
Instance Segmentation of Multiple Myeloma Cells Using Deep-Wise Data Augmentation and Mask R-CNN



Abstract
:1. Introduction
- (1)
- The number of nucleus and cytoplasm may vary from one cell to another;
- (2)
- The boundary of the plasma cells is fuzzy because the cytoplasm of the cell and the background of the image have a similar visual appearance;
- (3)
- In some situations, cells may be isolated as single cells, but sometimes they may be in clusters;
- (4)
- If cells are in clusters, they may be in three different conditions: (i) Nuclei of cells are touching; (ii) Cytoplasm of cells are touching; and (iii) Cytoplasm of one cell is touching the nucleus of other cells. In such cases, the computer-aided segmentation process will be more challenging because the cytoplasm and nucleus expose different colors;
- (5)
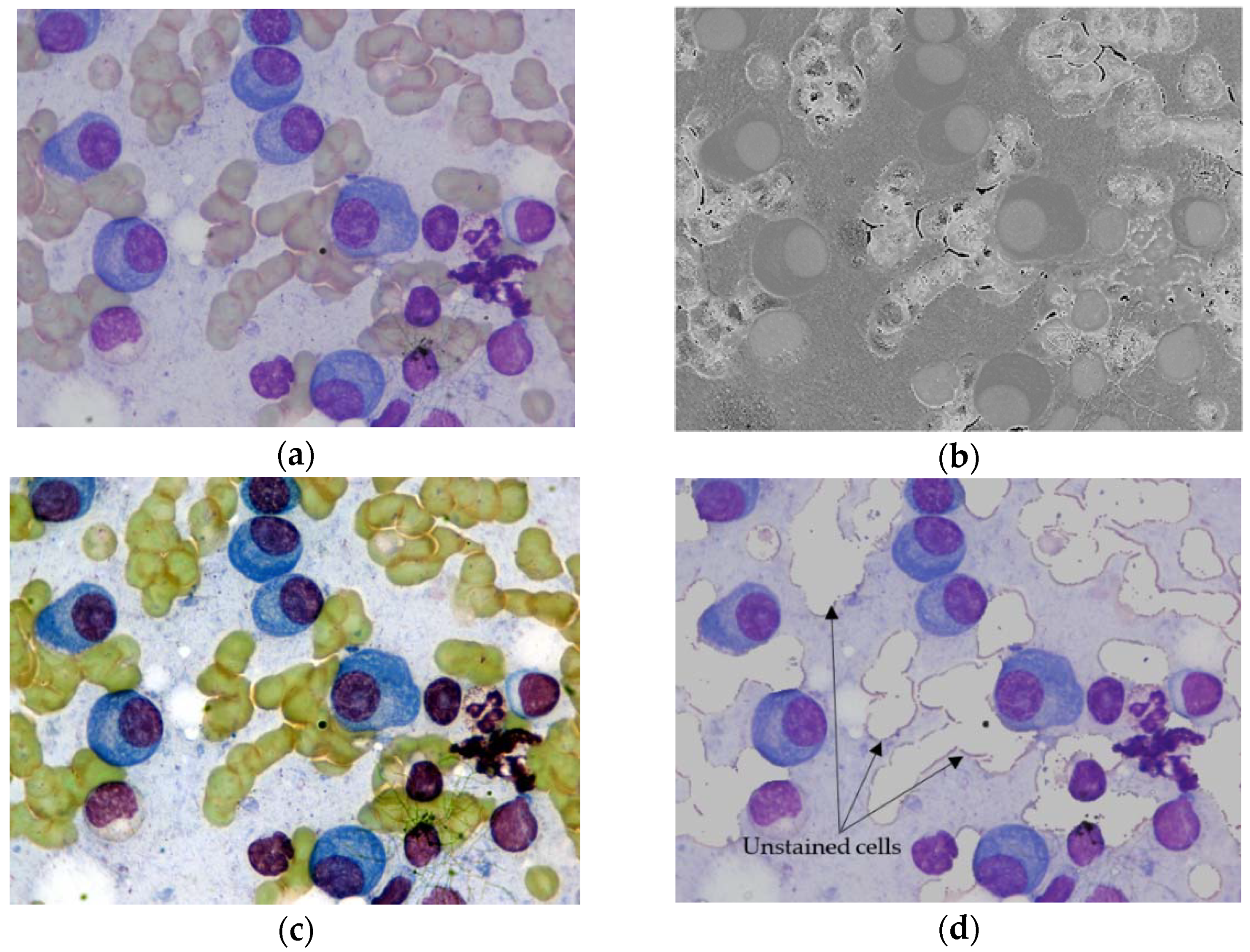
- In some situations, it is possible to have an unstained cell (for example, a red blood cell) underneath the cell of interest (MM plasma cell). In such cases, the color and shade of unstained cells may change and interfere with MM plasma cells. As a negative result, it may interfere with the detection and segmentation of cells of interest.
2. Related Works
3. Materials and Methods
3.1. Materials
3.2. Methods
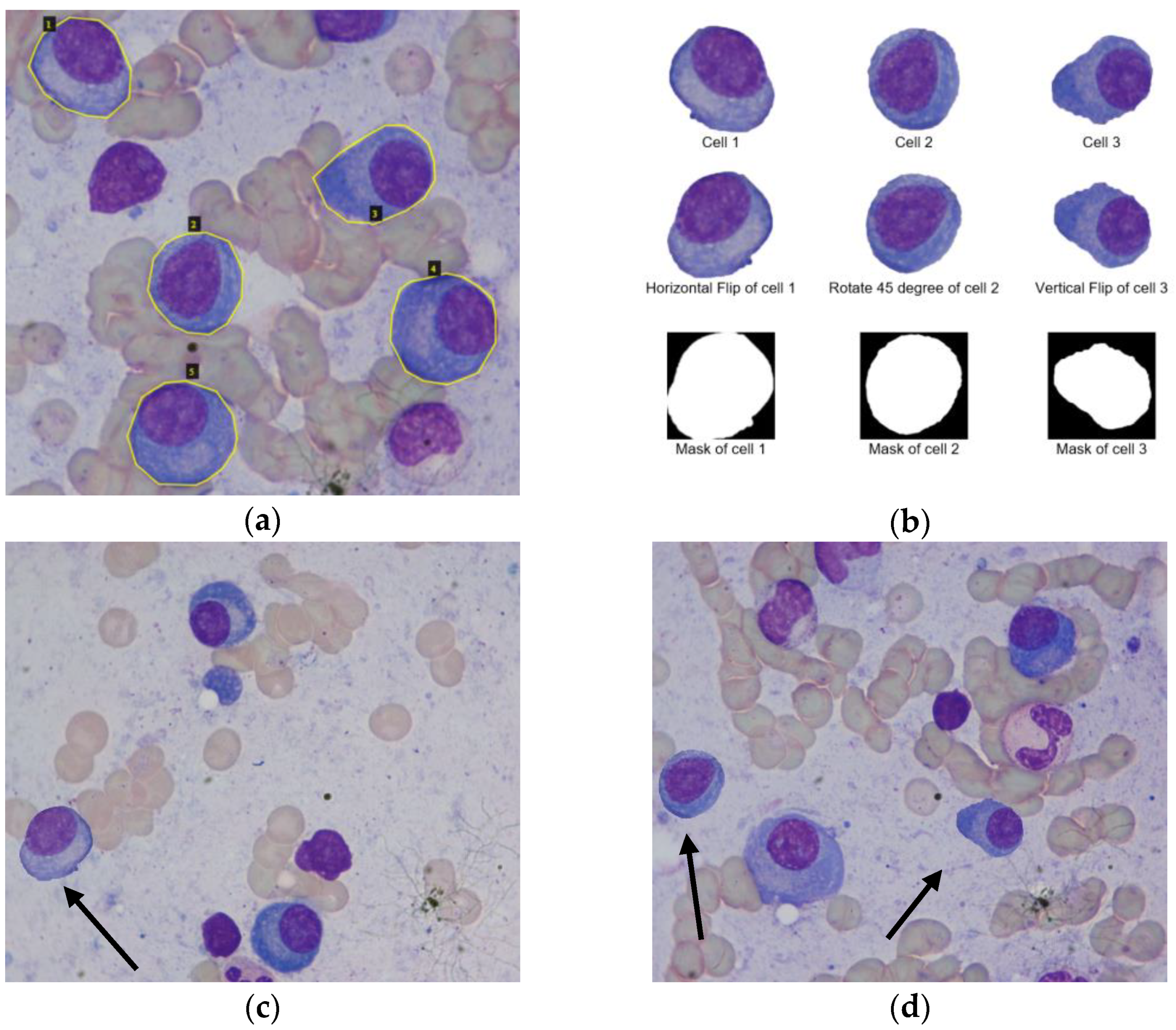
3.2.1. Segmentation of Stained Cells
3.2.2. Deep Wise Data Augmentation
3.2.3. Mask R-CNN
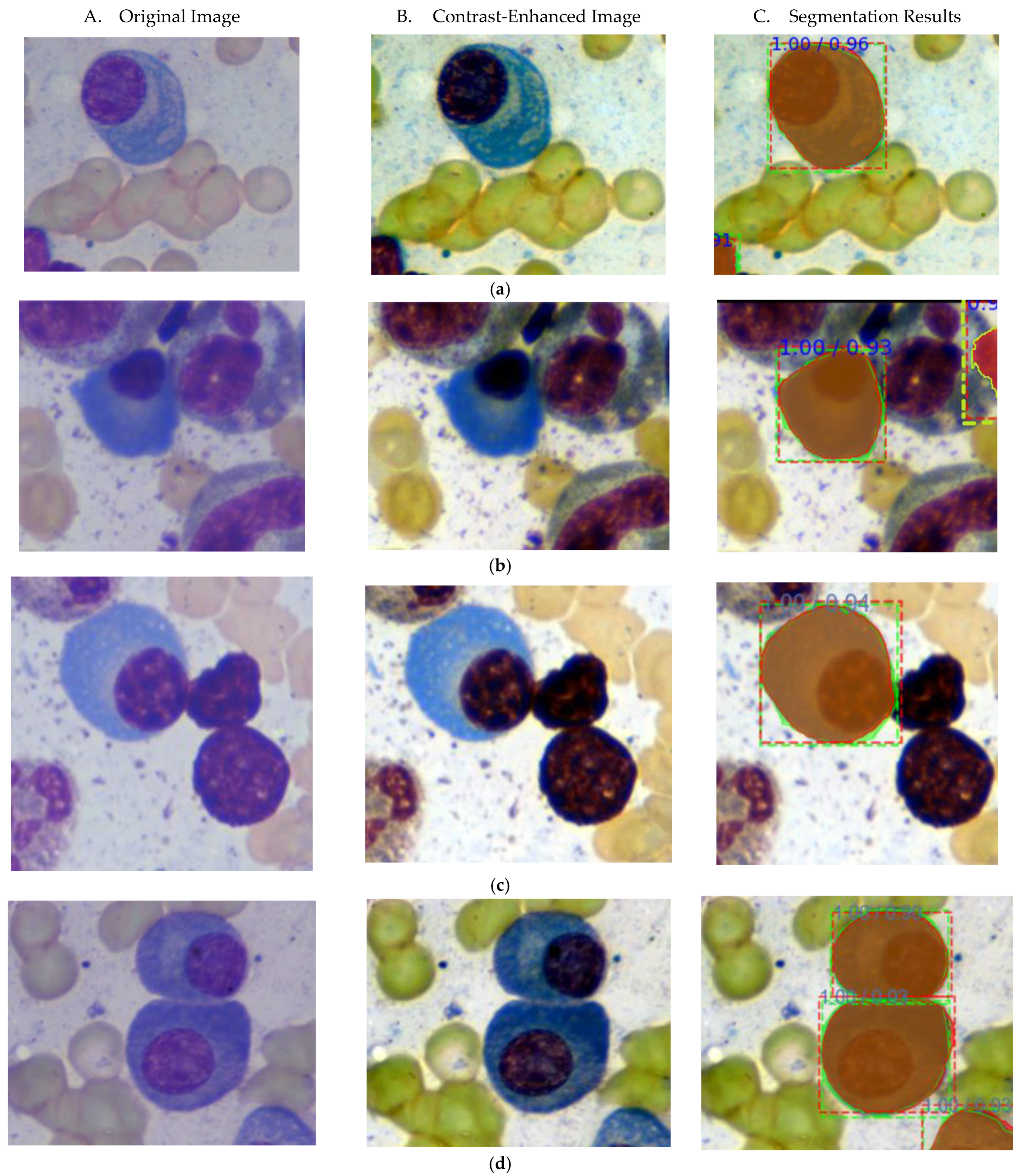
4. Experimental Results and Discussion
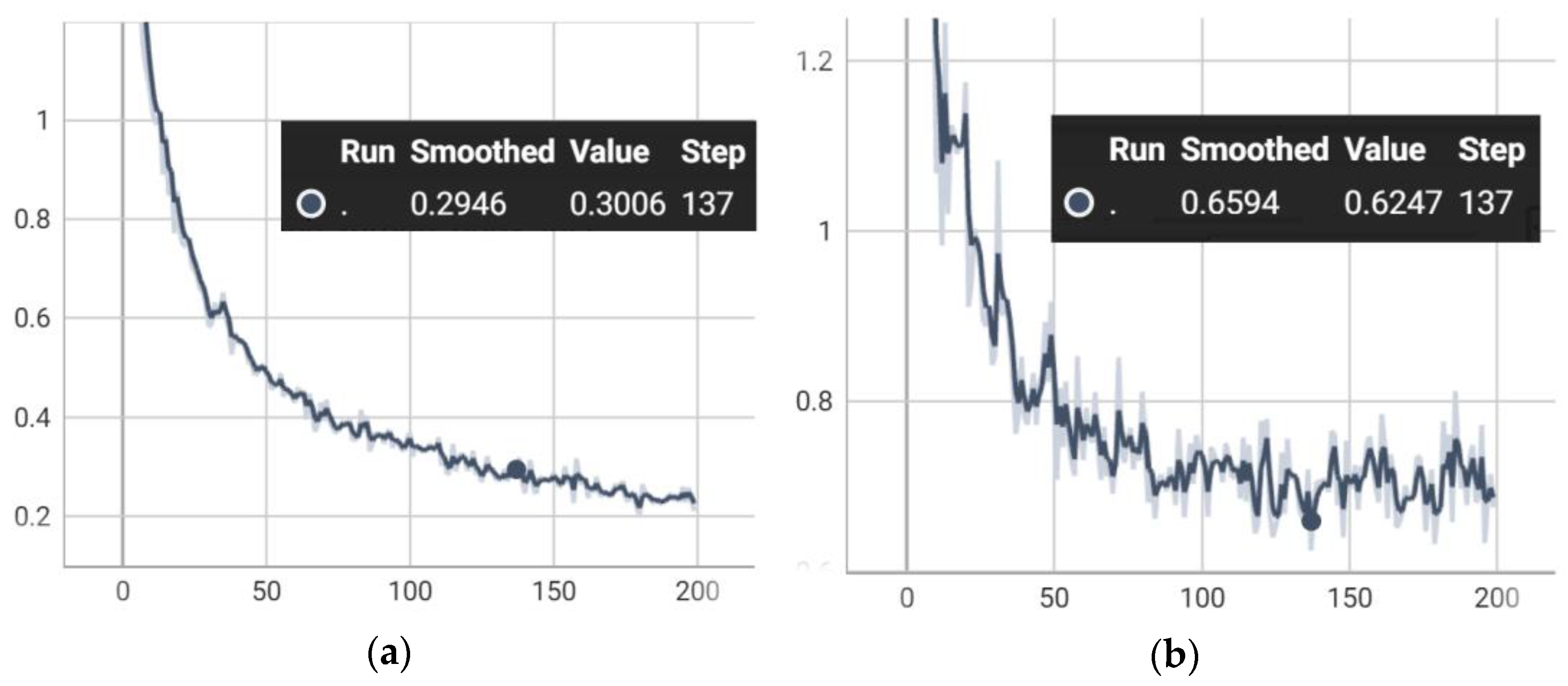
4.1. Ablation Study
4.2. Comparison with State-of-Art Methods
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bozorgpour, A.; Azad, R.; Showkatian, E. Multi-Scale Regional Attention Deeplab3+: Multiple Myeloma Plasma Cells Segmentation in Microscopic Images. arXiv 2021, arXiv:2105.06238. [Google Scholar]
- Padala, S.A.; Barsouk, A.; Barsouk, A.; Rawla, P.; Vakiti, A.; Kolhe, R.; Kota, V.; Ajebo, G.H. Epidemiology, Staging, and Management of Multiple Myeloma. Med. Sci. 2021, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Vyshnav, M.T.; Sowmya, V.; Gopalakrishnan, E.A.; Variyar, V.V.S.; Menon, V.K.; Soman, K.P. Deep Learning Based Approach for Multiple Myeloma Detection. In Proceedings of the 2020 11th International Conference on Computing, Communication and Networking Technologies (ICCCNT), Kharagpur, India, 1–3 July 2020; pp. 1–7. [Google Scholar]
- Gupta, A.; Mallick, P.; Sharma, O.; Gupta, R.; Duggal, R. PCSeg: Color Model Driven Probabilistic Multiphase Level Set Based Tool for Plasma Cell Segmentation in Multiple Myeloma. PLoS ONE 2018, 13, e0207908. [Google Scholar] [CrossRef] [PubMed]
- Saeedizadeh, Z.; Mehri Dehnavi, A.; Talebi, A.; Rabbani, H.; Sarrafzadeh, O.; Vard, A. Automatic Recognition of Myeloma Cells in Microscopic Images Using Bottleneck Algorithm, Modified Watershed and SVM Classifier: Automatic recognition of myeloma. J. Microsc. 2016, 261, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Gupta, A. MiMM_SBILab Dataset: Microscopic Images of Multiple Myeloma [Data Set]; The Cancer Imaging Archive: Little Rock, AR, USA, 2018; Available online: https://doi.org/10.7937/tcia.2019.pnn6aypl (accessed on 30 November 2021).
- Tehsin, S.; Zameer, S.; Saif, S. Myeloma Cell Detection in Bone Marrow Aspiration Using Microscopic Images. In Proceedings of the 2019 11th International Conference on Knowledge and Smart Technology (KST), Phuket, Thailand, 23–26 January 2019; pp. 57–61. [Google Scholar]
- Vuola, A.O.; Akram, S.U.; Kannala, J. Mask-RCNN and U-Net Ensembled for Nuclei Segmentation. arXiv 2019, arXiv:1901.10170. [Google Scholar]
- Gupta, A.; Duggal, R.; Gehlot, S.; Gupta, R.; Mangal, A.; Kumar, L.; Thakkar, N.; Satpathy, D. GCTI-SN: Geometry-Inspired Chemical and Tissue Invariant Stain Normalization of Microscopic Medical Images. Med. Image Anal. 2020, 65, 101788. [Google Scholar] [CrossRef] [PubMed]
- Gehlot, S.; Gupta, A.; Gupta, R. EDNFC-Net: Convolutional Neural Network with Nested Feature Concatenation for Nuclei-Instance Segmentation. In Proceedings of the ICASSP 2020—2020 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), Barcelona, Spain, 4–8 May 2020; pp. 1389–1393. [Google Scholar]
- Sahidan, S.I.; Mashor, M.Y.; Wahab, A.S.W.; Salleh, Z.; Ja’afar, H. Local and Global Contrast Stretching For Color Contrast Enhancement on Ziehl-Neelsen Tissue Section Slide Images. In 4th Kuala Lumpur International Conference on Biomedical Engineering 2008; Abu Osman, N.A., Ibrahim, F., Wan Abas, W.A.B., Abdul Rahman, H.S., Ting, H.-N., Eds.; IFMBE Proceedings; Springer: Berlin/Heidelberg, Germany, 2008; Volume 21, pp. 583–586. ISBN 978-3-540-69138-9. [Google Scholar]
- Dutta, A.; Zisserman, A. The VIA Annotation Software for Images, Audio and Video. In Proceedings of the 27th ACM International Conference on Multimedia (MM ’19), Nice, France, 21–25 October 2019. [Google Scholar]
- Zhang, L.; Wen, T.; Shi, J. Deep Image Blending. In Proceedings of the 2020 IEEE Winter Conference on Applications of Computer Vision (WACV), Snowmass Village, CO, USA, 1–5 March 2020; pp. 231–240. [Google Scholar]
- Zhang, L.; Wen, T.; Shi, J. Deep Image Blending. Available online: https://github.com/owenzlz/DeepImageBlending (accessed on 19 December 2021).
- He, K.; Gkioxari, G.; Dollar, P.; Girshick, R. Mask R-CNN. IEEE Trans. Pattern Anal. Mach. Intell. 2020, 42, 386–397. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Mean Precision | Mean Recall | Mean IOU |
|---|---|---|---|
| Original Mask R-CNN | 0.8769 | 0.8178 | 0.7689 |
| Original Augmented Mask R-CNN | 0.9426 | 0.8406 | 0.7465 |
| Original Mask R-CNN with Deep-wise data augmentation | 0.9464 | 0.8478 | 0.8721 |
| Contrast-enhanced Mask R-CNN | 0.8966 | 0.8333 | 0.7324 |
| Contrast-enhanced Augmented Mask R-CNN | 0.9843 | 0.8566 | 0.8879 |
| Contrast-enhanced Mask R-CNN with Deep-wise data augmentation | 0.9973 | 0.8631 | 0.9062 |
| Stained cell Mask R-CNN | 0.9389 | 0.7372 | 0.6478 |
| Stained cell Augmented Mask R-CNN | 0.9614 | 0.8130 | 0.7324 |
| Stained cell Mask R-CNN with Deep-wise data augmentation | 0.9632 | 0.8328 | 0.7348 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paing, M.P.; Sento, A.; Bui, T.H.; Pintavirooj, C. Instance Segmentation of Multiple Myeloma Cells Using Deep-Wise Data Augmentation and Mask R-CNN. Entropy 2022, 24, 134. https://doi.org/10.3390/e24010134
Paing MP, Sento A, Bui TH, Pintavirooj C. Instance Segmentation of Multiple Myeloma Cells Using Deep-Wise Data Augmentation and Mask R-CNN. Entropy. 2022; 24(1):134. https://doi.org/10.3390/e24010134
Chicago/Turabian StylePaing, May Phu, Adna Sento, Toan Huy Bui, and Chuchart Pintavirooj. 2022. "Instance Segmentation of Multiple Myeloma Cells Using Deep-Wise Data Augmentation and Mask R-CNN" Entropy 24, no. 1: 134. https://doi.org/10.3390/e24010134
APA StylePaing, M. P., Sento, A., Bui, T. H., & Pintavirooj, C. (2022). Instance Segmentation of Multiple Myeloma Cells Using Deep-Wise Data Augmentation and Mask R-CNN. Entropy, 24(1), 134. https://doi.org/10.3390/e24010134