7.1. Sadi Carnot’s Legacy
Carnot [
1] stated firmly on page 29 of his book regarding isothermal expansion, “When a gas increases in volume in geometrical progression, its
chaleur specifique increases in arithmetical progression”. His statement regarding the logarithmic increase in specific heat with volume at constant temperature is consistent with the decreasing Gibbs energy or increasing entropic energy (δ
sT) shown for stage 1 in
Table 1 and
Table 2, while internal energy (
cvT) remains constant. The heat absorbed isothermally from the hot source is regarded as consumed in the field energy sustaining the molecular orbits and maintaining the kinetic energy of the molecules as constant, allowing external work to be done via their pressure. This configurational entropic energy was also defined by Clausius in 1875 [
10] as work heat or the
ergal. There was no need for the editor Mendoza (see his foreword in Dover edition of Carnot’s book [
2]) to have rejected the significance of Carnot’s conclusion on page 29 of his memoir that the
chaleur specifique (specific heat) varied with the logarithm of the volume. Indeed, the editor of the Dover edition, Mendoza, claimed in 1960 that Carnot was mistaken, having been misled by faulty data produced by Delaroche and Bérard to calculate the effect of pressure on the specific heat of a gas. In fact, Mendoza’s criticism of their data was in error, as discussed next.
Moreover, Carnot’s hypothetical table on page 33 of pressure varying from 1/1024 to 1024 atmospheres labeling variations in specific heat with pressure is correct in principle —if Carnot’s chaleur specifique is interpreted as variation in the heat required for entropic energy (δsT) while work is performed—varying logarithmically with volume, decreasing Gibbs energy as a result of the increase in volume at constant temperature (stage 1=>2). When Carnot visualized the thermodynamic operation of the motrice de feu, he actually challenged the theory casting calorique as a diffusable fluid form of heat that could neither be created nor destroyed. He did this by proposing that calorique as heat could be temporarily exchanged with motive power in a reversible cycle. It is clear from what Carnot wrote that he did consider sensible heat or chaleur disappeared as internal work was performed in an adiabatic expansion.
At no point in his reflection does Carnot claim that the same quantity of heat as that introduced from the hot source is fully re-absorbed as caloric after the adiabatic fall (
chute de calorique) to the temperature of the cold sink, which was an error introduced by Clapeyron [
15]. On the contrary, on page 28 in the Mendoza edition [
2] (page 29 in the 1878 Gauthier-Villars reproduction of the original Bachelier 1824 edition), he clearly infers to the quantity of caloric
a, which is “necessary to maintain the temperature of the fluid constant during dilatation”, and that transferred from the hot source in stage 1 is not equal to the caloric
a′ that the gas abandons later as a result of its reduction of volume at lower temperature in stage 3, so
a −
a′ must have a positive value.
7.2. Caloric as Negative Gibbs Potential
To examine his analysis here in more detail, Carnot [
2] defines (p. 28, 30) the high-temperature phase of the cycle involving body A as consisting of two portions of caloric—that needed to maintain the temperature A in dilatation (
a) and that needed to restore the temperature of the fluid from that of body B to that of body A (
b). “The total caloric furnished by the body A will be expressed by
a +
b. The caloric transmitted by the fluid to the body B may also be divided in two parts: one,
b′, due to the cooling of the gas by the body B; the other,
a′, which the gas abandons as a result of its (isothermal) reduction in volume. The sum of these two quantities is
a′ +
b′; it should be equal to
a +
b, for, after a complete cycle of the operations, the gas is brought back exactly to its primitive state. It has been obliged to give up all the caloric which has been furnished to it”. So, we have:
or rather
At this point, Carnot leaves unsaid that Equation (37) gives the maximum work possible in a perfect heat engine, the difference between two similar logarithmic functions of changes in volume but differing only by temperature. However, he clearly identifies [
1] this link on page 22 with his precise statement: “La puissance motrice résultat …sera évidement la différence entre celle qui est produite par l’expansion du gaz, tandis qu’il se trouve á la temperature du corps A, et celle qui est consommeé pour comprimer ce gaz, tandis qu’il se trouve á la temperature du corps B”. Thus, he claims that the isothermal changes of volume at the different temperatures fully explains the potential to do motive work.
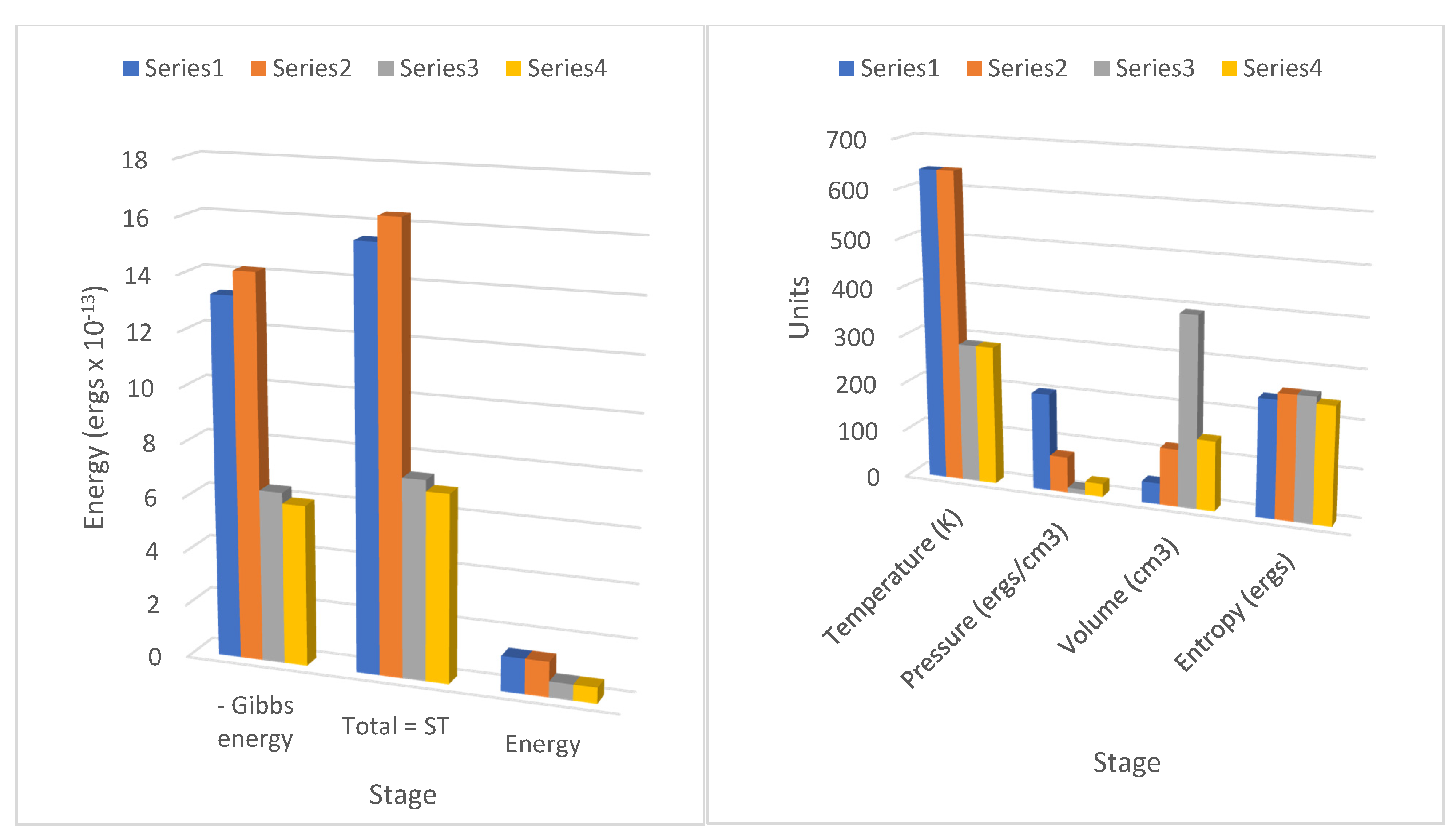
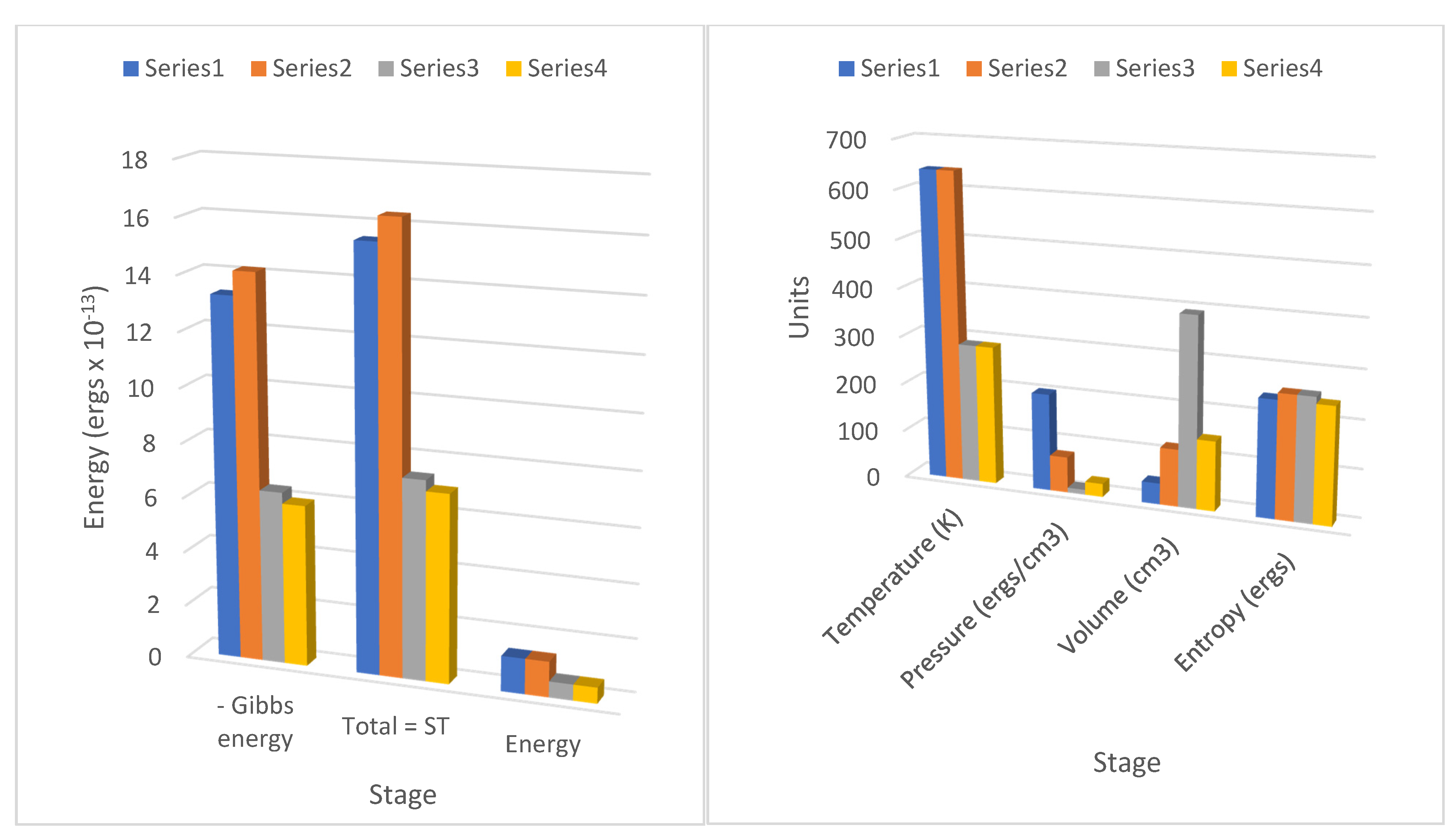
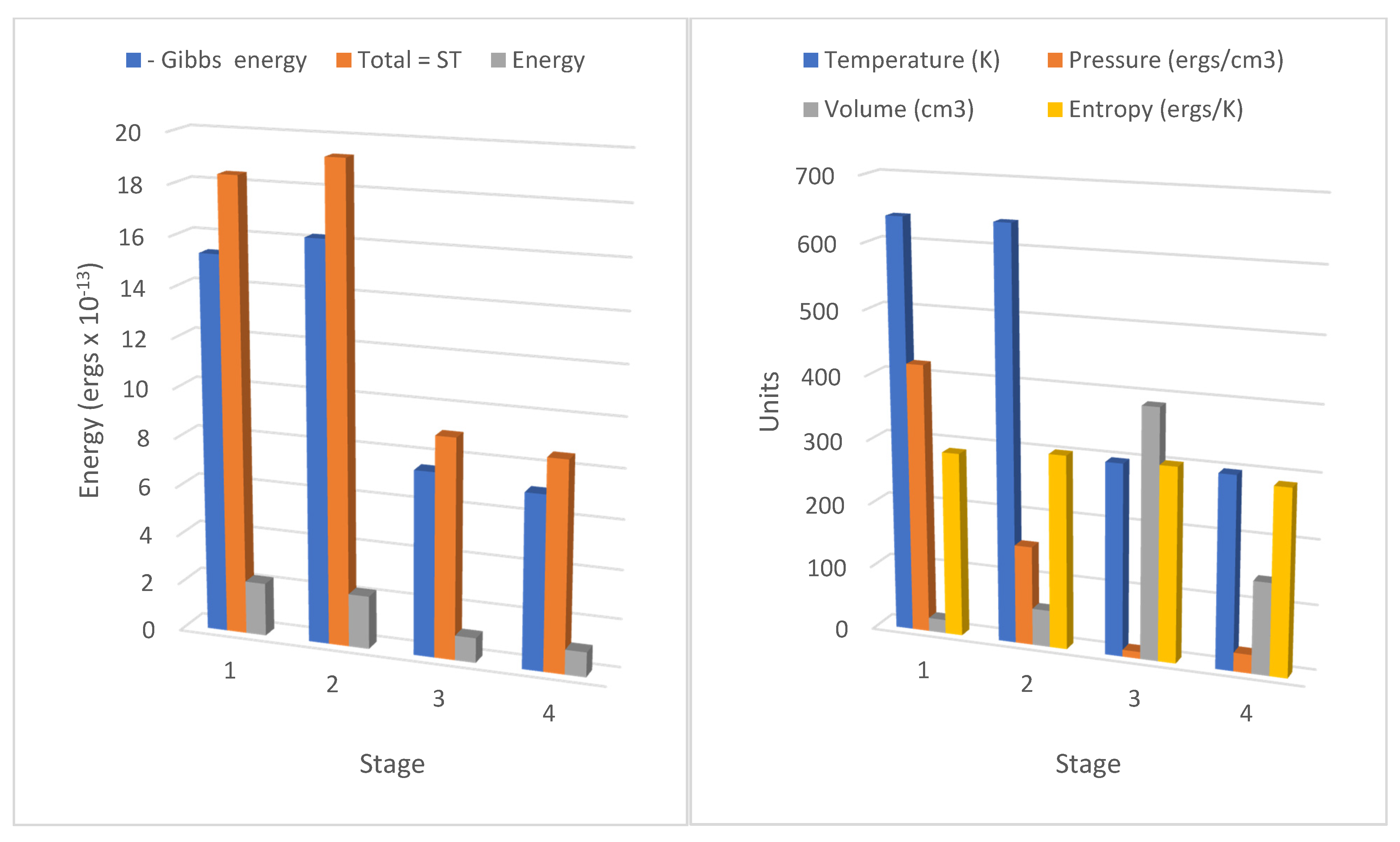
These terms can readily be identified in the results for the cycle stages 1–4 in
Table 1 and
Table 2, corresponding to the negative magnitudes of the Gibbs potential changes (or the Gibbs entropic energy values) in the columns 1=>2 (
a), 2=>3 (−
b′), 3=>4 (−
a′), and 4=>1 (
b). A typing error in his memoir on line 10 page 31 possibly first inserted in the 1878 edition of
b′ for
b, repeated in the Mendoza translation [
2], may have been confusing. Note that modern convention requires that the signs of
a′ and
b′ be made negative given that the Gibbs energy is increased by the extraction of heat during the adiabatic expansion and the isothermal compression by body B. From this analysis, we can now identify changes in Gibbs potential with Carnot’s changes in caloric, with no need to include changes in energy (δ
e) except in the stages where there is a change in temperature.
Carnot clearly understood that his term caloric related to the physical state of the working fluid with a meaning very similar to negative Gibbs potential or configurational entropic energy as calculated in action state theory. Whenever Carnot claims an increase in caloric of the working fluid, we can recognize absorption of radiant heat as increasing entropy and action and internal work of raising quantum states. Clausius refers to this reversible work as having consumed heat “nowhere present, it is consumed in the changes doing work”. Perhaps Clausius initially recognized Carnot’s perception as foreshadowing his ergal, being the internal work done on the engine’s working fluid. However, Clausius then seems to disregard the internal work implied for molecules further apart in the working fluid and decides to directly assign the heat required to external work.
Fortunately, despite Kelvin’s apparent misquotation from Clapeyron of Carnot’s theory implying that “all the heat from body A … during expansion has flowed into body B during compression” as work was done, in 1850, Clausius [
13] kindly corrected this false viewpoint. The German inventor of the thermodynamic concept of entropy recognized the value of Carnot’s principle of motive work depending on heat being transported from a hot source to a colder sink, using a working fluid that was unchanged in its state. To emphasize that heat and work were interchangeable, Clausius later [
15] abandoned the concept of latent heat, substituting ‘work-heat’ for increased entropy to account for the heat needed to overcome both the cohesion of molecules and expansion against an external pressure. Since these heat-absorbing processes are both reversible, heat disappears as work is performed, but work can reappear later as heat in a reversible Carnot cycle. Perhaps Clausius went too far in seeking to dispense with caloric when converting heat to work, many years before Planck and Einstein developed the theory of quanta.
It is surprising how the false statement from Clapeyron has persisted with highly skilled authors still continuing to claim that Carnot assumed all the heat given up by body A was transferred to body B [
16,
17]. Although Aumand et al. [
17] creditably provide a correct account based on various sources, they still propagate the error that Carnot equated
calorique to the property we now call entropy, which is one also fell into by the book editor Mendoza [
2]. As discussed above, Carnot clearly meant energy, requiring the product (
s ×
T) of entropy and temperature.
To put Carnot’s clear viewpoint to the contrary beyond doubt, his footnote in full on page 19 of the Mendoza edition of his book (page 20 of the 1878 Bachelier edition [
1]), misquoted by Kelvin and Clausius, actually states “We tacitly assume in our demonstration, that when a body has experienced any changes, and when after a certain number of transformations, it returns to precisely its original state, that is to that state considered in relation to density, to temperature, to mode of aggregation… I say that this body is found to contain the same quantity of heat (
chaleur) that it contained at first, or else that the quantities of heat absorbed or set free in these different transformations are exactly compensated. This fact has never been called into question. It was first admitted without reflection, and verified afterwards in many cases by experiments with the calorimeter. To deny it would be to overthrow the whole theory of heat to which it serves as a basis”. Thus, Carnot never inferred the equality of the heat provided from the hot source to that retrieved by the cold sink. Indeed, with his heat (caloric) function (
a −
a′) discussed earlier as work, Carnot was also assuming the first law of conservation of energy as heat and work as well as the second law of spontaneous development of entropy. In fact, except for heat (
chaleur or
calorique), all these terms remained to be defined.
It is clear from his tentative equations that Carnot was already thinking in terms of the entropy–temperature product of negative Gibbs energy that is a key part of the viewpoint presented here. This idea was nascent in the concept of
calorique he generally applied to the state of the working fluid and his pressure–volume form of the logarithmically variable
chaleur specifique indicating the heat required. We conclude that this term should not be confused with current definitions of specific heat or heat capacity that are purposely restricted to the internal energy or enthalpy (
Cv or
Cp). Furthermore, in Thurston’s 1890 (Macmillan) translation of Carnot’s book
Reflections on the Motive Power of Fire, the translator (pointed out by Mendoza [
2] Dover edition 1960) often used the same term heat for both of Carnot’s terms
calorique and
chaleur, providing lingering confusion regarding Carnot’s account. However, Carnot specifically states that while he is indifferent to the use of terms
chaleur or
calorique as a quantity of heat, he does reserve
chaleur as a measure for the sensible heat of fire and is consistent in using
calorique for changes in the state of the working fluid; we would consider the latter as variations in Gibbs energy or configurational entropic energy, not sensible heat.
At no stage is
calorique specifique referred to in Carnot’s text despite a comment by Mendoza in 1960 [
2] to the contrary, with
chaleur specifique used consistently. It must be remembered that entropy includes an internal energy or enthalpy component not relevant to Carnot’s
calorique. Carnot clearly distinguished this heat related to expansion and compression from the modern heat capacity where there are no such processes in measurement. So, he was also proposing the first law of the conservation of energy as well as the second law of increasing entropy as shown in Equation (16). Carnot even identified the internal energy or enthalpy as a separate entity of heat
U in his equation given here as (38), which is discussed in detail on page 43 of his memoir [
2].
where
s is “the quantity of heat (
chaleur) necessary to change the air that we have employed from the volume 1 and from the temperature zero (i.e., Celsius) to the volume
v and to the temperature
t, the difference between
s and
e will be the quantity of heat required to bring the air to the volume 1 from zero to
t. This quantity depends on
t alone; we will call it
U”. To obtain motive power, Carnot varies Equation (38) with temperature. Since he had already explained that the difference in heat capacity at constant pressure and that at constant volume was a constant independent of the working substance (i.e., in modern terms,
Cp −
Cv =
R), it is reasonable to conclude that
U represented the internal energy (
E) or the enthalpy (
H =
E +
RT), and thus, Equation (38) is closely analogous to Equations (16) and (17) that express the 2nd law of thermodynamics and statistical mechanics. Our modern version of this equation has the benefit of both the clarifying work of Clausius [
15] on correctly establishing the fundamental principles of the mechanical theory of heat, the statistical mechanics of Willard Gibbs [
8], and the quantum theory of Planck [
14] and even Einstein soon after 1900.
Referring to the equation for entropic energy (
ST) or total reversible heat requirement from Equation (11) above for a monatomic gas, we observe that for argon, Equation (39) applies at 1 atm external pressure.
Thus, expressing Equation (39) between two temperatures provides variations in Gibbs energy that allows the expression of motive power or rate of work. Thus, the similarity of Carnot’s Equation (38) and our equation for entropic energy (39) is evident.
In terms of the action method, the change in temperature within the logarithmic expression of (kTI)1/2 in adiabatic stages 2 and 4 exactly offsets the change in density and inertia (It = mr2), a function of volume, so the relative action mrv remains the same. In the adiabatic processes, the change in Gibbs or Helmholtz energies is a linear function of the change in temperature only, as is the internal energy. These changes in energy (E or H) in the adiabatic stages cannot result in net work, as stage 2=>3 is the reverse of stage 4=>1, ensuring that the working fluid returns to the same stage at the completion of each cycle. So, in Equation (38), taken with his conviction that as chaleur specifique or specific heat would also change with temperature, unlike the constant heat capacity now designated as Cv, we recognize that Carnot proposed the first version of the second law of thermodynamics.
7.3. Heat Capacity versus Specific Heat
So, was Carnot correct after all in his definition of specific heat as varying with pressure? On page 38 of the Dover edition [
2], Carnot performs a thought experiment confirming his viewpoint. He contends why a piston containing air first heated with
a units to 100 degrees at constant volume (
V1) and then, expanded with heat, added
b units at constant temperature to a larger volume (
V2) versus expanding first at 1 degree at constant temperature to the same volume
V2 with
b′ units and then heated to 100 degrees with
a′ units must be equivalent. “As the final result of these two processes is the same, the quantities of heat employed for both should be equal:
a′ is the quantity of heat required to cause the gas to rise from 1° to 100° when it occupies the larger volume, and a is the quantity of heat required when it occupies the smaller volume. The density of the air is less in the first case than the second and according to the experiments of MM Delaroche and Bérard, its capacity for heat should be a little greater”.
Carnot clearly considered that the measure of caloric content was a property of state, which is consistent with the future concept of entropy as a measure of heat content for both enthalpy and configurational energy as negative Gibbs energy; this must be the same initially and finally, assuming conditions for the cylinder contents are the same. The change in entropy per mole of argon is given by the following equation, with heat capacity
Cv for argon remaining the same throughout as 1.5
R. The energy change for an enclosed cylinder is equal to
Cvδ
T in both cases.
However, the quantity of caloric a required for heating at Vi includes an extra amount for the change of temperature from T1 to T2, indicating a change in state and entropy. By contrast, expanding with b units at the lower temperature of 1 degree Celsius involves less heat required, given that it equals RT ln(V2/V1).
Carnot [
2] continues on page 36, “The quantity
a′ being found to be greater than the quantity
a,
b should be greater than
b′. Consequently, generalizing the proposition, we should say: The quantity of heat due to change of volume of a gas is greater as the temperature is higher”. He also concludes: “The fall of caloric produces more motive power at inferior than at superior temperatures”, foreshadowing the benefits of having a heat sink for a given range of temperatures as close to absolute zero as possible, which is a hint regarding the third law of thermodynamics regarding zero entropy at absolute zero of temperature. However, Carnot had not completed his analysis of the effects of temperature versus pressure or system volume, and the truth of his conclusions depends on whether ratios of temperatures and volumes do depend on their relative magnitude.
Modern textbooks assume that the factors
Cv and
Cp for heat capacity of gases are constants, although this is not true at either high or low temperatures because of quantum effects. If we consider 3.5
R as the heat capacity of air
Cp given its predominant composition of nitrogen, the difference in entropic energy as negative Gibbs energy −
G per mole between 298.15 and 297.15 K at the standard pressure of 1 atmosphere can be calculated as follows.
So, the variation in heat content as enthalpy shows very little change as a result of the increase in Gibbs energy (ca. 0.24%), which is so small it can almost be overlooked. If the pressure is increased to 64 atmospheres, the specific volume (
a3) for an ideal gas at the same temperature will be 1/64 that at 1 atmosphere, so separation of the molecules will be one-quarter of that at one atmosphere, with a decrease in the translational moment of inertia to one-sixteenth. Will this change the Gibbs work factor and the heat capacity measured as the change in entropic energy between 298.15 and 297.15 K?
In fact, assuming ideal gas behavior, the change in entropy between these two temperatures will be exactly the same as before, despite the increased density, since the relative translational and rotational actions will be the same as before.
Thus, the heat capacity is very little affected by marginal changes in pressure, irrespective of the density. With this understanding, Carnot’s inclusion of the heat exchanged isothermally in reversible work processes as his chaleur specifique or specific heat, now recognized as negative Gibbs potential, has validity. Note in these calculations that with pressure constant at one atmosphere, the temperature and specific volume vary inversely, or the product of temperature and number density (N) is constant. If we allow pressure to vary with the lower temperature, holding number density constant so that the moment of inertia of N2 is constant, heat capacity will vary proportional to kln(T2/T1)5/2 or 3.49160k per molecule or 29.02917 J per mole per degree Kelvin, which is the accepted value for Cp of N2.
By contrast, holding temperature constant, as in stage 1 of the Carnot cycle, the product of pressure and volume is constant (
PV =
RT), and the specific heat varies with the logarithm of the specific volume (or pressure), just as Carnot proposed in his table. This understanding of his term for specific heat is also relevant for the amount of heat consumed or released in stages 2 and 4 during adiabatic expansion and compression. As the entropic energy changes (
Table 1, line 16;
Table 2, line 31), some eight to ten times more specific heat is consumed or released to the fluid as Carnot’s caloric, depending on the logarithmic function of temperature and volume compared with the change in internal energy or enthalpy. So, we can conclude that in the same terms as Carnot hypothesized, the specific heat does vary with the volume, the temperature, or the pressure.
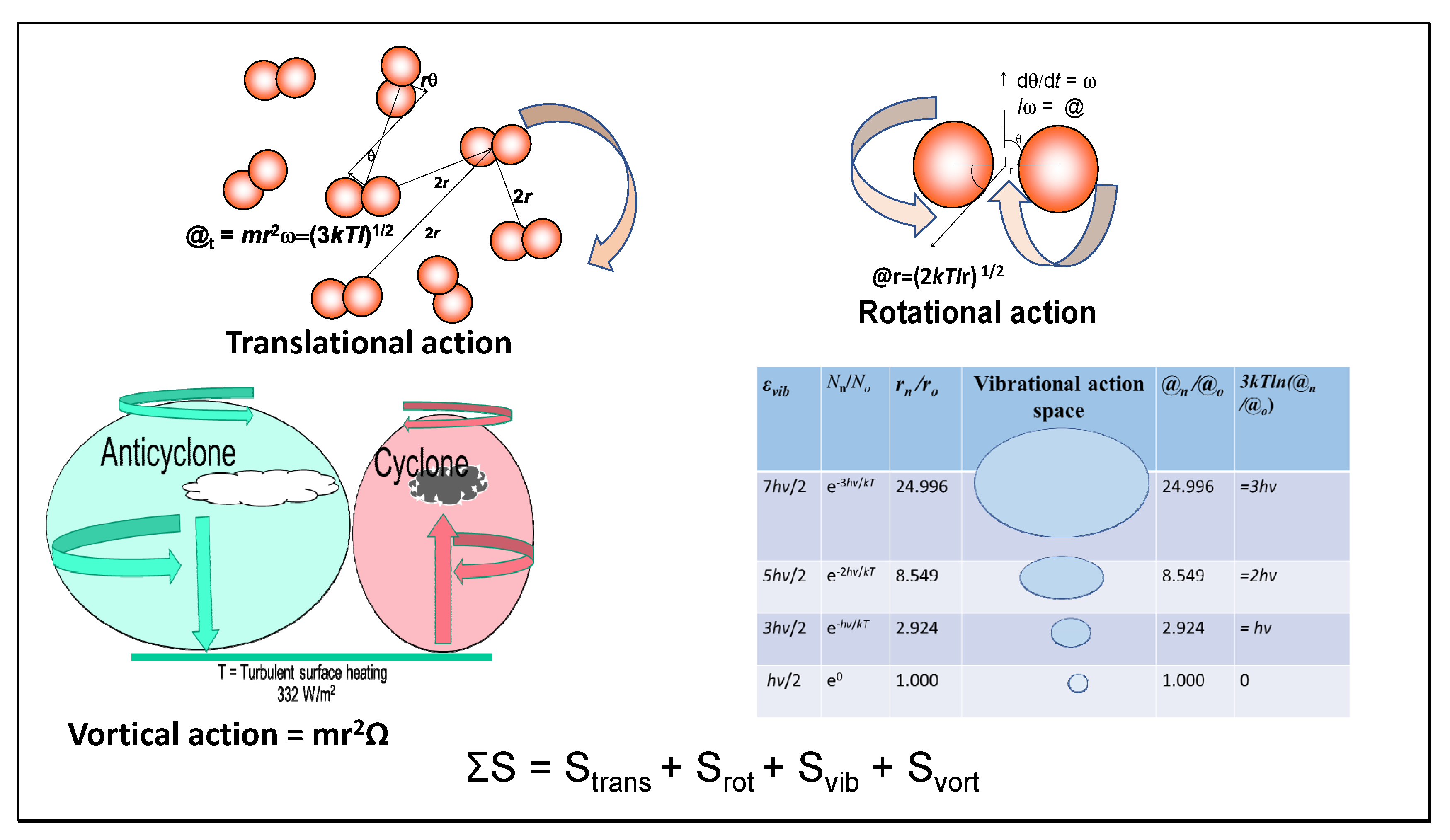
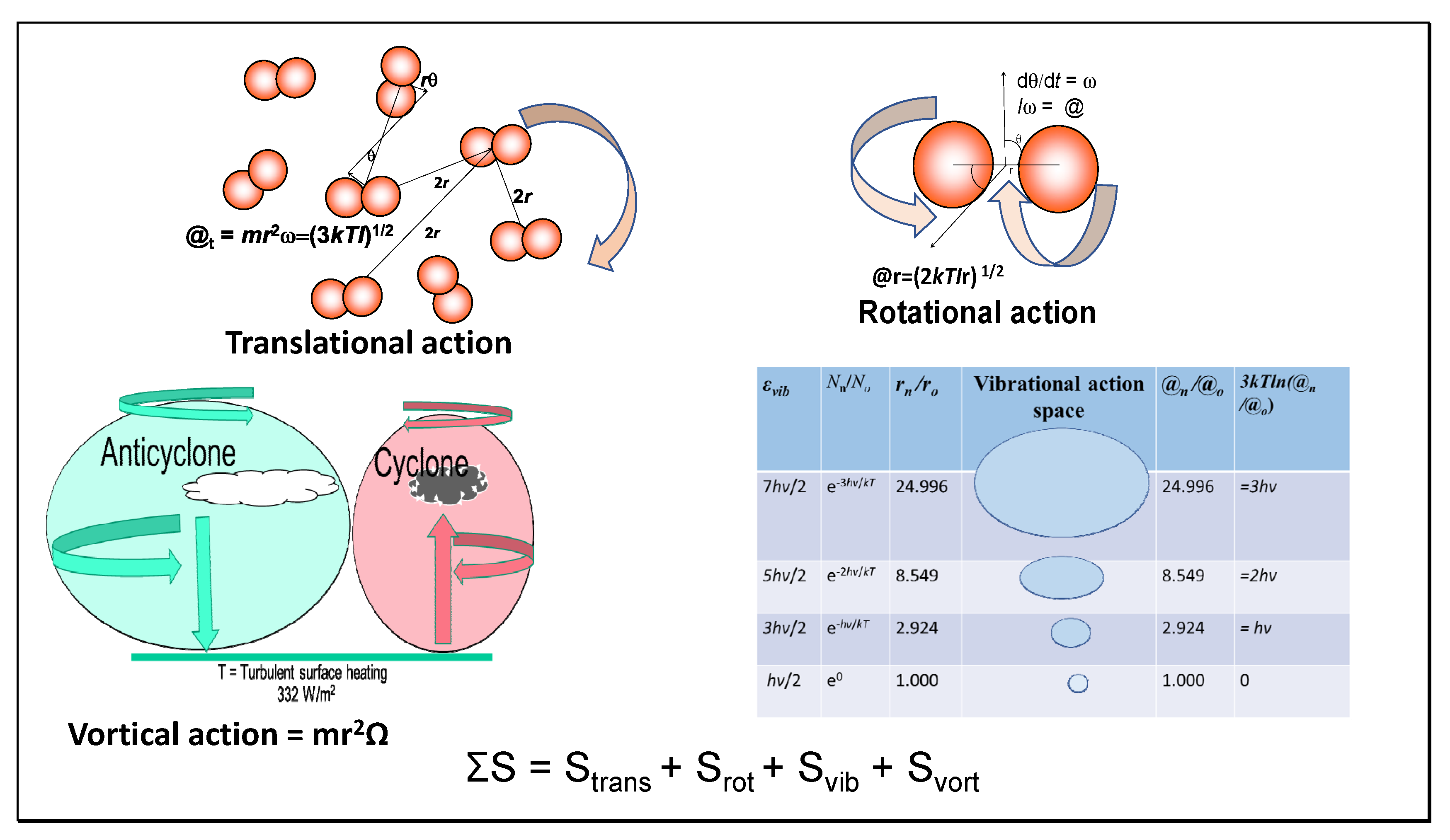
7.4. Quantum State Numbers
For both translational and rotational actions, mean values for quantum numbers (n
t) supporting the molecular morphology of the field have been calculated in the tables, including
Table 3, where these are summarized. These mean quantum numbers are calculated simply from the ratio of the action values (@
t, @
r) with Planck’s quantum of action (
ħ). It is a feature of such translational quantum states that their magnitude decreases with the quantum number, as pointed out by Schrödinger [
9], with only the levels very near the average energy occupied, explaining why the Maxwell–Boltzmann distribution has a sharp maximum. The average occupation number of the quantum cells in
Table 1 and
Table 2 approaches one in a million.
Note that none of the translational or rotational quantum states in heat engines correspond to extremely cold temperatures, so the quantum microstates are non-degenerate. This contrasts with the size of vibrational quanta that vary little with temperature, even down to absolute zero. Most texts in discussing quantum states are considering vibration, with the greatest population in the ground state, misleadingly for translational states. In
Table 3, average quantum occupation numbers and magnitudes are shown. These are all exceeded by
kT by at least an order of magnitude, even for rotation, indicating a lack of interaction between quantum particles as molecules of working fluid. As a symmetrical molecule lacking a dipole moment, nitrogen is proposed to exchange rotational quanta resonantly, with negligible net emission. The translational n
t3 and rotational quantum j
r2 products are also shown. For isentropic states, these are expected to be equal as adiabatic, although a small variation after four significant figures is shown in the table. Heat engines might function by irradiation with resonant quanta of specific long wavelength, varying according to the physical stage in the cycle, providing more efficient work than hitherto achieved. Such an experimental model should now be tested.
It is suggested that translational quanta are released gradually in molecular decelerations during elastic collisions. When recovering their velocities, the field quanta would be reabsorbed, only momentarily being freed. In effect, the Gibbs potential of separate molecules oscillates, being at a minimum nearest zero when colliding molecules are closest. In his discussion of Brownian motion, Einstein referred to forces acting between molecules in which this quantum field can participate. Action requires that all relative molecular motions be complemented by sustaining field energy related to configurational entropy. Yet the heat required for a molecular ensemble to reversibly reach a given temperature sustaining the enthalpy includes very significant heat contributions from changes in state such as melting or vaporization. This heat is regarded as consumed as work when the system reaches a given state under current environmental conditions of pressure and temperature. Whether such heat causing reversible changes of state disappears as work is performed or is consumed as field energy sustaining a molecular scaffold that also sustains external work (e.g., lifting a weight against gravity) is irrelevant; any such energy under given conditions can reappear as heat. This was Carnot’s conclusion.
The heat added or extracted in the isothermal stages in the cycle can be considered as changing the density of field quanta supporting the molecular field in expansion or compression. Since the internal and kinetic energy in these isothermal stages is constant, it has no overall role in the work performed. Action mechanics suggests that this irrelevance for internal energy may also be true of the two adiabatic stages, although the variation in the Gibbs energy in adiabatic stage 2=>3 (
b′) of dilatation is greater than that in stage 4=>1 (
b), the difference also being equal to the maximum work performed. This is a result of the higher quantum number for the former process and the reduced quantum number in stage 4=>1 after heat is lost isothermally in stage 3=>4. Obviously, the molecular kinetic energy dictates the momentum and pressure on the piston, but this would not be possible without the intensity of the quanta exerting the primary pressure on molecules with impulses at the speed of light. This is a basic statement of action resonance theory [
4].
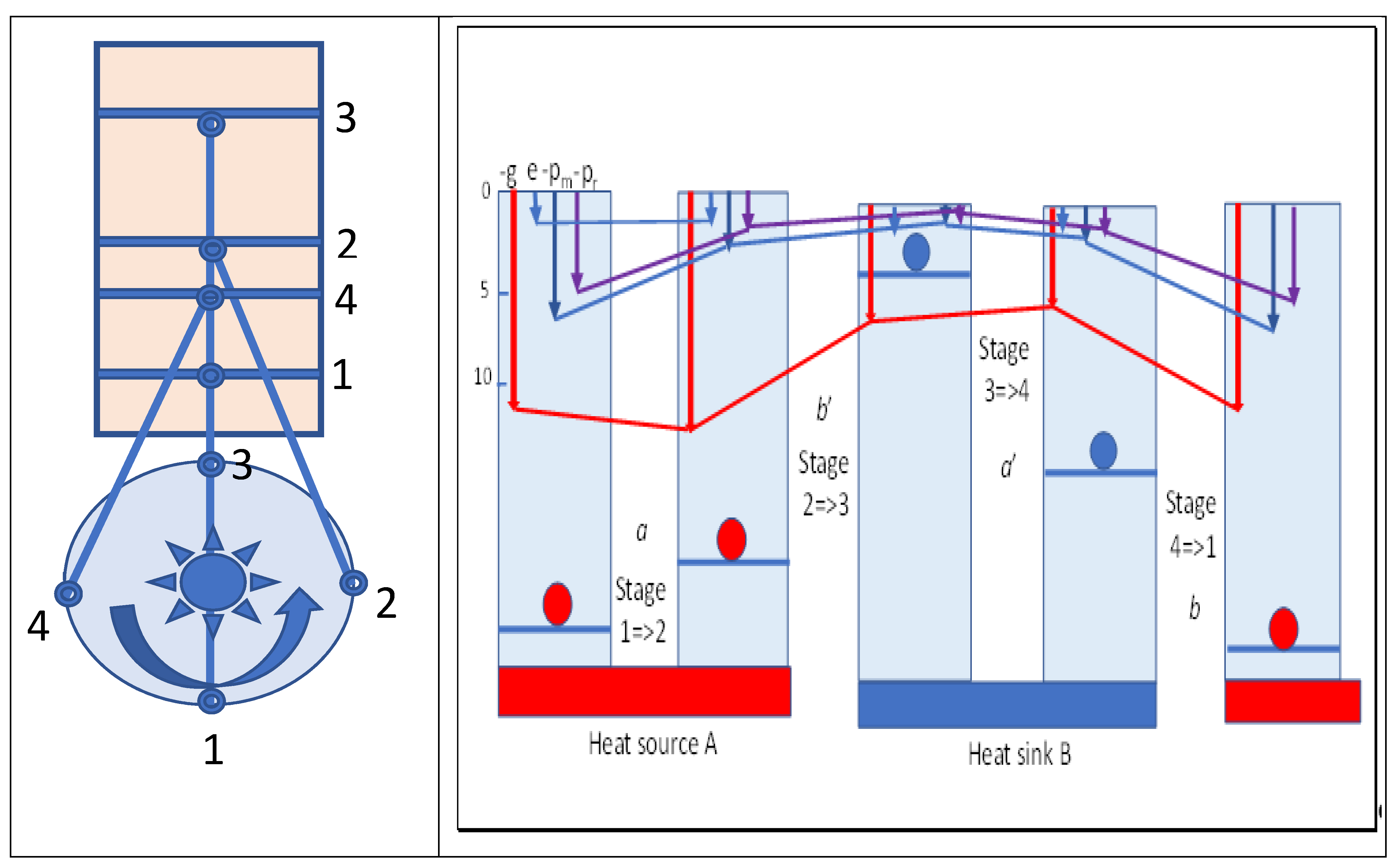
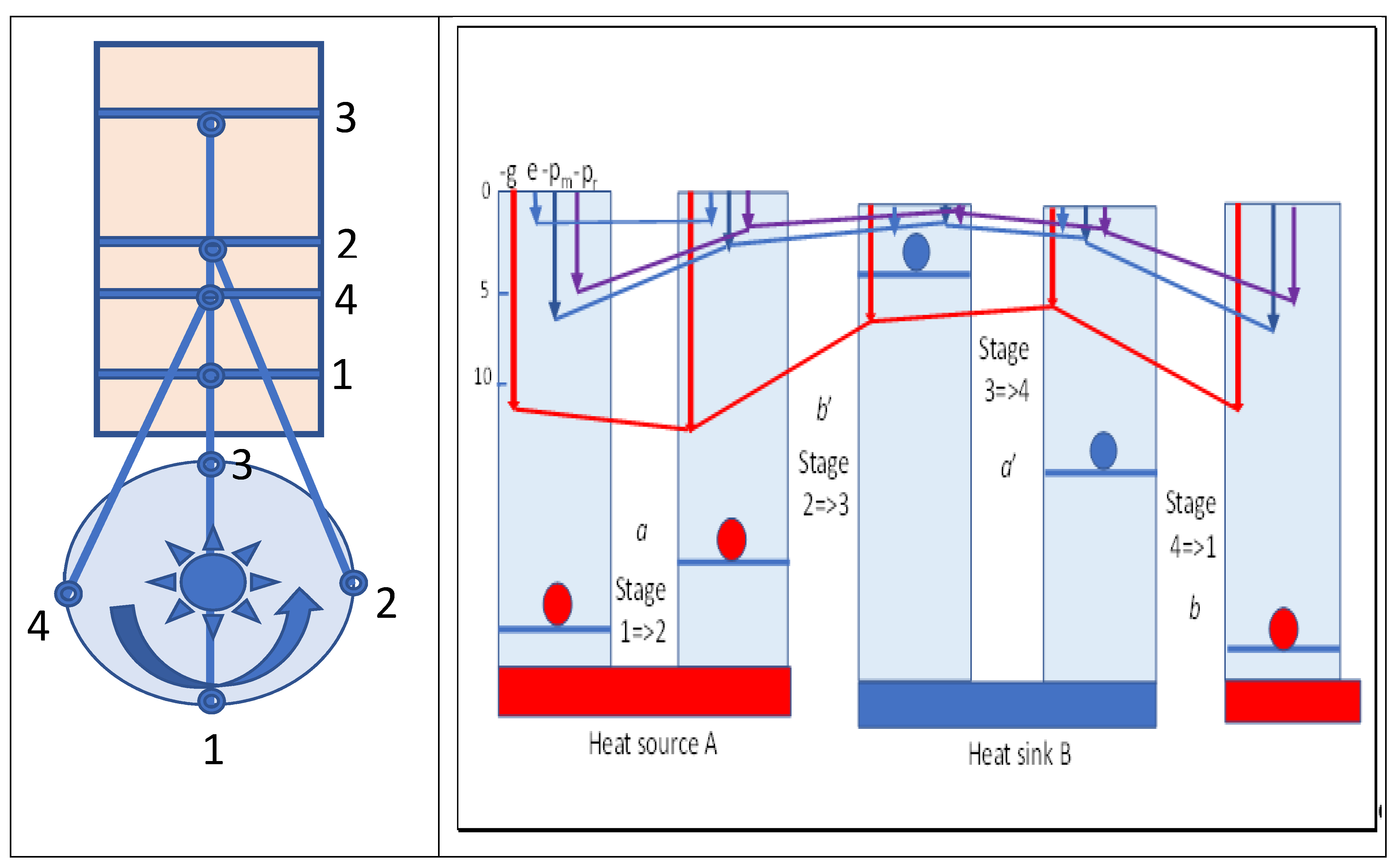
Figure 4 summarizes various variables and their changes in Carnot cycles. A flywheel is indicated that would be required to provide reversibility in the cycle. From maximum compression in stage 1, heat flows spontaneously from the hot source as marginal cooling occurs in expansion of the cylinder, as driven by the flywheel. Once the hot source is removed, adiabatic expansion doing external work continues, cooling the working fluid. The increased volume compensates for the decreased temperature with no change in action and entropy, but with a fall in Gibbs energy, which is shown as the red line in
Figure 4. Reversing these stages by reversing the flywheel would absorb heat at a low temperature from body B, compressing the volume to reach the temperature of the hot source and slightly exceeding it. Then, heat is released to the hot source body A, acting as a heat pump from cold B to hot A.
If the working fluid expands irreversibly into a vacuum following heating by body A, as in a Joule or Gay-Lussac expansion, no work would be performed so no cooling need occur. That is, the cubic action (mrv)3 would increase proportional to the increase in volume (r3), but the temperature would remain constant, with no effect on mean molecular velocity (v). While the Gibbs energy would decrease by the increase in entropic energy on expansion, the action field would contain the same amount of field energy as before, since no heat is needed given the vacuum, but with a larger quantum number of smaller quanta nt cubed. Thus, the size of the associated quanta must be diminished by the same amount as the radial separation is increased. The frequency of impulses would be decreased by the increased radial separation, but the torques developed would remain the same. Viewing kinetic energy as the statistical consequence of mean value of torques exerted in exchanges of quanta, the temperature will remain the same if well insulated from the environment at large.
This Joule expansion also has relevance for the famous Gibbs paradox, explaining realistically why combining two equal volumes of the same gas isothermally does not increase the total entropy, even if the two identical volumes of gas fully diffuse into one another by Brownian motion. Obviously, the mean negative Gibbs energy per molecule dependent on the mean action mrv, which was previously the same in both volumes of gas at the same pressure, will remain identical after the diffusion. This is an objective solution to the paradox that does not involve knowledge by an observer that all molecules are identical, assuming they are. However, if the two volumes of the same ideal gas differ isotopically in the number of nucleons, then both isotopes will have increased entropy on mixing and decreased Gibbs energy or chemical potential. The two isotopic species are considered to exist independently in their separate action fields. However, these differences are usually ignored in thermodynamics where calculations use averages.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}