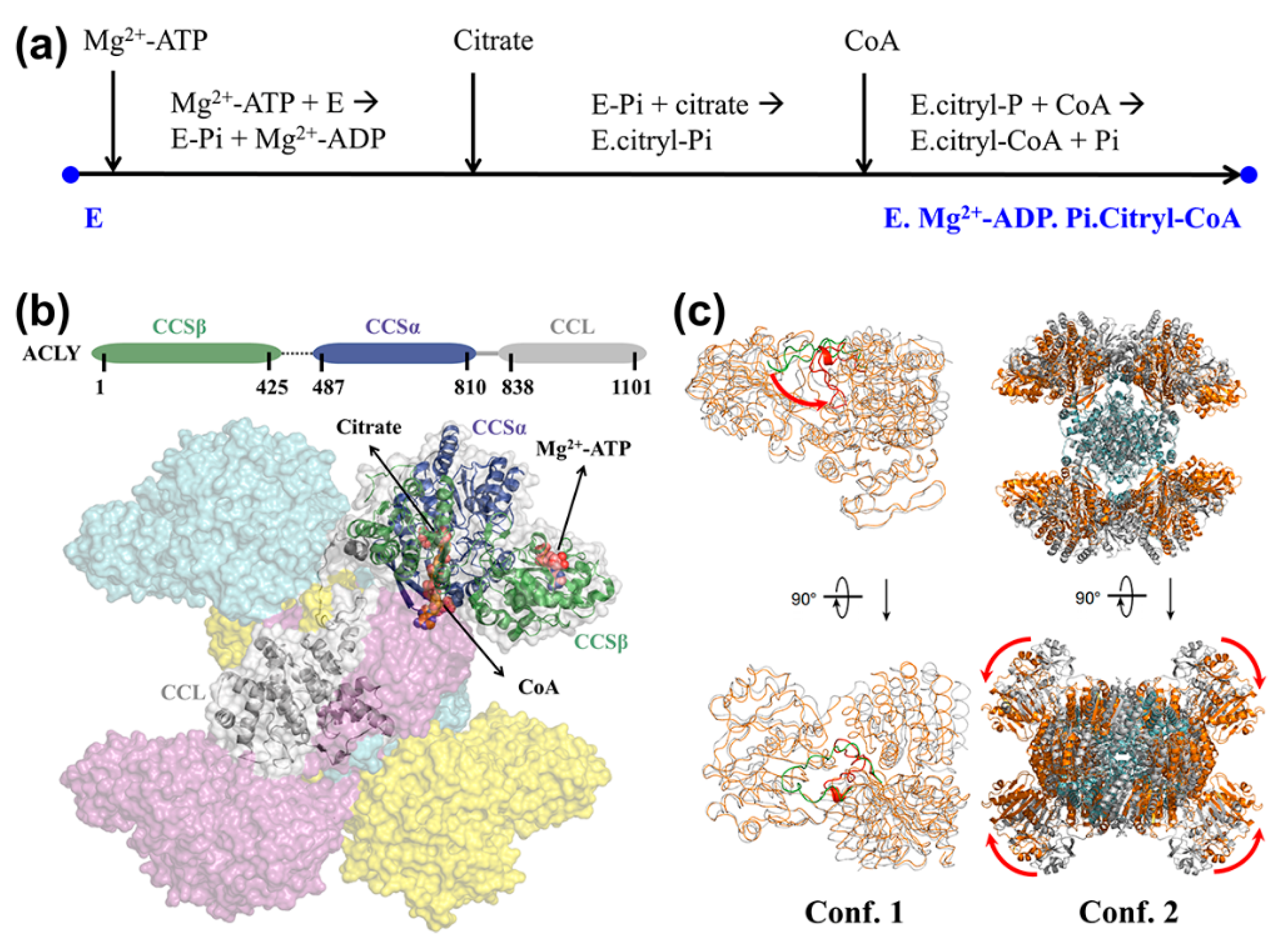
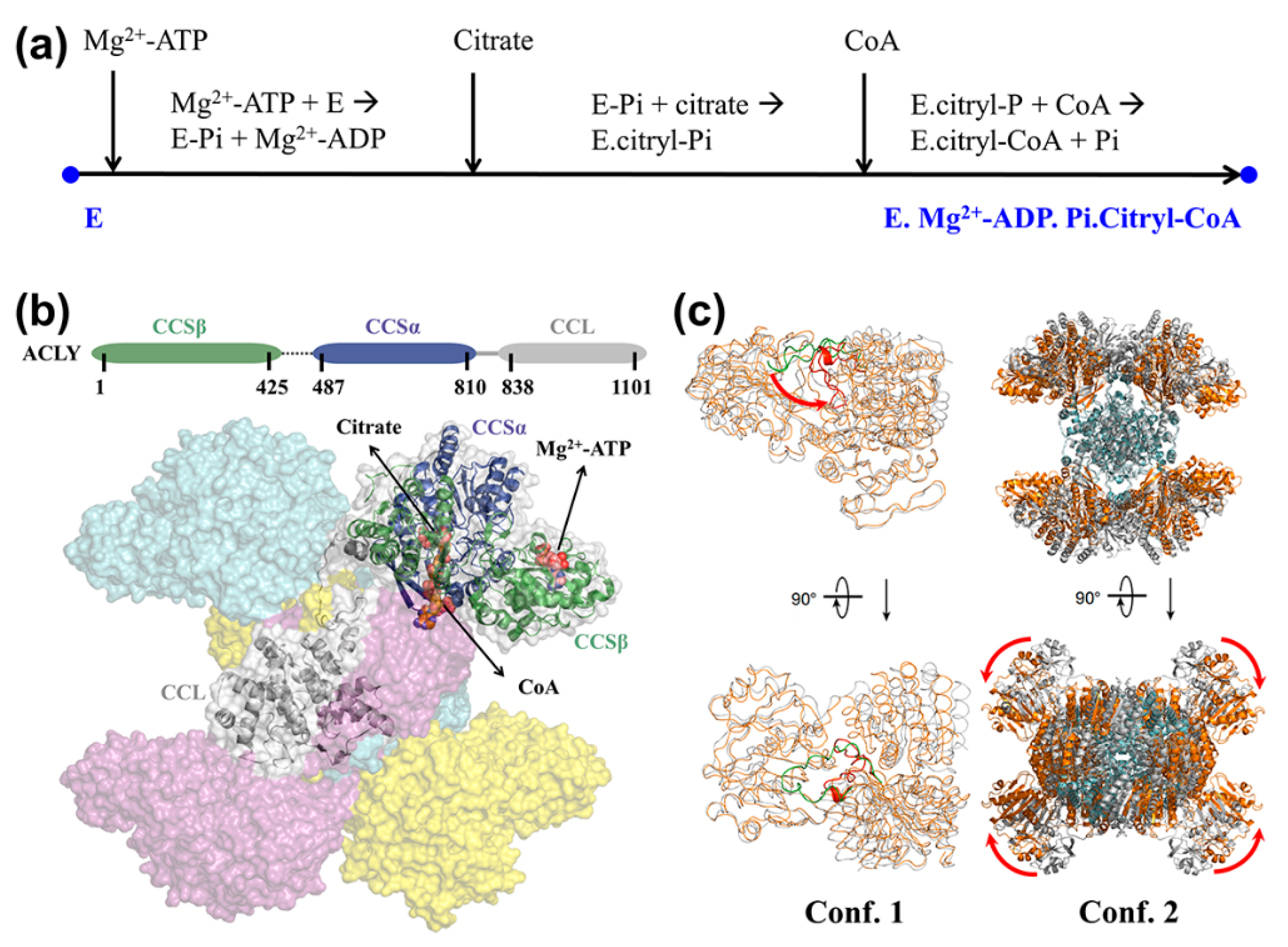
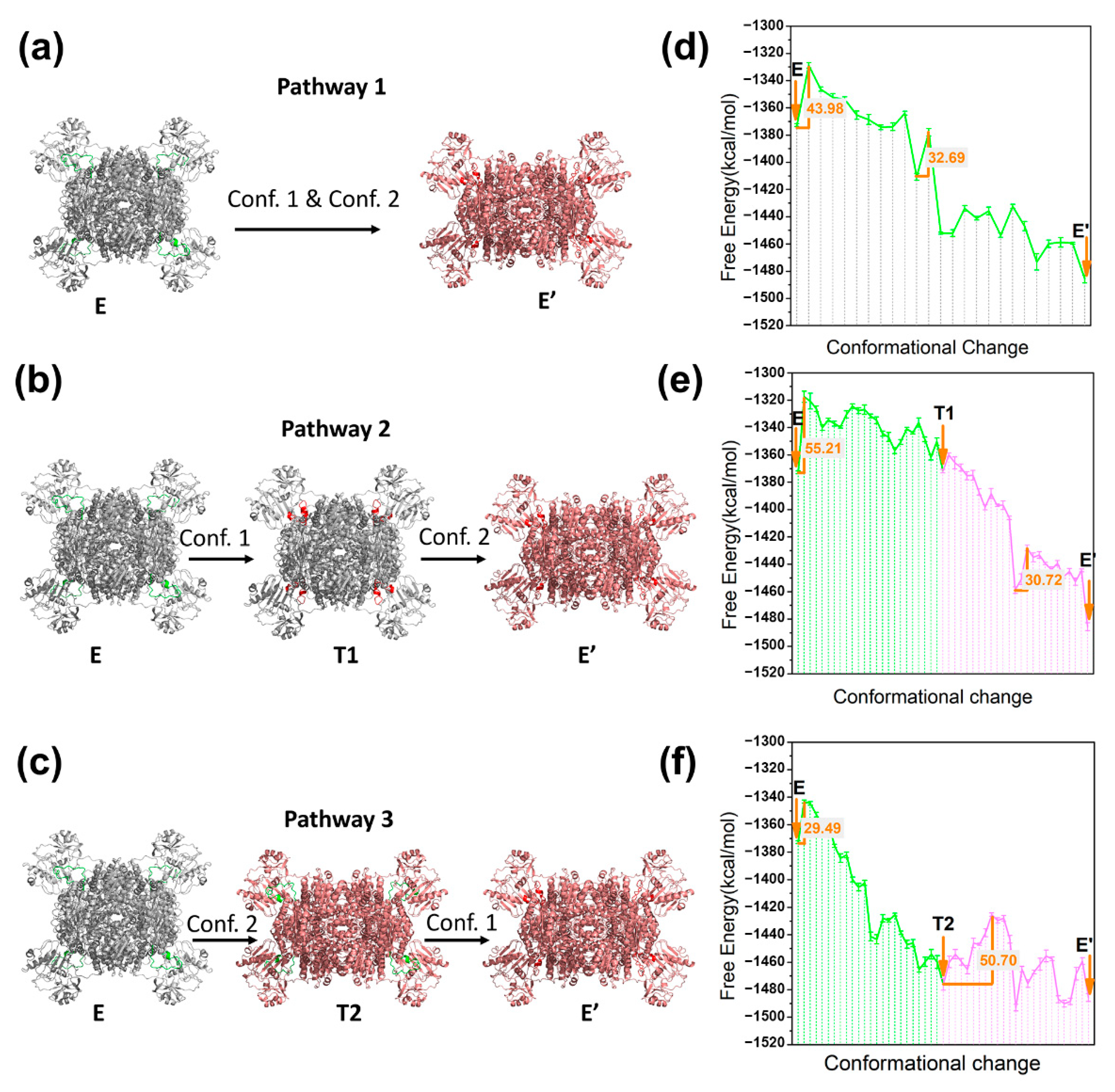
4. Application to Enzyme System
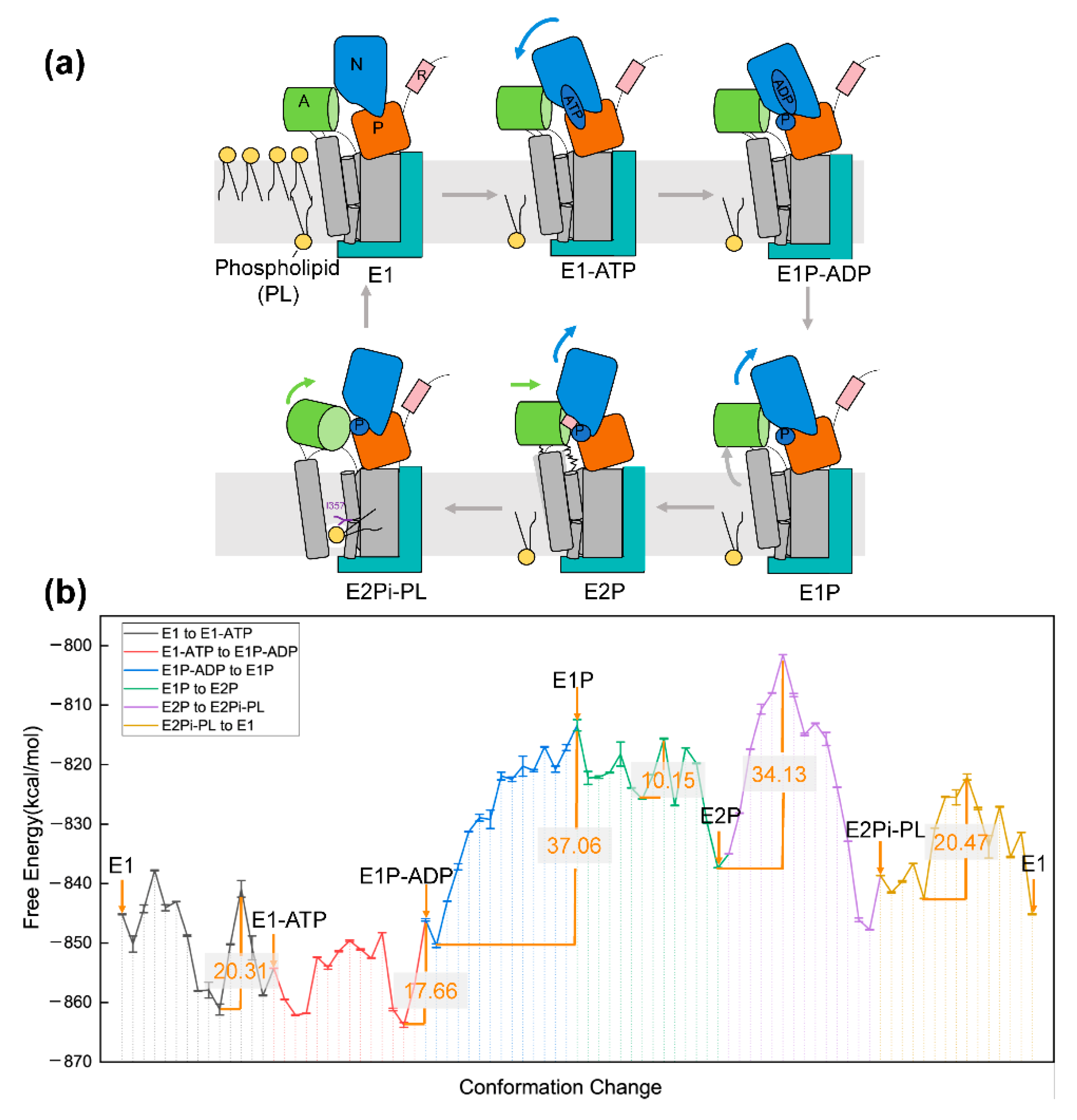
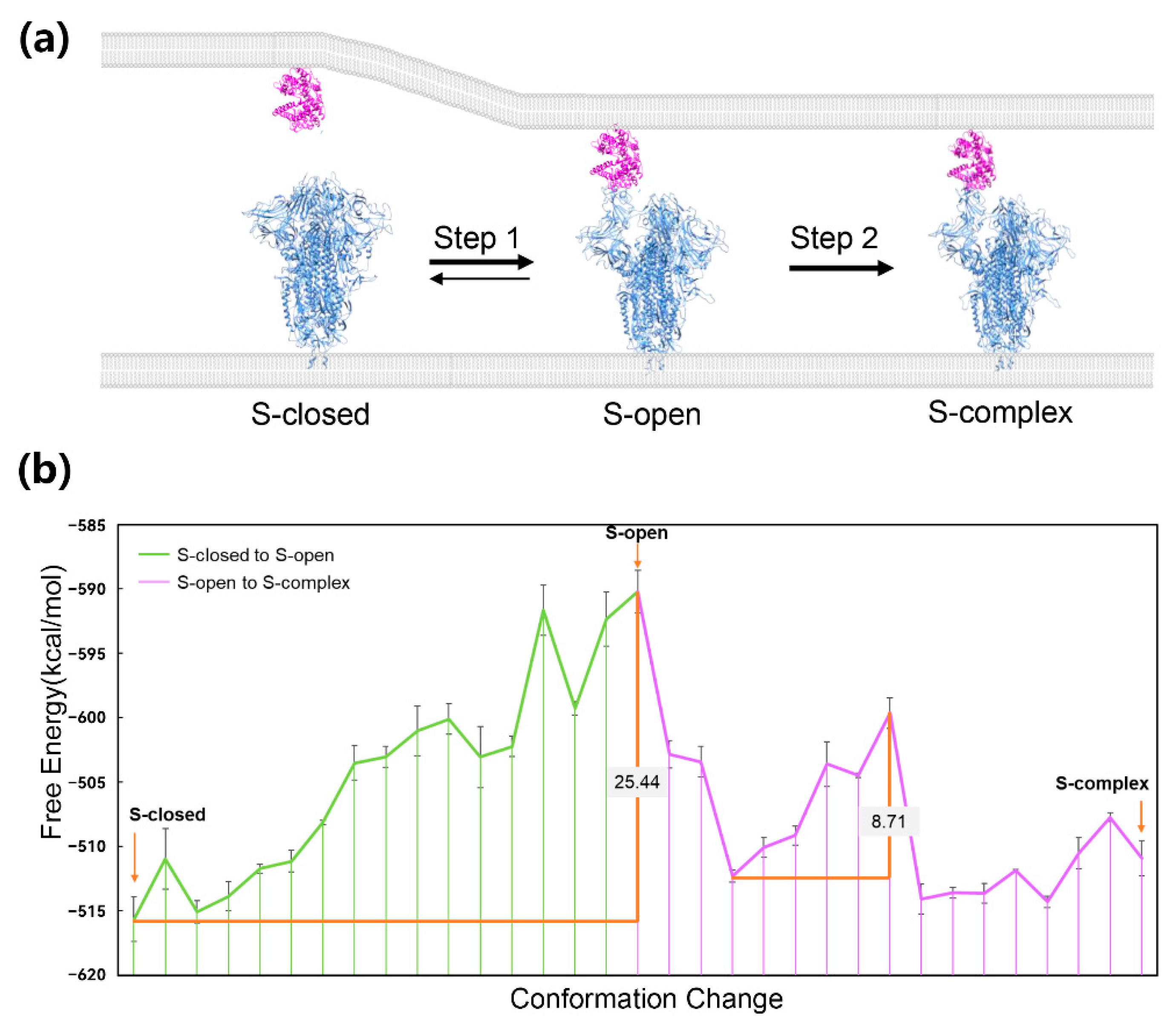
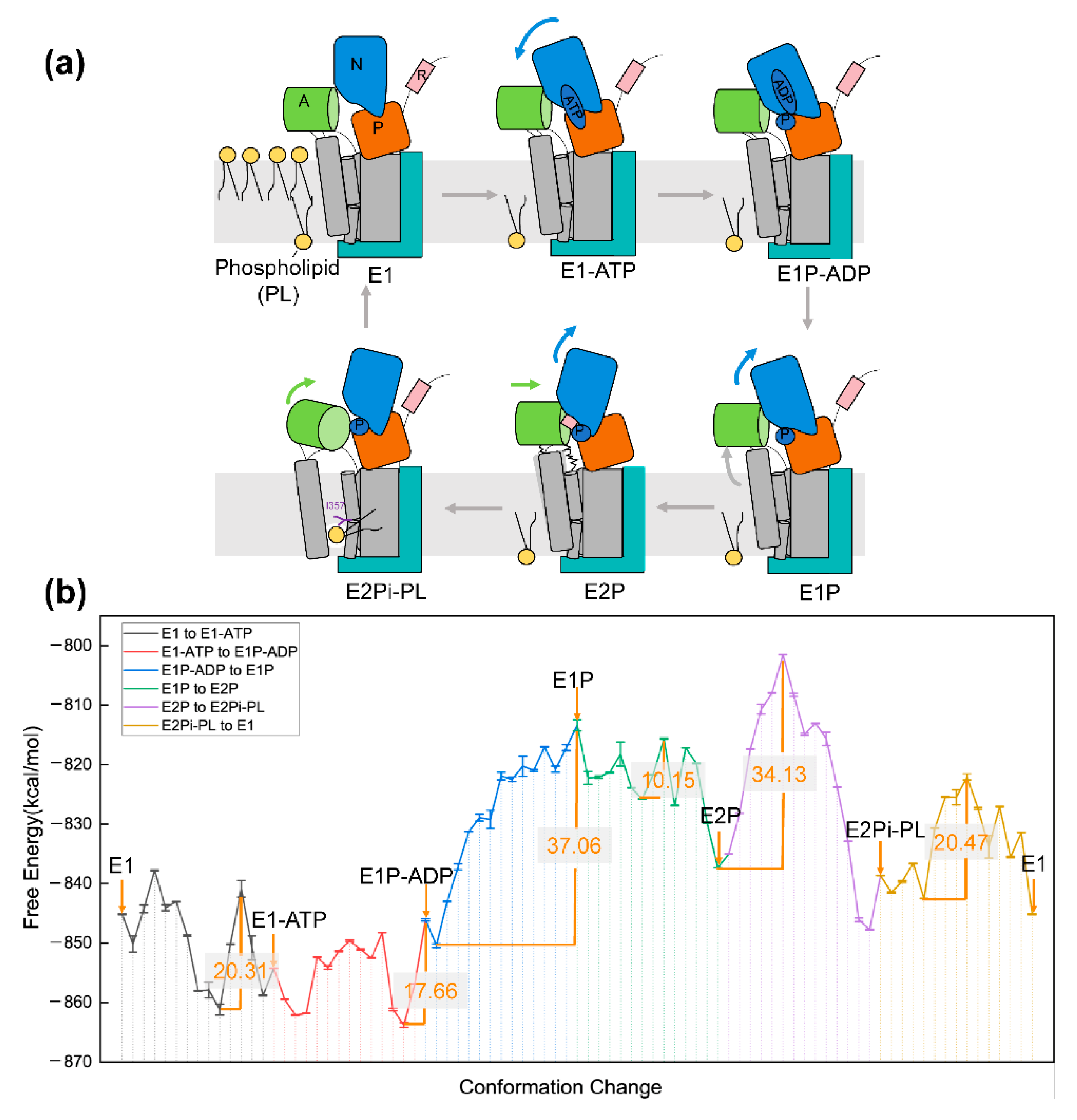
5. Application to Transmembrane Protein System
6. Conclusions and Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Piccolino, M. Biological machines: From mills to molecules. Nat. Rev. Mol. Cell Biol. 2000, 1, 149–152. [Google Scholar] [CrossRef]
- Iino, R.; Kinbara, K.; Bryant, Z. Introduction: Molecular motors. Chem. Rev. 2020, 120, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.S.; Kooistra, A.J.; Munk, C.; Heydenreich, F.M.; Veprintsev, D.B.; Bouvier, M.; Babu, M.M.; Gloriam, D.E. GPCR activation mechanisms across classes and macro/microscales. Nat. Struct. Mol. Biol. 2021, 28, 879–888. [Google Scholar] [CrossRef]
- Hauer, B. Embracing nature’s catalysts: A viewpoint on the future of biocatalysis. ACS Catal. 2020, 10, 8418–8427. [Google Scholar] [CrossRef]
- Micheletti, C.; Banavar, J.R.; Maritan, A. Conformations of proteins in equilibrium. Phys. Rev. Lett. 2001, 87, 088102. [Google Scholar] [CrossRef] [Green Version]
- Helliwell, J.R. Combining X-rays, neutrons and electrons, and NMR, for precision and accuracy in structure–function studies. Acta Crystallogr. A 2021, 77, 173–185. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-grained protein models and their applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [Green Version]
- De Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schafer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef]
- Davtyan, A.; Schafer, N.P.; Zheng, W.; Clementi, C.; Wolynes, P.G.; Papoian, G.A. AWSEM-MD: Protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J. Phys. Chem. B 2012, 116, 8494–8503. [Google Scholar] [CrossRef] [Green Version]
- Liwo, A.; Baranowski, M.; Czaplewski, C.; Golas, E.; He, Y.; Jagiela, D.; Krupa, P.; Maciejczyk, M.; Makowski, M.; Mozolewska, M.A.; et al. A unified coarse-grained model of biological macromolecules based on mean-field multipole-multipole interactions. J. Mol. Model. 2014, 20, 2306. [Google Scholar] [CrossRef] [Green Version]
- Kolinski, A. Protein modeling and structure prediction with a reduced representation. Acta Biochim. Polym. 2004, 51, 349–371. [Google Scholar] [CrossRef] [Green Version]
- Messer, B.M.; Roca, M.; Chu, Z.T.; Vicatos, S.; Kilshtain, A.V.; Warshel, A. Multiscale simulations of protein landscapes: Using coarse-grained models as reference potentials to full explicit models. Proteins 2010, 78, 1212–1227. [Google Scholar] [CrossRef] [Green Version]
- Levitt, M.; Warshel, A. Computer simulation of protein folding. Nature 1975, 253, 694–698. [Google Scholar] [CrossRef]
- Vicatos, S.; Rychkova, A.; Mukherjee, S.; Warshel, A. An Effective Coarse-Grained Model for Biological Simulations: Recent Refinements and Validations. Proteins 2014, 82, 1168–1185. [Google Scholar] [CrossRef]
- Vorobyov, I.; Kim, I.; Chu, Z.T.; Warshel, A. Refining the treatment of membrane proteins by coarse-grained models. Proteins 2016, 84, 92–117. [Google Scholar] [CrossRef] [Green Version]
- Schopf, P.; Warshel, A. Validating Computer Simulations of Enantioselective Catalysis; Reproducing the Large Steric and Entropic Contributions in Candida Antarctica Lipase B. Proteins 2014, 82, 1387–1399. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Bora, R.P.; Warshel, A. Torque, chemistry and efficiency in molecular motors: A study of the rotary-chemical coupling in F1-ATPase. Q. Rev. Biophys. 2015, 48, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Warshel, A. Brønsted slopes based on single-molecule imaging data help to unveil the chemically coupled rotation in F1-ATPase. Proc. Natl. Acad. Sci. USA 2015, 112, 14121–14122. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Warshel, A. Dissecting the role of the γ-subunit in the rotary-chemical coupling and torque generation of F1-ATPase. Proc. Natl. Acad. Sci. USA 2015, 112, 2746–2751. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Warshel, A. Electrostatic Origin of the Mechanochemical Rotary Mechanism and the Catalytic Dwell of F1-Atpase. Proc. Natl. Acad. Sci. USA 2011, 108, 20550. [Google Scholar] [CrossRef] [Green Version]
- Bai, C.; Warshel, A. Revisiting the protonmotive vectorial motion of F0-ATPase. Proc. Natl. Acad. Sci. USA 2019, 116, 19484–19489. [Google Scholar] [CrossRef] [Green Version]
- Bai, C.; Asadi, M.; Warshel, A. The catalytic dwell in ATPases is not crucial for movement against applied torque. Nat. Chem. 2020, 12, 1187–1192. [Google Scholar] [CrossRef]
- Mukherjee, S.; Warshe, A. Realistic Simulations of the Coupling between the Protomotive Force and the Mechanical Rotation of the F-0-Atpase. Proc. Natl. Acad. Sci. USA 2012, 109, 14876. [Google Scholar] [CrossRef] [Green Version]
- Rychkova, A.; Mukherjee, S.; Bora, R.P.; Warshel, A. Simulating the Pulling of Stalled Elongated Peptide from the Ribosome by the Translocon. Proc. Natl. Acad. Sci. USA 2013, 110, 10195. [Google Scholar] [CrossRef] [Green Version]
- Adamczyk, A.J.; Warshel, A. Converting Structural Information into an Allosteric-Energy-Based Picture for Elongation Factor Tu Activation by the Ribosome. Proc. Natl. Acad. Sci. USA 2011, 108, 9827. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.K.; Xiang, Y.; Kato, M.; Warshel, A. What Are the Roles of Substrate-Assisted Catalysis and Proximity Effects in Peptide Bond Formation by the Ribosome? Biochemistry 2005, 44, 11307. [Google Scholar] [CrossRef]
- Alhadeff, R.; Warshel, A. A free-energy landscape for the glucagon-like peptide 1 receptor GLP1R. Proteins 2020, 88, 127–134. [Google Scholar] [CrossRef]
- Bai, C.; Wang, J.; Mondal, D.; Du, Y.; Ye, R.D.; Warshel, A. Exploring the activation process of the β2AR-Gs complex. J. Am. Chem. Soc. 2021, 143, 11044–11051. [Google Scholar] [CrossRef]
- Alhadeff, R.; Vorobyov, I.; Yoon, H.W.; Warshel, A. Exploring the free-energy landscape of GPCR activation. Proc. Natl. Acad. Sci. USA 2018, 115, 10327–10332. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.S.; Chu, Z.T.; Warshel, A. Microscopic and semimicroscopic calculations of electrostatic energies in proteins by the POLARIS and ENZYMIX programs. J. Comput. Chem. 1993, 14, 161–185. [Google Scholar] [CrossRef]
- Beroza, P.; Fredkin, D.R.; Okamura, M.Y.; Feher, G. Protonation of interacting residues in a protein by a Monte Carlo method: Application to lysozyme and the photosynthetic reaction center of Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA 1991, 88, 5804–5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dryga, A.; Chakrabarty, S.; Vicatos, S.; Warshel, A. Coarse grained model for exploring voltage dependent ion channels. Biochim. Biophys. Acta 2012, 1818, 303–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.Z.; Hwang, J.K.; Warshel, A. Using simplified protein representation as a reference potential for all-atom calculations of folding free energy. Theor. Chem. Acc. 1999, 103, 77–80. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [Green Version]
- Schlitter, J.; Engels, M.; Krüger, P. Targeted molecular dynamics: A new approach for searching pathways of conformational transitions. J. Mol. Graph. 1994, 12, 84–89. [Google Scholar] [CrossRef]
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, M.I.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-COV: A comparative overview. Infez. Med. 2020, 28, 174–184. [Google Scholar]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef]
- Fallon, L.; Belfon, K.A.A.; Raguette, L.; Wang, Y.; Stepanenko, D.; Cuomo, A.; Guerra, J.; Budhan, S.; Varghese, S.; Corbo, C.P.; et al. Free Energy Landscapes from SARS-CoV-2 Spike Glycoprotein Simulations Suggest that RBD Opening Can Be Modulated via Interactions in an Allosteric Pocket. J. Am. Chem. Soc. 2021, 143, 11349–11360. [Google Scholar] [CrossRef]
- Wu, Y.; Qian, R.; Yang, Y.; Sheng, Y.; Li, W.; Wang, W. Activation Pathways and Free Energy Landscapes of the SARS-CoV-2 Spike Protein. ACS Omega 2021, 6, 23432–23441. [Google Scholar] [CrossRef]
- Ray, D.; Le, L.; Andricioaei, I. Distant residues modulate conformational opening in SARS-CoV-2 spike protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2100943118. [Google Scholar] [CrossRef]
- Govind Kumar, V.; Ogden, D.S.; Isu, U.H.; Polasa, A.; Losey, J.; Moradi, M. Prefusion spike protein conformational changes are slower in SARS-CoV-2 than in SARS-CoV-1. J. Biol. Chem. 2022, 298, 101814. [Google Scholar] [CrossRef]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-Time Conformational Dynamics of SARS-CoV-2 Spikes on Virus Particles. Cell Host Microbe 2020, 28, 880–891.e8. [Google Scholar] [CrossRef]
- Sztain, T.; Ahn, S.H.; Bogetti, A.T.; Casalino, L.; Goldsmith, J.A.; Seitz, E.; Mccool, R.S.; Kearns, F.L.; Acosta-Reyes, F.; Maji, S.; et al. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat. Chem. 2021, 13, 963–968. [Google Scholar] [CrossRef]
- Bai, C.; Warshel, A. Critical Differences between the Binding Features of the Spike Proteins of SARS-CoV-2 and SARS-CoV. J. Phys. Chem. B 2020, 124, 5907–5912. [Google Scholar] [CrossRef]
- Bai, C.; Wang, J.; Chen, G.; Zhang, H.; An, K.; Xu, P.; Du, Y.; Ye, R.D.; Saha, A.; Zhang, A.; et al. Predicting Mutational Effects on Receptor Binding of the Spike Protein of SARS-CoV-2 Variants. J. Am. Chem. Soc. 2021, 143, 17646–17654. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; Mcguire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; Mclellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Mayer, M.P.; Schröder, H.; Rüdiger, S.; Paal, K.; Laufen, T.; Bukau, B. Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat. Struct. Biol. 2000, 7, 586–593. [Google Scholar]
- Callender, R.; Dyer, R.B. The dynamical nature of enzymatic catalysis. Acc. Chem. Res. 2015, 48, 407–413. [Google Scholar] [CrossRef]
- Granchi, C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 2018, 157, 1276–1291. [Google Scholar] [CrossRef]
- Verschueren, H.G.; Blanchet, C.; Felix, J.; Dansercoer, A.; De Vos, D.; Bloch, Y.; Van Beeumen, J.; Svergun, D.; Gutsche, I.; Savvides, S.N.; et al. Structure of ATP citrate lyase and the origin of citrate synthase in the Krebs cycle. Nature 2019, 568, 571–575. [Google Scholar] [CrossRef]
- Bazilevsky, G.A.; Affronti, H.C.; Wei, X.; Campbell, S.L.; Wellen, K.E.; Marmorstein, R. ATP-citrate lyase multimerization is required for coenzyme-A substrate binding and catalysis. J. Biol. Chem. 2019, 294, 7259–7268. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Leit, S.; Kuai, J.; Therrien, E.; Rafi, S.; Harwood, H.J., Jr.; Delabarre, B.; Tong, L. An allosteric mechanism for potent inhibition of human ATP-citrate lyase. Nature 2019, 568, 566–570. [Google Scholar] [CrossRef]
- Fan, F.; Williams, H.J.; Boyer, J.G.; Graham, T.L.; Zhao, H.; Lehr, R.; Qi, H.; Schwartz, B.; Raushel, F.M.; Meek, T.D. On the catalytic mechanism of human ATP citrate lyase. Biochemistry 2012, 51, 5198–5211. [Google Scholar] [CrossRef]
- Mbaye, M.N.; Hou, Q.; Basu, S.; Teheux, F.; Pucci, F.; Rooman, M. A comprehensive computational study of amino acid interactions in membrane proteins. Sci. Rep. 2019, 9, 12043. [Google Scholar] [CrossRef] [Green Version]
- Giliberti, V.; Badioli, M.; Nucara, A.; Calvani, P.; Ritter, E.; Puskar, L.; Aziz, E.F.; Hegemann, P.; Schade, U.; Ortolani, M.; et al. Heterogeneity of the Transmembrane Protein Conformation in Purple Membranes Identified by Infrared Nanospectroscopy. Small 2017, 13, 1701181. [Google Scholar] [CrossRef]
- Timcenko, M.; Dieudonné, T.; Montigny, C.; Boesen, T.; Lyons, J.; Lenoir, G.; Nissen, P. Structural Basis of Substrate-Independent Phosphorylation in a P4-ATPase Lipid Flippase. J. Mol. Biol. 2021, 433, 167062. [Google Scholar] [CrossRef]
- Bevers, E.M.; Williamson, P.L. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef]
- Kato, U.; Inadome, H.; Yamamoto, M.; Emoto, K.; Kobayashi, T.; Umeda, M. Role for Phospholipid Flippase Complex of ATP8A1 and CDC50A Proteins in Cell Migration. J. Biol. Chem. 2013, 288, 4922–4934. [Google Scholar] [CrossRef] [Green Version]
- Leventis, P.A.; Grinstein, S. The Distribution and Function of Phosphatidylserine in Cellular Membranes. Annu. Rev. Biophys. 2010, 39, 407. [Google Scholar] [CrossRef]
- Emoto, K.; Kobayashi, T.; Yamaji, A.; Aizawa, H.; Yahara, I.; Inoue, K.; Umeda, M. Redistribution of phosphatidylethanolamine at the cleavage furrow of dividing cells during cytokinesis. Proc. Natl. Acad. Sci. USA 1996, 93, 12867. [Google Scholar] [CrossRef] [Green Version]
- Zwaal, R.F.A.; Schroit, A.J. Pathophysiologic Implications of Membrane Phospholipid Asymmetry in Blood Cells. Blood 1997, 89, 1121–1132. [Google Scholar] [CrossRef]
- Muthusamy, B.P.; Natarajan, P.; Zhou, X.; Graham, T.R. Linking phospholipid flippases to vesicle-mediated protein transport. Biochim. Biophys. Acta 2009, 1791, 612–619. [Google Scholar] [CrossRef] [Green Version]
- Eijnde, S.M.V.D.; Hoff, M.; Reutelingsperger, C.P.M.; Heerde, W.L.V.; Henfling, M.E.; Vermeij-Keers, C.; Schutte, B.; Borgers, M.; Ramaekers, F.C. Transient expression of phosphatidylserine at cell-cell contact areas is required for myotube formation. J. Cell Sci. 2001, 114, 3631–3642. [Google Scholar] [CrossRef]
- Baldridge, R.D.; Graham, T.R. Two-gate mechanism for phospholipid selection and transport by type IV P-type ATPases. Proc. Natl. Acad. Sci. USA 2013, 110, E358–E367. [Google Scholar] [CrossRef] [Green Version]
- Vestergaard, A.L.; Coleman, J.A.; Lemmin, T.; Mikkelsen, S.A.; Molday, L.L.; Vilsen, B.; Molday, R.S.; Dal Peraro, M.; Andersen, J.P. Critical roles of isoleucine-364 and adjacent residues in a hydrophobic gate control of phospholipid transport by the mammalian P4-ATPase ATP8A2. Proc. Natl. Acad. Sci. USA 2014, 111, E1334–E1343. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; You, Q.; Jain, B.K.; Duan, H.D.; Kovach, A.; Graham, T.R.; Li, H. Transport mechanism of P4 ATPase phosphatidylcholine flippases. eLife 2020, 9, e62163. [Google Scholar] [CrossRef]
- Hiraizumi, M.; Yamashita, K.; Nishizawa, T.; Nureki, O. Cryo-EM structures capture the transport cycle of the P4-ATPase flippase. Science 2019, 365, 1149–1155. [Google Scholar] [CrossRef]
- Segawa, K.; Kurata, S.; Nagata, S. The CDC50A extracellular domain is required for forming a functional complex with and chaperoning phospholipid flippases to the plasma membrane. J. Biol. Chem. 2018, 293, 2172–2182. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.S.; Costa, S.R.; Duelli, A.S.; Andersen, P.A.; Poulsen, L.R.; Stanchev, L.D.; Gourdon, P.; Palmgren, M.; Gunther Pomorski, T.; Lopez-Marques, R.L. Phospholipid flipping involves a central cavity in P4 ATPases. Sci. Rep. 2017, 7, 17621. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, S.; Ueno, H.; Shaikh, A.R.; Umemura, M.; Kamiya, M.; Ito, Y.; Ikeguchi, M.; Komoriya, Y.; Iino, R.; Noji, H. Molecular Mechanism of ATP Hydrolysis in F1-ATPase Revealed by Molecular Simulations and Single-Molecule Observations. J. Am. Chem. Soc. 2012, 134, 8447–8454. [Google Scholar] [CrossRef]
- Zhou, Y.; Ojeda-May, P.; Nagaraju, M.; Pu, J. Toward Determining ATPase Mechanism in ABC Transporters: Development of the Reaction Path-Force Matching QM/MM Method. Methods Enzymol. 2016, 577, 185–212. [Google Scholar]
- Van Der Velden, L.M.; Van De Graaf, S.F.J.; Klomp, L.W.J. Biochemical and cellular functions of P4 ATPases. Biochem. J. 2010, 431, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, H.; Nishizawa, T.; Segawa, K.; Nureki, O.; Fujiyoshi, Y.; Nagata, S.; Abe, K. Transport Cycle of Plasma Membrane Flippase ATP11C by Cryo-EM. Cell Rep. 2020, 32, 108208. [Google Scholar] [CrossRef]
- Kamerlin, S.C.L.; Vicatos, S.; Dryga, A.; Warshel, A. Coarse-Grained (Multiscale) Simulations in Studies of Biophysical and Chemical Systems. Annu. Rev. Phys. Chem. 2011, 62, 41–64. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral. Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Warshel, A.; Weiss, R.M. Empirical valence bond calculations of enzyme catalysis. Ann. N. Y. Acad. Sci. 1981, 367, 370–382. [Google Scholar] [CrossRef]
- Sham, Y.Y.; Chu, Z.T.; Tao, H.; Warshel, A. Examining methods for calculations of binding free energies: LRA, LIE, PDLD-LRA, and PDLD/S-LRA calculations of ligands binding to an HIV protease. Proteins 2000, 39, 393–407. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EForm2MC 1 | EScaled size 2 | EHydro 3 | EVDW 4 | E-DG UF 5 | EPOLAR 6 | Etotal 7 | STD 8 | |
|---|---|---|---|---|---|---|---|---|
| Spike protein | ||||||||
| S-closed | −421.71 | 578.35 | −683.64 | −49.01 | 4.38 | −44.01 | −615.65 | 1.74 |
| S-open | −424.53 | 578.35 | −658.64 | −46.55 | 4.38 | −43.21 | −590.20 | 1.66 |
| S-complex | −426.12 | 578.35 | −677.86 | −46.51 | 4.38 | −43.13 | −610.89 | 1.37 |
| ACLY tetramer | ||||||||
| E | −990.30 | 706.79 | −1014.88 | −57.54 | 1.07 | −17.92 | −1372.78 | 1.01 |
| E’ | −1035.79 | 706.79 | −1073.75 | −63.33 | 1.07 | −20.71 | −1485.72 | 2.93 |
| T1 | −983.19 | 706.79 | −1021.02 | −57.24 | 1.07 | −18.36 | −1371.95 | 1.37 |
| T2 | −1038.84 | 706.79 | −1061.91 | −63.20 | 1.07 | −19.93 | −1476.02 | 4.30 |
| P4-ATPase | ||||||||
| E1 | −57.31 | 254.49 | −960.92 | −20.74 | 0.96 | −61.64 | −845.16 | 0.10 |
| E1-ATP | −77.34 | 254.49 | −945.38 | −21.01 | 0.96 | −65.96 | −854.24 | 0.04 |
| E1P-ADP | −61.12 | 254.49 | −959.66 | −21.05 | 0.96 | −59.73 | −846.11 | 0.21 |
| E1P | −27.82 | 254.49 | −952.45 | −20.94 | 0.96 | −67.63 | −813.39 | 0.96 |
| E2P | −50.63 | 254.49 | −946.14 | −23.20 | 0.96 | −70.50 | −835.02 | 0.02 |
| E2Pi-PL | −40.35 | 254.49 | −967.79 | −21.09 | 0.96 | −64.88 | −838.66 | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, D.; An, K.; Zhang, H.; Xu, P.; Bai, C. Application of Coarse-Grained (CG) Models to Explore Conformational Pathway of Large-Scale Protein Machines. Entropy 2022, 24, 620. https://doi.org/10.3390/e24050620
Shi D, An K, Zhang H, Xu P, Bai C. Application of Coarse-Grained (CG) Models to Explore Conformational Pathway of Large-Scale Protein Machines. Entropy. 2022; 24(5):620. https://doi.org/10.3390/e24050620
Chicago/Turabian StyleShi, Danfeng, Ke An, Honghui Zhang, Peiyi Xu, and Chen Bai. 2022. "Application of Coarse-Grained (CG) Models to Explore Conformational Pathway of Large-Scale Protein Machines" Entropy 24, no. 5: 620. https://doi.org/10.3390/e24050620
APA StyleShi, D., An, K., Zhang, H., Xu, P., & Bai, C. (2022). Application of Coarse-Grained (CG) Models to Explore Conformational Pathway of Large-Scale Protein Machines. Entropy, 24(5), 620. https://doi.org/10.3390/e24050620