Application of Biological Domain Knowledge Based Feature Selection on Gene Expression Data



Abstract
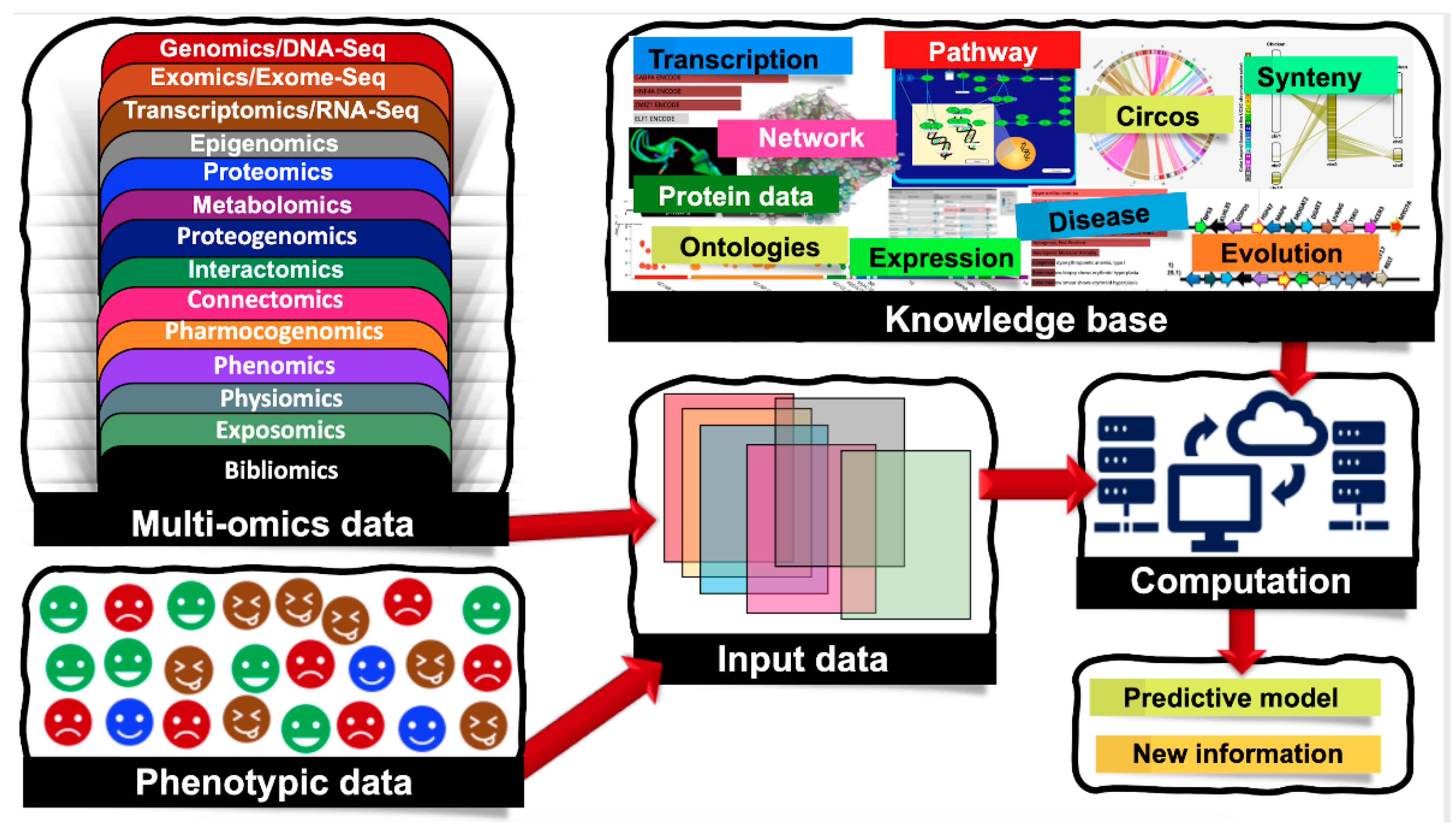
1. Introduction
2. Gene Selection Approaches for Gene Expression Datasets
2.1. Traditional Gene Selection
2.2. Integrative Gene Selection
3. Grouping and Ranking of the Genes for Classification Problem
3.1. Traditional Approach of Feature/Gene Selection Using a Classifier
SVM-RFE (Support Vector Machines with Recursive Feature Elimination)
- Train the classifier on the given data;
- Assign rank for each feature as its weight;
- Remove one feature or percentage (10%) with the smallest weight;
- Repeat steps 1–3 until reaching a predefined number of genes.
3.2. Biological Domain Knowledge Based ML Approaches
3.2.1. SVM-RCE (Support Vector Machines with Recursive Cluster Elimination)
3.2.2. SVM-RNE (Support Vector Machines with Recursive Network Elimination)
3.2.3. MaTE
3.2.4. CogNet
3.2.5. MiRcorrNet
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Zitnik, M.; Nguyen, F.; Wang, B.; Leskovec, J.; Goldenberg, A.; Hoffman, M.M. Machine learning for integrating data in biology and medicine: Principles, practice, and opportunities. Inf. Fusion 2019, 50, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. Poznan Pol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Fiala, C.; Diamandis, E.P. Mutations in normal tissues—some diagnostic and clinical implications. BMC Med. 2020, 18, 283. [Google Scholar] [CrossRef]
- Sheng, Q.; Zhao, S.; Li, C.-I.; Shyr, Y.; Guo, Y. Practicability of detecting somatic point mutation from RNA high throughput sequencing data. Genomics 2016, 107, 163–169. [Google Scholar] [CrossRef]
- Veer, L.J.V.; Laura, J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002. [Google Scholar] [CrossRef]
- Chou, C.; Chang, N.; Shrestha, S.; Hsu, S.; Lin, Y.; Lee, W.; Yang, C.; Hong, H.; Wei, T.; Tu, S.; et al. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016, 44. [Google Scholar] [CrossRef]
- Bellazzi, R.; Zupan, B. Towards knowledge-based gene expression data mining. J. Biomed. Inform. 2007, 40, 787–802. [Google Scholar] [CrossRef]
- Falcon, S.; Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 2007, 23, 257–258. [Google Scholar] [CrossRef]
- Consortium, T.G.O. Gene ontology: Tool for the unification of biology. Gene Ontol. Consort. 2000, 25, 25–29. [Google Scholar]
- Kustra, R.; Zagdanski, A. Incorporating Gene Ontology in Clustering Gene Expression Data. In Proceedings of the 19th IEEE Symposium on Computer-Based Medical Systems (CBMS’06), Salt Lake City, UT, USA, 22–23 June 2006; pp. 555–563. [Google Scholar] [CrossRef]
- Azuaje, F.; Dopazo, J. (Eds.) Data Analysis and Visualization in Genomics and Proteomics; John Wiley: Hoboken, NJ, USA, 2005. [Google Scholar]
- Perscheid, C.; Grasnick, B.; Uflacker, M. Integrative Gene Selection on Gene Expression Data: Providing Biological Context to Traditional Approaches. J. Integr. Bioinform. 2019, 16. [Google Scholar] [CrossRef] [PubMed]
- Bellman, R. Adaptive Control Processes: A Guided Tour. (A RAND Corporation Research Study); Princeton University Press: London, UK, 1961. [Google Scholar]
- Lazar, C.; Taminau, J.; Meganck, S.; Steenhoff, D.; Coletta, A.; Molter, C.; de Schaetzen, V.; Duque, R.; Bersini, H.; Nowe, A. A survey on filter techniques for feature selection in gene expression microarray analysis. IEEEACM Trans. Comput. Biol. Bioinform. IEEE ACM 2012, 9, 1106–1119. [Google Scholar] [CrossRef]
- Inza, I.; Larrañaga, P.; Blanco, R.; Cerrolaza, A.J. Filter versus wrapper gene selection approaches in DNA microarray domains. Artif. Intell. Med. 2004, 31, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Fang, O.H.; Mustapha, N.; Sulaiman, M.N. An integrative gene selection with association analysis for microarray data classification. Intell. Data Anal. 2014, 18, 739–758. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]
- Qi, J.; Tang, J. Integrating gene ontology into discriminative powers of genes for feature selection in microarray data. In Proceedings of the 2007 ACM symposium on Applied computing—SAC’07, Seoul, Korea, 11–15 March 2007; p. 430. [Google Scholar] [CrossRef]
- Papachristoudis, G.; Diplaris, S.; Mitkas, P.A. SoFoCles: Feature filtering for microarray classification based on Gene Ontology. J. Biomed. Inform. 2010, 43, 1–14. [Google Scholar] [CrossRef]
- Raghu, V.K.; Ge, X.; Chrysanthis, P.K.; Benos, P.V. Integrated Theory-and Data-Driven Feature Selection in Gene Expression Data Analysis. In Proceedings of the 2017 IEEE 33rd International Conference on Data Engineering (ICDE), San Diego, CA, USA, 19–22 April 2017; pp. 1525–1532. [Google Scholar] [CrossRef]
- Quanz, B.; Park, M.; Huan, J. Biological pathways as features for microarray data classification. In 2nd International Workshop on Data and Text Mining in Bioinformatics—DTMBIO’08; ACM Press: Napa Valley, CA, USA, 2008; p. 5. [Google Scholar] [CrossRef]
- Mitra, S.; Ghosh, S. Feature Selection and Clustering of Gene Expression Profiles Using Biological Knowledge. IEEE Trans. Syst. Man Cybern. Part C Appl. Rev. 2012, 42, 1590–1599. [Google Scholar] [CrossRef]
- Ghosh, S.; Mitra, S. Gene selection using biological knowledge and fuzzy clustering. In Proceedings of the 2012 IEEE International Conference on Fuzzy Systems, Brisbane, Australia, 10–15 June 2012; pp. 1–9. [Google Scholar] [CrossRef]
- Acharya, S.; Saha, S.; Nikhil, N. Unsupervised gene selection using biological knowledge: Application in sample clustering. BMC Bioinform. 2017, 18, 513. [Google Scholar] [CrossRef]
- Yousef, M.; Jung, S.; Showe, L.C.; Showe, M.K. Recursive Cluster Elimination (RCE) for classification and feature selection from gene expression data. BMC Bioinform. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Abdallah, L.; Allmer, J. maTE: Discovering expressed interactions between microRNAs and their targets. Bioinformatics 2019, 35, 4020–4028. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Ketany, M.; Manevitz, L.; Showe, L.C.; Showe, M.K. Classification and biomarker identification using gene network modules and support vector machines. BMC Bioinform. 2009, 10, 337. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.; Niekerk, A.V. Feature clustering and ranking for selecting stable features from high dimensional remotely sensed data. Int. J. Remote Sens. 2018, 39, 8934–8949. [Google Scholar] [CrossRef]
- Lazzarini, N.; Bacardit, J. RGIFE: A ranked guided iterative feature elimination heuristic for the identification of biomarkers. BMC Bioinform. 2017. [Google Scholar] [CrossRef]
- Deshpande, G.; Li, Z.; Santhanam, P.; Coles, C.D.; Lynch, M.E.; Hamann, S.; Hu, X. Recursive cluster elimination based support vector machine for disease state prediction using resting state functional and effective brain connectivity. PLoS ONE 2010, 5, e14277. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, L.; Chen, G. Joint Covariate Detection on Expression Profiles for Identifying MicroRNAs Related to Venous Metastasis in Hepatocellular Carcinoma. Sci. Rep. 2017, 7, 5349. [Google Scholar] [CrossRef]
- Johannes, M.; Brase, J.; Fröhlich, H.; Gade, S.; Gehrmann, M.; Fälth, M.; Sültmann, H.; Beißbarth, T. Integration of pathway knowledge into a reweighted recursive feature elimination approach for risk stratification of cancer patients. Bioinformatics 2010. [Google Scholar] [CrossRef]
- Yousef, M.; Bakir-Gungor, B.; Jabeer, A.; Goy, G.; Qureshi, R.; Showe, L.C. Recursive Cluster Elimination based Rank Function (SVM-RCE-R) implemented in KNIME. F1000Research 2020, 9, 1255. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME—The Konstanz Information Miner. SIGKDD Explor. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Zycinski, G.; Barla, A.; Squillario, M.; Sanavia, T.; di Camillo, B.; Verri, A. Knowledge Driven Variable Selection (KDVS)—A new approach to enrichment analysis of gene signatures obtained from high-throughput data. Source Code Biol. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Ulgen, E.; Ozisik, O.; Sezerman, O.U. CogNet: Classification of gene expression data based on ranked active-subnetwork-oriented KEGG pathway enrichment analysis. PeerJ 2020. [Google Scholar] [CrossRef]
- Yousef, M.; Goy, G.; Mitra, R.; Eischen, C.M.; Amhar, J.; Burcu, B. miRcorrNet: Integrated microRNA Gene Expression and mRNA Expression Based Machine Learning combined with Features Grouping and Ranking. Unpublished Work. 2020; in submit. [Google Scholar]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster Analysis and Display of Genome-Wide Expression Patterns; National Academy of Sciences: Washington, DC, USA, 1998; Volume 95. [Google Scholar]
- Wang, J.; Li, H.; Zhu, Y.; Yousef, M.; Nebozhyn, M.; Showe, M.; Showe, L.; Xuan, J.; Clarke, R.; Wang, Y. VISDA: An open-source caBIGTM analytical tool for data clustering and beyond. Bioinformatics 2007, 23. [Google Scholar] [CrossRef]
- Guyon, J.W.I.; Stephen, B.; Vladimir, V. Gene Selection for Cancer Classification using Support Vector Machines, Machine Learning. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Nacu, S.; Critchley-Thorne, R.; Lee, P.; Holmes, S. Gene expression network analysis and applications to immunology. Bioinformatics 2007, 23, 850–858. [Google Scholar] [CrossRef]
- Sain, S.R.; Vapnik, V.N. The Nature of Statistical Learning Theory. Technometrics 1996. [Google Scholar] [CrossRef]
- Duan, K.-B.; Rajapakse, J.C.; Wang, H.; Azuaje, F. Multiple SVM-RFE for Gene Selection in Cancer Classification With Expression Data. IEEE Trans. Nanobiosci. 2005, 4, 228–234. [Google Scholar] [CrossRef]
- Das, P.; Roychowdhury, A.; Das, S.; Roychoudhury, S.; Tripathy, S. sigFeature: Novel Significant Feature Selection Method for Classification of Gene Expression Data Using Support Vector Machine and t Statistic. Front. Genet. 2020, 11, 247. [Google Scholar] [CrossRef]
- Xu, C.; Jackson, S.A. Machine learning and complex biological data. Genome Biol. 2019, 20, 76. [Google Scholar] [CrossRef]
- Ulgen, E.; Ozisik, O.; Sezerman, O.U. PathfindR: An R package for comprehensive identification of enriched pathways in omics data through active subnetworks. Front. Genet. 2019, 10, 858. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Tool Name | Incorporated Biological Knowledge | Methodology | Advantage/Disadvantage | Ref |
|---|---|---|---|---|
| N/A | GO | Rank the genes uses information gain (IF) incorporated with Gene Ontology GO terms | A: The novelty of this work is to evaluate genes based on not only their individual discriminative powers but also the powers of GO terms that annotate them. | [21] |
| N/A | GO | χ2, ReliefF, or IG | A: Including biological knowledge in the gene selection process improves results. | [22] |
| N/A | Combines KEGG and GO terms | Utilizes graphical causal modeling IG as an initial filter search for GO and KEGG annotations’ frequent items | A: Method is capable of intelligently selecting genes for learning effective causal networks. D: No significant improvement in accuracy. | [18] |
| N/A | KEGG, DisGeNET, and further genetic meta information | Gene–disease association score from DisGeNET Gene distance metrics | [23] | |
| N/A | KEGG pathways | Uses these pathways as features for further pattern mining | A: Reduce the dimension of the data by transforming to KEGG feature space. A: Improved performance over different traditional approaches. | [24] |
| N/A | Gene ontology (GO) | Randomized search (CLARANS) | A: Reducing the dimension dramatically. | [25] |
| SVM-RCE | Genes related are correlated | SVM and K-means | A: Discover significant of clusters. D: Might lose important genes because they were in lower-ranked clusters. | [28,36] |
| SVM-RNE | GXNA for creating subnetworks from gene expression | SVM, GXNA | A: Reducing the dimension of the data by considering subnetworks. D: The subnetworks are created as a prediction of the gene expressions data. | [30] |
| maTE | microRNA genes targets | Random forest groups the genes that associated with microRNA | A: A novel approach of integrating microRNA into gene expression. D: The size of the groups might be large and might rank these groups highly as a result of that. | [29] |
| CogNet | Random forest, based on pathFindR tool | A: Improve the results of the pathFindR tool by ranking its groups. | [39] | |
| miRcorrNet | Random forest based on the correlation with miRN expressions | A: Novel approach for integrating miRNE and mRNA expressions using machine learning. | [40] |
| MicroRNA Group Name | Target Genes List |
|---|---|
| HSA-MIR-147A | VEGFA, ACVR1C, MCM3, NDUFA4, PSMA3, HIF3A, SLC22A3, MCM3, NDUFA4, PSMA3, HIF3A, VEGFA, ACVR1C, MCM3, NDUFA4, PSMA3, HIF3A, SLC22A3 |
| HSA-MIR-18B-5P | ESR1, MDM2, CTGF, TNRC6B, HIF1A, SMAD2, FOXN1, IGF1, IGF1, CTGF, HIF1A, SMAD2, FOXN1, ESR1, MDM2, CTGF, TNRC6B, HIF1A, SMAD2, FOXN1, IGF1, IGF1 |
| HSA-MIR-19B-3P | BACE1, PTEN, PTEN, PTEN, ATXN1, HIPK3, ARID4B, MYLIP, ESR1, KAT2B, SOCS1, BCL2L11, BCL2L11, TGFBR2, TGFBR2, BMPR2, BMPR2, TLR2, PPP2R5E, PPP2R5E, CYP19A1, GCM1, HIPK1, SMAD4, MYCN, MXD1, BCL3, DNMT1, TNFAIP3, PKNOX1, MTUS1, PITX1, PTEN, PTEN, PTEN, ATXN1, ESR1, NCOA3, KAT2B, SOCS1, TGFBR2, BMPR2, CUL5, TLR2, HIPK1, MXD1, BCL3, TNFAIP3, MTUS1, PITX1, BACE1, PTEN, PTEN, PTEN, PTEN, ATXN1, HIPK3, ARID4B, MYLIP, ESR1, NCOA3, KAT2B, SOCS1, BCL2L11, BCL2L11, TGFBR2, TGFBR2, BMPR2, BMPR2, CUL5, TLR2, PPP2R5E, PPP2R5E, CYP19A1, GCM1, HIPK1, SMAD4, MYCN, MXD1, BCL3, DNMT1, TNFAIP3, PKNOX1, MTUS1, PITX1 |
| HSA-MIR-210-5P | CFB |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousef, M.; Kumar, A.; Bakir-Gungor, B. Application of Biological Domain Knowledge Based Feature Selection on Gene Expression Data. Entropy 2021, 23, 2. https://doi.org/10.3390/e23010002
Yousef M, Kumar A, Bakir-Gungor B. Application of Biological Domain Knowledge Based Feature Selection on Gene Expression Data. Entropy. 2021; 23(1):2. https://doi.org/10.3390/e23010002
Chicago/Turabian StyleYousef, Malik, Abhishek Kumar, and Burcu Bakir-Gungor. 2021. "Application of Biological Domain Knowledge Based Feature Selection on Gene Expression Data" Entropy 23, no. 1: 2. https://doi.org/10.3390/e23010002
APA StyleYousef, M., Kumar, A., & Bakir-Gungor, B. (2021). Application of Biological Domain Knowledge Based Feature Selection on Gene Expression Data. Entropy, 23(1), 2. https://doi.org/10.3390/e23010002