
Statistical Inference for Ergodic Algorithmic Model (EAM), Applied to Hydrophobic Hydration Processes



Abstract
1. Introduction
2. Results
Statistical Inference: User-Friendly Functions
- (i)
- Subdividing the apparent partition function into thermal {T-PF} and motive {M-PF} partition functions;
- (ii)
- Determining iceberg formation or iceberg reduction in a niche within the field of motive partition function;
- (iii)
- Considering two types of water clusters WI and WII, together with free molecules WIII;
- (iv)
- Determining the number ±ξw of water clusters WI (phase change) from curvatures of the binding potential functions α) RlnKdual = {f(1/T)*g(T)} and β) RTlnKdual = {f(T)*g(lnT)}, calculated from sets of equilibrium constants measured at different temperatures and treated by thermal equivalent dilution principle;
- (v)
- assigning to WI the role of solvent (implicit solvent);
- (vi)
- attributing any change of configuration entropy of WII and WIII to the motive partition function {M-PF} (solute) and not to the thermal partition function {T-PF} of implicit solvent.
3. Discussion
3.1. Water in Thermal and Chemical Denaturation
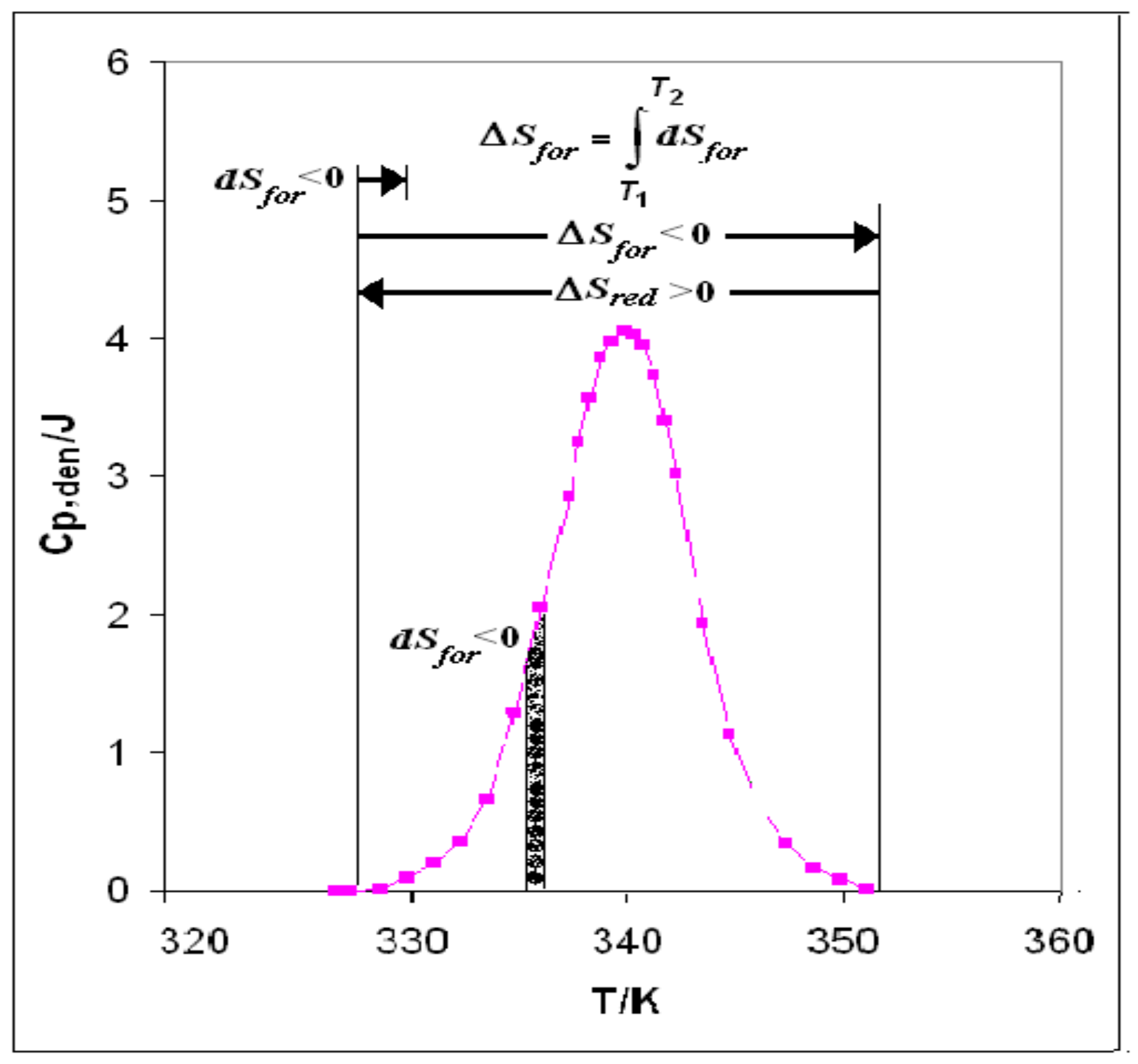
- (1)
- Start: The heat supplied to the system generates melting of some clusters of water WI to give WII +WIII, creating the niche wherein iceberg is formed. In fact, the creation of the niche with iceberg reduces the solvent (WI) volume and produces negative entropy (dSfor < 0), thus beginning to cancel the positive entropic contribution of protein folding (ΔSred + dSfor).
- (2)
- Scanning: The process of heat supply continues until the integral entropy:T2
ΔSfor = ∫ dSfor
T1cancels completely the positive entropy of folding and causes disruption of the hydrophobic bonds, at least those around the active site, that had been keeping the native protein folded. - (3)
- Final: At this stage (ΔSred + ΔSfor = 0), the whole positive entropy contribution produced by folding is cancelled: the disruption of every hydrophobic bond is completed, and the denatured state has become the stable one. The denaturation process consists in the disruption, through iceberg creation and negative entropy production, of the hydrophobic bonds that had been keeping the chains folded in the native protein.
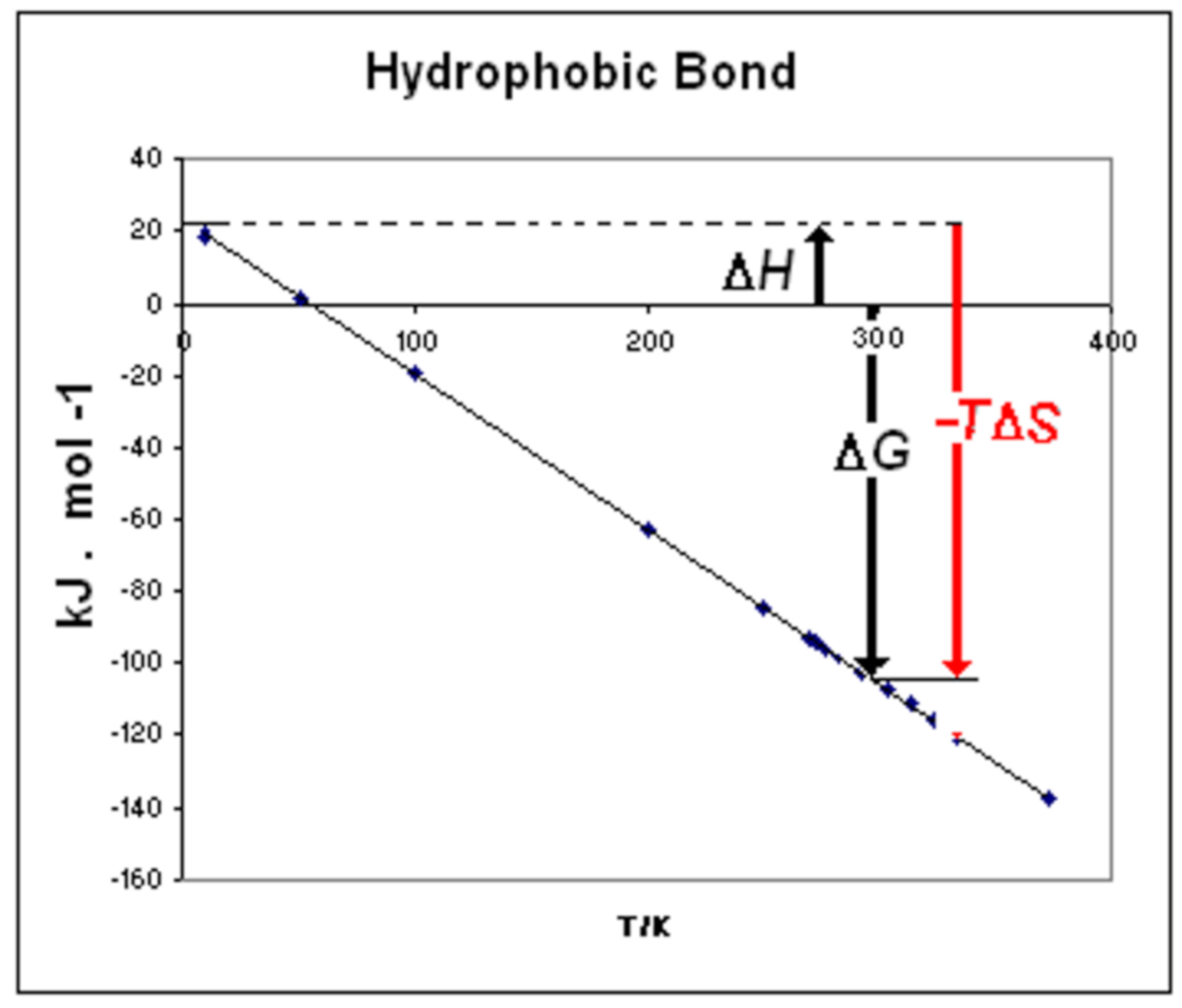
3.2. Motive Free Energy and Iceberg Formation/Reduction
3.3. Null Thermal Free Energy
3.4. Water WI, WII, WIII, and Hydrophobic Bond
3.5. Water WI: Implicit Solvent
- (i)
- is developing in the solute motive partition function {M-PF}, and
- (ii)
- is the driving force forming hydrophobic bonds between solute units.
3.6. From Ergodic Algorithmic Model to Computer Chemistry
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Symbols
- PDT = Potential Distribution Theorem
- EAM = Ergodic Algorithmic Model
- {DS-PF} = Dual Structure Partition Function
- {M-PF} = Motive Partition Function (Solute)
- {T-PF} = Thermal Partition Function (Solvent)
- Kdual = experimental equilibrium constant
- Kdual = ζth · Kmot = product partition function
- Kmot = motive equilibrium constant (solute, density entropy, REME ensemble,)
- ζth = 1 = thermal partition function (solvent, intensity entropy, NoremE ensemble)
- DMSGN = dimethionine derivative of chymotrypsinogen A
- Cp,w = 75.36 J·K−1mol−1 = molar heat capacity for liquid water
- Δsp,w = Δhp,w/T = Cp,w = 75.36 J·K−1mol−1 entropy change for WI → WII + WIII in pure water
- ΔCp,hydr = heat capacity in hydrophobic hydration processes
- Class A = hydr. hydration process with reaction A(ξwWI → ξwWII(iceberg) + ξwWIII
- Class B = hydr. hydration process with reaction B(−ξwWIII − ξwWII(iceberg) → ξwWI
- WI (solvent), WII (iceberg), and WIII = types of water
- nw = number of water molecules WIII in a hydrophobic hydration process (ξw = |nw|)
- ±ξw = (ξw = |nw|) = ±absolute pseudo-stoichiometric number of water molecules WIII
- TED = thermal equivalent dilution principle (−Rdln{X}n = n Cp,X d(lnT)
- ΔHdual = experimental enthalpy (ΔHdual = ΔHmot + ΔHth = ΔH0(ξw = 0) + ΔHfor + ξw Cp,wT)
- ΔSdual = experimental entropy (ΔSdual = ΔSmot + ΔSth = ΔS0(ξw = 0) + ΔSfor + ξw Cp.wlnT)
- ΔHth = thermal enthalpy (ΔHth = ΔCp,hydr·T) (in Class A: ΔHth > 0, in Class B: ΔHth < 0)
- ΔSth = thermal entropy (ΔSth = ΔCp,hydr·ln T). (in Class A: ΔSth > 0, in Class B: ΔSth < 0)
- ΔGth = thermal free energy (−ΔGth/T = 0)
- ΔH0 = experimental enthalpy from ΔHapp extrapolated to T = 0
- ΔHmot ≡ ΔH0 = motive enthalpy:
- in Class A: ΔHmot = ΔH0(ξw = 0) + ΔHfor
- in Class B: ΔHmot = ΔH0(ξw = 0) + ΔHred
- ΔHfor = ξw·Δhfor < 0 = enthalpy change for iceberg formation (Class A)
- Δhfor>A = −22.2 ± 0.7 kJ·mol–1·ξw–1 mean unitary (for ξw = 1) enthalpy chg. For iceberg formation
- ΔHred = enthalpy change for iceberg reduction (Class B)
- <Δhfor>B = +22.2 ± 0.7 kJ·mol–1·ξw–1 mean unitary (for ξw = 1) enthalpy change for iceberg reduction
- ΔS0 = experimental entropy from ΔSdual extrapolated to lnT = 0
- ΔSmot ≡ ΔS0 = motive entropy:
- in Class A: ΔSmot = ΔS0(ξw = 0) + ΔSfor
- in Class B: ΔSmot = ΔS0(ξw = 0) + ΔSred
- ΔSfor = ξw Δsfor < 0 = entropy change for iceberg formation (Class A)
- <Δsfor> A = −445 ± 3 J·K−1·mol−1·ξw−1, mean unitary (for ξw = 1) entropy change for iceberg formation
- ΔSred = ξw Δsred > 0 = entropy change for iceberg reduction (Class B)
- <Δsred> B = +445 ± 3 J·K−1·mol−1·ξw−1. mean unitary (for ξw = 1) entropy change for iceberg reduction
- ΔGmot = motive free energy (ΔGmot = ΔHmot –TΔSmot ≡ ΔH0–TΔS0)
- –ΔG°/RT = ln K
- R ln Kdual = f(1/T)*g(T), (α) observed convoluted Binding Potential Function in entropy unit
- RTlnKdual = f(T)*g(lnT), (β) observed convoluted Binding Potential Function in enthalpy unit
- ΔHj = difference between enthalpy levels in reacting REME Ensemble
- ΔVsolvent = change of solvent volume
- VWI = volume of one unit of water cluster WI
- ΔVsolvent > 0 = −Vcav = change of solvent volume (Vcav = −ξwVWI = iceberg reduction)
- R = 8.31451 J·K−1· mol−1, gas constant
- ΔH0(ξw = 0) = ΔHfor extrapolated to null iceberg
- ΔS0(ξw = 0) = ΔSfor extrapolated to null iceberg
- ξw–1 = unitary function, for ξw = 1
- Td = denaturation temperature
- TH = temperature at ΔHapp = 0 (Tmin in Class A or Tmax in Class B)
- TS = temperature at ΔSapp = 0 (Tmin in Class A or Tmax in Class B)
- NoremE = Non-reacting molecule Ensemble (small m = molecule)
- REME = Reacting Mole Ensemble (capital M = Mole)
- niche = portion of water WI losing rigidity to form water WII, free water WIII and iceberg
References
- Fisicaro, E.; Compari, C.; Braibanti, A. Entropy/enthalpy compensation: Hydrophobic effect, micelles and protein complexes. Phys. Chem. Chem. Phys. 2004, 6, 4156–4166. [Google Scholar] [CrossRef]
- Fisicaro, E.; Compari, C.; Duce, E.; Biemmi, M.; Peroni, M.; Braibanti, A. Thermodynamics of micelle formation in water, hydrophobic processes and surfactant self-assemblies. Phys. Chem. Chem. Phys. 2008, 10, 3903–3914. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, E.; Compari, C.; Braibanti, A. Hydrophobic hydration processes. General thermodynamic model by thermal equivalent dilution determinations. Biophys. Chem. 2010, 151, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, E.; Compari, C.; Braibanti, A. Hydrophobic hydration processes. Biophys. Chem. 2011, 156, 51–67. [Google Scholar] [CrossRef]
- Fisicaro, E.; Compari, C.; Braibanti, A. Hydrophobic Hydration Processes. I: Dual-Structure Partition Function for Biphasic Aqueous Systems. ACS Omega 2018, 3, 15043–15065. [Google Scholar] [CrossRef]
- Lumry, R. Bioenergetics and Thermodynamics: Model Systems; Braibanti, A., Ed.; Reidel: Dordrecht, The Netherlands, 1980; p. 405. [Google Scholar]
- Braibanti, A.; Fisicaro, E.; Compari, C. Thermal Equivalent Dilution. J. Phys. Chem. B 1998, 102, 8357–8359. [Google Scholar] [CrossRef]
- Ben Naim, A. Solvation Thermodynamics; Plenum Press: New York, NY, USA, 1987. [Google Scholar]
- Beck, T.T.; Paulatis, M.E.; Pratt, L.R. The Potential Distribution Theorem and Models of Molecular Solutions; Cambridge University Press: New York, NY, USA, 2012. [Google Scholar]
- Shiao, D.F.; Lumry, R.; Fahey, J. Chymotrypsinogen family of proteins. XI. Heat-capacity changes accompanying reversible thermal unfolding of proteins. J. Am. Chem. Soc. 1971, 93, 2024–2035. [Google Scholar] [CrossRef]
- Lum, K.; Chandler, D.; Weeks, J.D. Hydrophobicity at Small and Large Length Scales. J. Phys. Chem. B 1999, 103, 4570–4577. [Google Scholar] [CrossRef]
- Ten Wolde, P.R.; Sun, S.X.; Chandler, D. Model of a fluid at small and large length scales and the hydrophobic effect. Phys. Rev. E 2002, 65, 011201. [Google Scholar] [CrossRef]
- Fisicaro, E.; Compari, C.; Braibanti, A. Hydrophobic Hydration Processes: Intensity Entropy and Null Thermal Free Energy and Density Entropy and Motive Free Energy. ACS Omega 2019, 4, 19526–19547. [Google Scholar] [CrossRef]
- Lee, B.; Graziano, G. A Two-State Model of Hydrophobic Hydration That Produces Compensating Enthalpy and Entropy Changes. J. Am. Chem. Soc. 1996, 118, 5163–5168. [Google Scholar] [CrossRef]
- Benzinger, T.H. Thermodynamics, chemical reactions, and molecular biology. Nature 1971, 229, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. A View of the Hydrophobic Effect. J. Phys. Chem. B 2002, 106, 521–533. [Google Scholar] [CrossRef]
- Privalov, P.L.; Griko, Y.V.; Venyaminov, S.Y.; Kutyshenko, V.P. Cold denaturation of myoglobin. J. Mol. Biol. 1986, 190, 487–498. [Google Scholar] [CrossRef]
- Prabhu, N.V.; Sharp, K.A. Heat capacity in proteins. Annu. Rev. Phys. Chem. 2005, 56, 521–548. [Google Scholar] [CrossRef] [PubMed]
- Wyman, J.; Gill, S.J. Binding and Linkage: Functional Chemistry of Biological Macromolecules; University Science Books: Mill Valley, CA, USA, 1990. [Google Scholar]
- Hayne, D.T. Biological Thermodynamics; Cambridge University Press: Cambridge, UK, 2001. [Google Scholar]
- Maibaum, L.; Dinner, A.R.; Chandler, D. Micelle formation and the hydrophobic effect. J. Phys. Chem. B 2004, 108, 6778–6781. [Google Scholar] [CrossRef]
- Edsall, J.T.; Gutfreund, H. Biothermodynamics: The Study of Biochemical Processes at Equilibrium; Wiley: Chichester, UK, 1983. [Google Scholar]
- Tanford, C. The Hydrophobic Effect: Formation of Micelles and Biological Membranes; John Wiley & Sons Inc.: New York, NY, USA, 1973. [Google Scholar]
- Irudayam, S.J.; Henchman, R.H. Solvation theory to provide a molecular interpretation of the hydrophobic entropy loss of noble-gas hydration. J. Phys. Condens. Matter 2010, 22, 284108/1–284108/17. [Google Scholar] [CrossRef]
- Pratt, L.R.; Laviolette, R.A. Quasi-chemical theories of associated liquids. Mol. Phys. 1998, 94, 909–915. [Google Scholar]
- Pratt, L.R.; LaViolette, R.A.; Gomez, M.A.; Gentile, M.E. Quasi-Chemical Theory for the Statistical Thermodynamics of the Hard-Sphere Fluid. J. Phys. Chem. B 2001, 105, 11662–11668. [Google Scholar] [CrossRef]
- Talhout, R.; Villa, A.; Mark, A.E.; Engberts, J.B.F.N. Understanding Binding Affinity: A Combined Isothermal Titration Calorimetry/Molecular Dynamics Study of the Binding of a Series of Hydrophobically Modified Benzamidinium Chloride Inhibitors to Trypsin. J. Am. Chem. Soc. 2003, 125, 10570–10579. [Google Scholar] [CrossRef]
- Rezus, Y.L.A.; Bakker, H.J. Observation of Immobilized Water Molecules around Hydrophobic Groups. Phys. Rev. Lett. 2007, 99, 148301. [Google Scholar] [CrossRef] [PubMed]
- Qvist, J.; Halle, B. Thermal Signature of Hydrophobic Hydration Dynamics. J. Am. Chem. Soc. 2008, 130, 10345–10353. [Google Scholar] [CrossRef] [PubMed]
- Laage, D.; Stirnemann, G.; Hynes, J.T. Why Water Reorientation Slows without Iceberg Formation around Hydrophobic Solutes. J. Phys. Chem. B 2009, 113, 2428–2435. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Motive Function | Equation | R2 Factor | At Null Iceberg (ξw = 0) | St. dev. |
|---|---|---|---|---|
| ΔHmot = | +13.124 − 31687 nw | 0.983 | ΔH0(ξw = 0) = −31.7 kJ·mol−1 | 2.1 |
| ΔSmot = | −82.681 − 445.44 nw | 0.993 | ΔS0(ξw = 0) = −82.7 J·K− 1·mol−1 | 5.4 |
| Transformations | |||
| Class A | A(ξwWI → ξwWII(iceberg) + ξwWIII) | iceberg formation | |
| Class B | B(−ξwWIII − ξwWII (iceberg) → ξwWI) | iceberg reduction | |
| Thermodynamic Functions | |||
| Class A | |||
| Apparent | Motive | Iceberg | Thermal |
| ΔHdual = ΔH0(ξw = 0) + ΔHfor + ΔHth | ΔHmot = ΔH0(ξw = 0) + ΔHfor | ΔHfor = ξw·Δhfor < 0 | ΔHth = +ξw Cp,w T |
| ΔSdual = ΔS0(ξw = 0) + ΔSfor + ΔSth | Δsmot = ΔS0(ξw = 0) + ΔSfor | ΔSfor = ξw·Δsfor < 0 | ΔSth = +ξw Cp,w lnT |
| Class B | |||
| Apparent | Motive | Iceberg | Thermal |
| ΔHdual = ΔH0(ξw = 0) + ΔHred + ΔHth | ΔHmot = ΔH0(ξw = 0) + ΔHred | ΔHred = ξw·Δhred > 0 | ΔHth = −ξw Cp,w T |
| ΔSdual = ΔS0(ξw = 0) + ΔSred + ΔSth | ΔSmot = ΔS0(ξw = 0) + ΔSred | ΔSred = ξw·Δsred > 0 | ΔSth = −ξw Cp,w lnT |
| Class A. Unitary Enthalpy Function: Δhfor | |||
| Process | Δhfor | Unit | ξw range |
| Gas dissolut. | −21.6 | kJ·mol−1·ξw−1 | 2–6 |
| Liquid dissol. | −23.3 | kJ·mol−1·ξw−1 | 2.7–5.4 |
| Protein denat. | −22.1 | kJ·mol−1·ξw−1 | 80–140 |
| Carbox Proton.(*) | −21.8 | kJ·mol−1·ξw−1 | 1.8–2.3 |
| Class A. Unitary Entropy Function: Δsfor | |||
| Process | Δsfor | Unit | ξw range |
| Gas dissolut. | −445.4 | J·K−1mol−1·ξw−1 | 2–6 |
| Gas (Privalov) | −450 | J·K−1mol−1·ξw−1 | 2–6 |
| Liquid dissol. | −447 | J·K−1mol−1·ξw−1 | 2.7–5.4 |
| Protein denat. | −428.5 | J·K−1mol−1·ξw−1 | 80–140 |
| Carbox Proton.(*) | −442.6 | J·K−1mol−1·ξw−1 | 1.8–2.3 |
| Class B. Unitary Enthalpy Function: Δhred | |||
| Process | Δhred | Unit | ξw range |
| Micelle | +23.2 | kJ·mol−1·ξw−1 | 4–19 |
| Bio-complx | +24.3 | kJ·mol−1·ξw−1 | 19–189 |
| Benz.Cl-tryps. | +23.41 | kJ·mol−1·ξw−1 | 5.3–11.3 |
| Class B. Unitary Entropy Function: Δsred | |||
| Process | Δhred | Unit | ξw range |
| Micelle | −428±33 | J·K−1mol−1·ξw−1 | 4–19 |
| Bio-complx | −436.2 | J·K−1mol−1·ξw−1 | 19–189 |
| Benz.Cl-tryps. | −434.4 | J·K−1mol−1·ξw−1 | 5.3–11.3 |
| Analysis within Classes | ||
| Class A: iceberg formation | Unit | Relative error |
| <Δhfor>A = −22.7 ± 0.7 | kJ·mol−1 ·ξw−1 | ±3.1% |
| <Δsfor>A = −445 ± 3 | J·K−1·mol−1·ξw−1 | ±0.7% |
| Class B: iceberg reduction | Unit | Relative error |
| <Δhred>B = +23.7 ± 0.6 | kJ·mol−1 ·ξw−1 | ±2.51% |
| <Δsred> B = +432 ± 4 | J·K−1·mol−1·ξw−1 | ±0.9% |
| Comparison among Classes | ||
| Enthalpy | Entropy | |
| <Δhfor>A = −22.7 ± 0.7 kJ·mol−1·ξw−1 | <Δsfor> A = −445 ± 3 J·K−1·mol−1·ξw−1 | |
| <Δhred>B = +23.7 ± 0.6 kJ·mol−1·ξw−1 | <Δsred> B = +432 ± 4 J·K−1·mol−1·ξw−1 | |
| mean abs.value <Δh>A,B = 22.95 ± 0.75 | mean abs.value <Δs> A,B = 438.5 ± 6.5 | |
| mean sd: (0.72 + 0.62) 1/2 = 0.92 | mean sd: (32 + 42) 1/2 = 5 | |
| Student’s ratio: 0.75/0.92 = 0.815 | Student’s ratio: 6.5/5 = 1.3 | |
| Hypothesis: absolute values in Class A and B are equal: hypothesis accepted (Mean in Class A = mean in Class B with sign reversed) | ||
| Process | Motive Function | Disaggregation Equation | ΔGmot(298) | Unit | ξw Range | <ξw> Mean |
|---|---|---|---|---|---|---|
| Class A | ||||||
| Gas Dissolut. | ΔHmot | −17.7 − 21.6 ξw | kJ·mol−1 | |||
| ΔSmot | −86.4(!) − 445.4ξw | J·K−1mol−1 | ||||
| ΔGmot(298) | 8.04(**) + 111.133ξw | +452.4 | kJ·mol−1 | 2–6 | 4 | |
| Liq. Dissolut. | ΔHmot | +4.6 − 23.3 ξw | kJ·mol−1 | |||
| ΔSmot | −0.5(!!) − 447 ξw | J·K−1mol−1 | ||||
| ΔGmot(298) | 4.74 (**) + 109.9 ξw | +437.9 | kJ·mol−1 | 2.7–5.4 | 4 | |
| Protein denat. | ΔHmot | +211.82 − 22.5 ξw | kJ·mol−1 | |||
| ΔSmot | +415 − 428.5 ξw | J·K−1mol−1 | ||||
| ΔGmot(298) | −88.15(**) + 105.04ξw | +8319 | kJ·mol−1 | 80–140 | 120 | |
| Protonation | ΔHmot | +0.1 − 21.8 ξw | kJ·mol−1 | |||
| ΔSmot | +104 − 442.6 ξw | J K−1·mol−1 | ||||
| ΔGmot(298) | −30.9(**) + 104.6ξw | +1783 | kJ·mol−1 | 1.8–2.3 | 2.1 | |
| Class B | ||||||
| Protein fold. | ΔHmot | −211.82 + 22.5ξw | kJ·mol−1 | |||
| ΔSmot | −415.8 + 424.2ξw | J K−1·mol−1 | ||||
| ΔGmot(298) | +88.15 − 103.9ξw | −8281 | kJ·mol−1 | 80–140 | 120 | |
| Micelle form. | ΔHmot | −3.97 + 23.13 ξw | kJ·mol−1 | |||
| ΔSmot | +10.2 + 428 ξw | J K−1·mol−1 | ||||
| ΔGmot(298) | –7.01(**) − 104.4ξw | –1569 | kJ·mol−1 | 4–19 | 15 | |
| Deprotonat. | ΔHmot | –0.1 − 21.8ξw | kJ·mol−1 | |||
| ΔSmot | −104 + 442.6ξw | J K−1·mol−1 | ||||
| ΔGmot(298) | +30.9(**) − 104.6ξw | −1783 | kJ·mol−1 | 1.8–2.3 | 2.1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisicaro, E.; Compari, C.; Braibanti, A. Statistical Inference for Ergodic Algorithmic Model (EAM), Applied to Hydrophobic Hydration Processes. Entropy 2021, 23, 700. https://doi.org/10.3390/e23060700
Fisicaro E, Compari C, Braibanti A. Statistical Inference for Ergodic Algorithmic Model (EAM), Applied to Hydrophobic Hydration Processes. Entropy. 2021; 23(6):700. https://doi.org/10.3390/e23060700
Chicago/Turabian StyleFisicaro, Emilia, Carlotta Compari, and Antonio Braibanti. 2021. "Statistical Inference for Ergodic Algorithmic Model (EAM), Applied to Hydrophobic Hydration Processes" Entropy 23, no. 6: 700. https://doi.org/10.3390/e23060700
APA StyleFisicaro, E., Compari, C., & Braibanti, A. (2021). Statistical Inference for Ergodic Algorithmic Model (EAM), Applied to Hydrophobic Hydration Processes. Entropy, 23(6), 700. https://doi.org/10.3390/e23060700