Set-Wise Differential Interaction between Copy Number Alterations and Gene Expressions of Lower-Grade Glioma Reveals Prognosis-Associated Pathways

Abstract
1. Introduction
2. Materials and Methods
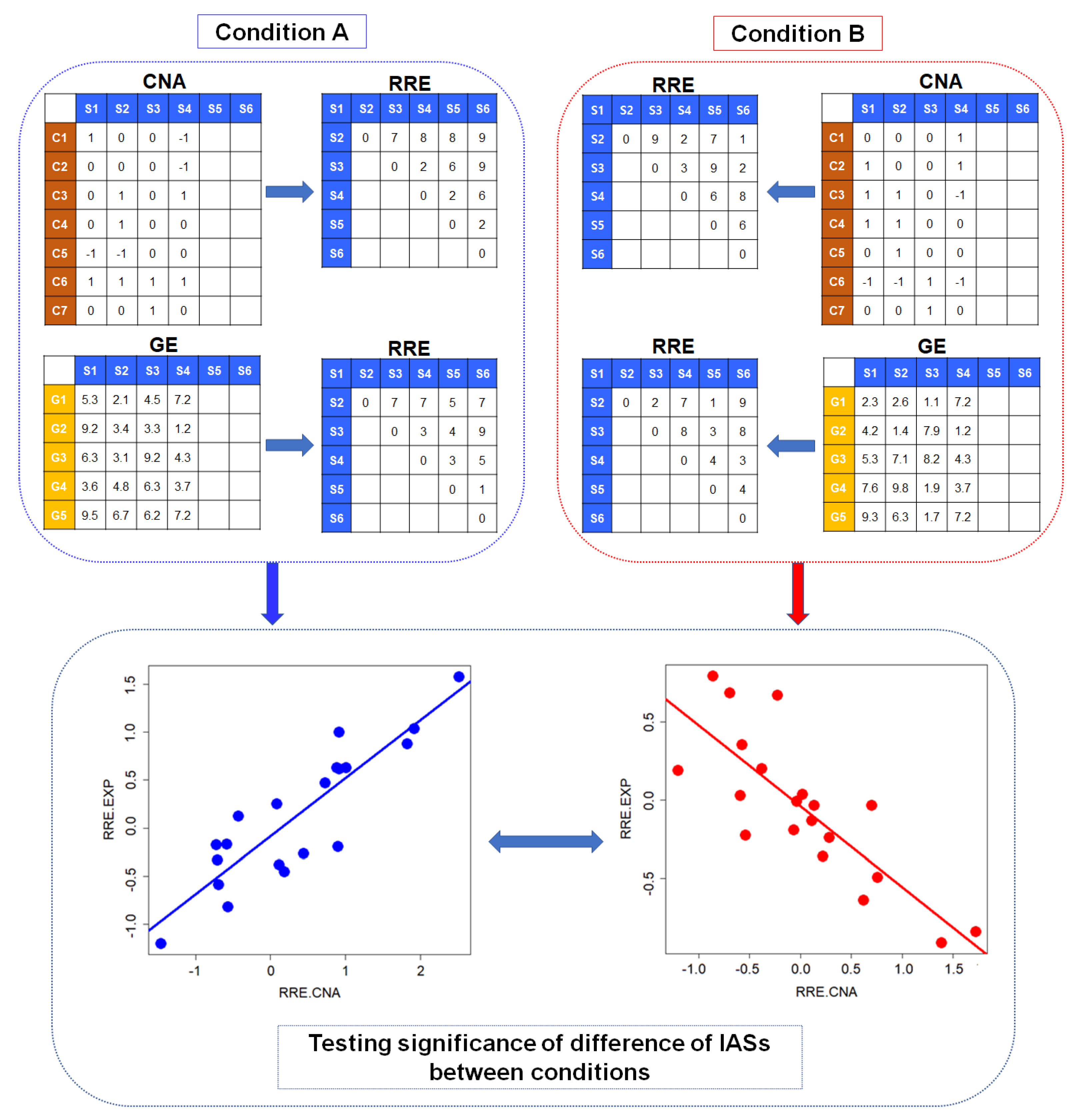
2.1. Identification of Conditional Change of Interactions between Set-Wise CNAs and GEs
2.2. Simulation Analysis
2.3. Analysis of TCGA-Multiomics Data
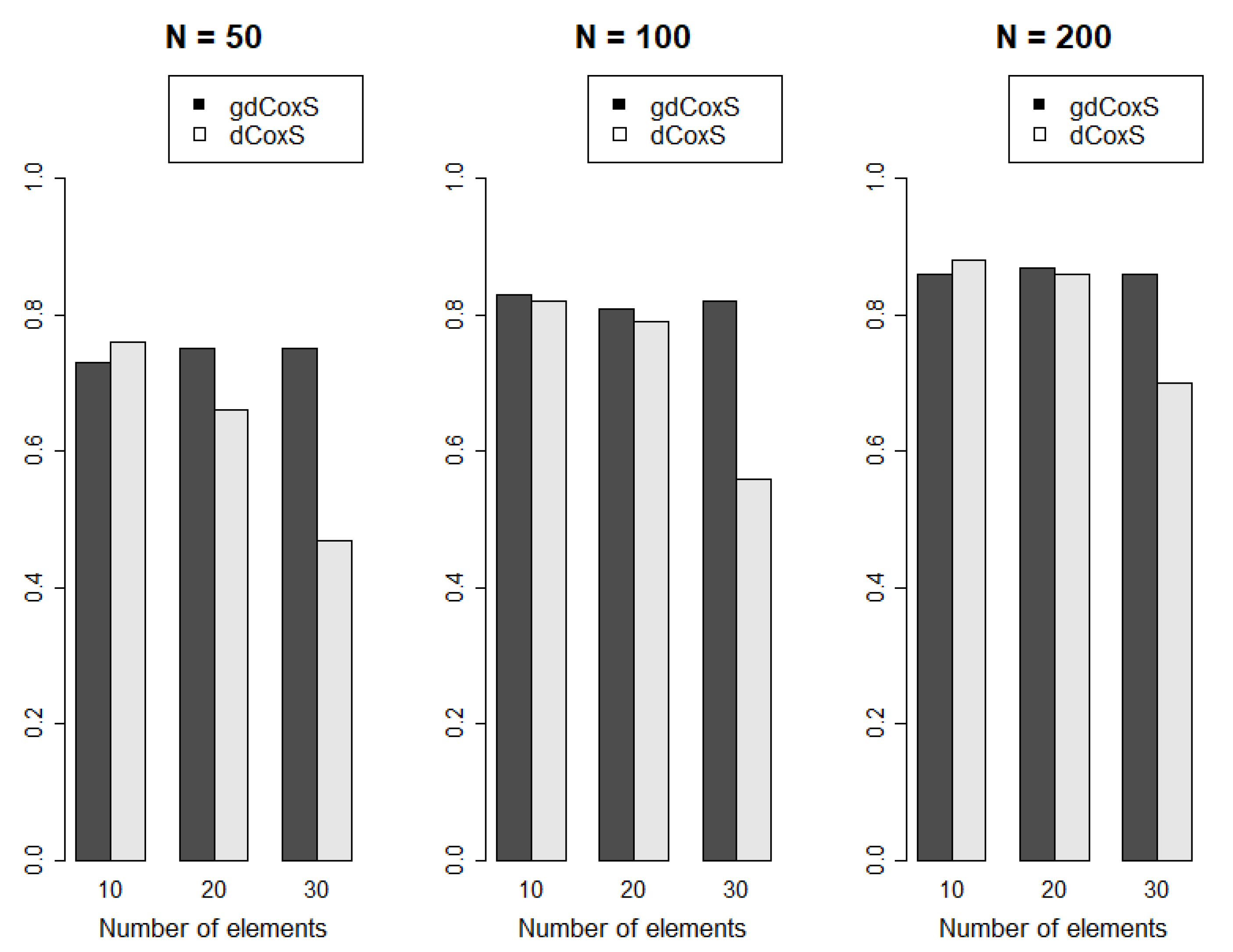
2.4. Comparison with Single Gene-Wise CNA Expression Analysis
3. Results
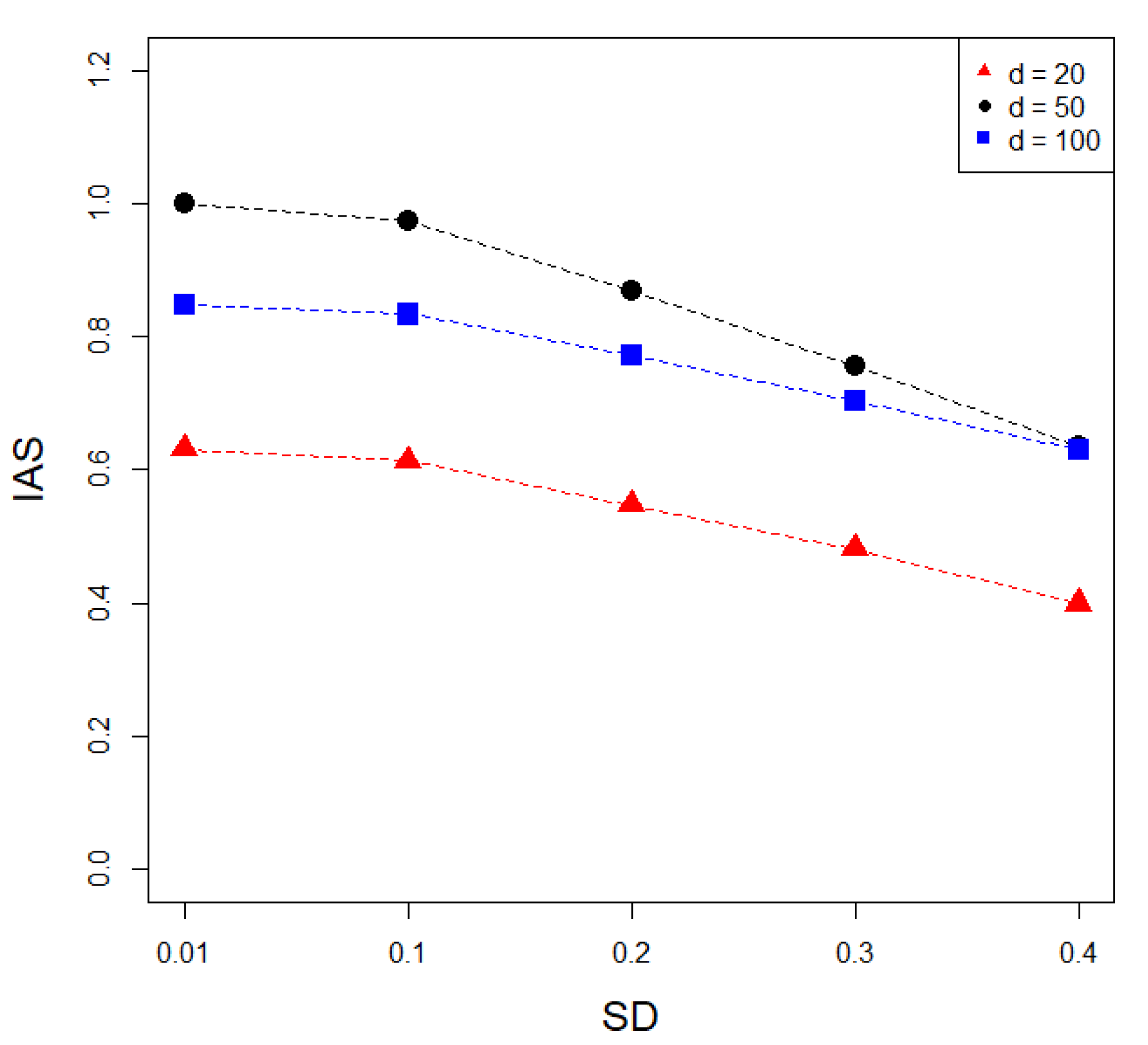
3.1. Simulation Analysis Results
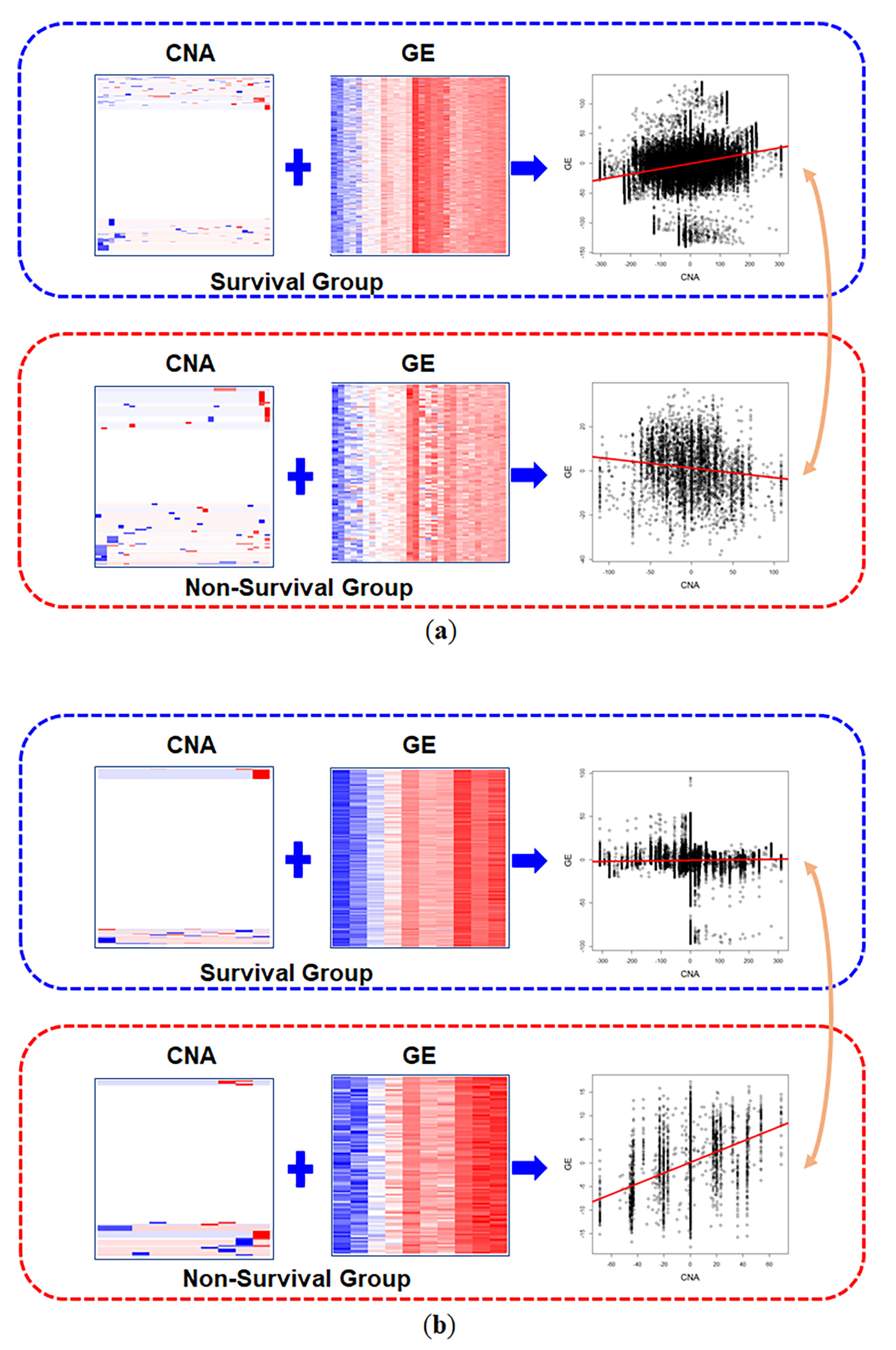
3.2. Real Data Analysis
4. Discussion
Supplementary Materials
Funding
Conflicts of Interest
References
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Varella-Garcia, M.; Cappuzzo, F. Predictive value of EGFR and HER2 overexpression in advanced non-small-cell lung cancer. Oncogene 2009, 28, S32–S37. [Google Scholar] [CrossRef]
- Ono, M.; Kuwano, M. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin. Cancer Res. 2006, 12, 7242–7251. [Google Scholar] [CrossRef]
- Yang, L.; Li, Y.; Wei, Z.; Chang, X. Coexpression network analysis identifies transcriptional modules associated with genomic alterations in neuroblastoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2341–2348. [Google Scholar] [CrossRef]
- Chang, X.; Zhao, Y.; Hou, C.; Glessner, J.; McDaniel, L.; Diamond, M.A.; Thomas, K.; Li, J.; Wei, Z.; Liu, Y.; et al. Common variants in MMP20 at 11q22.2 predispose to 11q deletion and neuroblastoma risk. Nat. Commun. 2017, 8, 569. [Google Scholar] [CrossRef]
- Lopez, G.; Conkrite, K.L.; Doepner, M.; Rathi, K.S.; Modi, A.; Vaksman, Z.; Farra, L.M.; Hyson, E.; Noureddine, M.; Wei, J.S.; et al. Somatic structural variation targets neurodevelopmental genes and identifies SHANK2 as a tumor suppressor in neuroblastoma. Genome Res. 2020, 30, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Pinkel, D.; Segraves, R.; Sudar, D.; Clark, S.; Poole, I.; Kowbel, D.; Collins, C.; Kuo, W.L.; Chen, C.; Zhai, Y.; et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 1998, 20, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Vauhkonen, H.; Bohling, T.; Mertens, F.; Mandahl, N.; Knuutila, S. Gene copy number changes in dermatofibrosarcoma protuberans—A fine-resolution study using array comparative genomic hybridization. Cytogenet. Genome Res. 2006, 115, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Yim, S.H.; Kwon, M.S.; Kim, T.M.; Shin, S.H.; Kang, H.M.; Lee, C.; Chung, Y.J. Recurrent genomic alterations with impact on survival in colorectal cancer identified by genome-wide array comparative genomic hybridization. Gastroenterology 2006, 131, 1913–1924. [Google Scholar] [CrossRef]
- Stransky, N.; Vallot, C.; Reyal, F.; Bernard-Pierrot, I.; de Medina, S.G.; Segraves, R.; de Rycke, Y.; Elvin, P.; Cassidy, A.; Spraggon, C.; et al. Regional copy number-independent deregulation of transcription in cancer. Nat. Genet. 2006, 38, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Staaf, J.; Torngren, T.; Rambech, E.; Johansson, U.; Persson, C.; Sellberg, G.; Tellhed, L.; Nilbert, M.; Borg, A. Detection and precise mapping of germline rearrangements in BRCA1, BRCA2, MSH2, and MLH1 using zoom-in array comparative genomic hybridization (aCGH). Hum. Mutat. 2008, 29, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Nowak, N.J.; Pacchia, A.L.; Morrison, C. Chromosome 11 genomic changes in parathyroid adenoma and hyperplasia: Array CGH, FISH, and tissue microarrays. Genes Chromosomes Cancer 2008, 47, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Pitea, A.; Kondofersky, I.; Sass, S.; Theis, F.J.; Mueller, N.S.; Unger, K. Copy number aberrations from Affymetrix SNP 6.0 genotyping data-how accurate are commonly used prediction approaches? Brief. Bioinform. 2018, 21, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Yau, C.; Mouradov, D.; Jorissen, R.N.; Colella, S.; Mirza, G.; Steers, G.; Harris, A.; Ragoussis, J.; Sieber, O.; Holmes, C.C. A statistical approach for detecting genomic aberrations in heterogeneous tumor samples from single nucleotide polymorphism genotyping data. Genome Biol. 2010, 11, R92. [Google Scholar] [CrossRef]
- Sun, W.; Wright, F.A.; Tang, Z.; Nordgard, S.H.; Van Loo, P.; Yu, T.; Kristensen, V.N.; Perou, C.M. Integrated study of copy number states and genotype calls using high-density SNP arrays. Nucleic Acids Res. 2009, 37, 5365–5377. [Google Scholar] [CrossRef]
- Van Loo, P.; Nordgard, S.H.; Lingjærde, O.C.; Russnes, H.G.; Rye, I.H.; Sun, W.; Weigman, V.J.; Marynen, P.; Zetterberg, A.; Naume, B.; et al. Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 16910–16915. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC 2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- De Tayrac, M.; Etcheverry, A.; Aubry, M.; Saikali, S.; Hamlat, A.; Quillien, V.; Le Treut, A.; Galibert, M.D.; Mosser, J. Integrative genome-wide analysis reveals a robust genomic glioblastoma signature associated with copy number driving changes in gene expression. Genes Chromosomes Cancer 2009, 48, 55–68. [Google Scholar] [CrossRef]
- Louhimo, R.; Lepikhova, T.; Monni, O.; Hautaniemi, S. Comparative analysis of algorithms for integration of copy number and expression data. Nat. Methods 2012, 9, 351–355. [Google Scholar] [CrossRef]
- Lahti, L.; Schäfer, M.; Klein, H.U.; Bicciato, S.; Dugas, M. Cancer gene prioritization by integrative analysis of mRNA expression and DNA copy number data: A comparative review. Brief. Bioinform. 2013, 14, 27–35. [Google Scholar] [CrossRef]
- Menezes, R.X.; Boetzer, M.; Sieswerda, M.; van Ommen, G.J.; Boer, J.M. Integrated analysis of DNA copy number and gene expression microarray data using gene sets. BMC Bioinform. 2009, 10, 203. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhu, J.; Bergamaschi, A.; Han, W.; Noh, D.Y.; Pollack, J.R.; Wang, P. Regularized multivariate regression for identifying master predictors with application to integrative genomics study of breast cancer. Ann. Appl. Stat. 2010, 4, 53–77. [Google Scholar] [CrossRef] [PubMed]
- Lahti, L.; Myllykangas, S.; Knuutila, S.; Kaski, S. Dependency detection with similarity constraints. In Proceedings of the 2009 IEEE International Workshop on Machine Learning for Signal Processing, Grenoble, France, 1–4 September 2009; IEEE: Piscataway, NJ, USA, 2009; pp. 89–94. [Google Scholar]
- Waaijenborg, S.; Zwinderman, A.H. Penalized canonical correlation analysis to quantify the association between gene expression and DNA markers. BMC Proc. 2007, 1, S122. [Google Scholar] [CrossRef] [PubMed]
- Parkhomenko, E.; Tritchler, D.; Beyene, J. Genome-wide sparse canonical correlation of gene expression with genotypes. BMC Proc. 2007, 1, S119. [Google Scholar] [CrossRef]
- Cho, S.B.; Kim, J.; Kim, J.H. Identifying set-wise differential co-expression in gene expression microarray data. BMC Bioinform. 2009, 10, 109. [Google Scholar] [CrossRef]
- Schäfer, J.; Strimmer, K. A Shrinkage Approach to Large-Scale Covariance Matrix Estimation and Implications for Functional Genomics. Stat. Appl. Genet. Mol. Biol. 2005, 4, 32. [Google Scholar] [CrossRef]
- Segal, E.; Friedman, N.; Kaminski, N.; Regev, A.; Koller, D. From signatures to models: Understanding cancer using microarrays. Nat. Genet. 2005, 37, S38–S45. [Google Scholar] [CrossRef]
- Zhang, Y.; Parmigiani, G.; Johnson, W. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; Narasimhan, B.; Chu, G. Impute: Imputation for Microarray Data; R Package Version 1.62.0; GitHub, Inc.: San Francisco, CA, USA, 2020. [Google Scholar]
- Bolstad, B. preprocessCore: A Collection of Pre-Processing Functions, R Package Version 1.50.0; Available online: https://github.com/bmbolstad/preprocessCore (accessed on 14 July 2020).
- England, B.; Huang, T.; Karsy, M. Current understanding of the role and targeting of tumor suppressor p53 in glioblastoma multiforme. Tumour. Biol. 2013, 34, 2063–2074. [Google Scholar] [CrossRef]
- Krex, D.; Mohr, B.; Appelt, H.; Schackert, H.K.; Schackert, G. Genetic analysis of a multifocal glioblastoma multiforme: A suitable tool to gain new aspects in glioma development. Neurosurgery 2003, 53, 1377–1384. [Google Scholar] [CrossRef]
- Djuzenova, C.S.; Fiedler, V.; Memmel, S.; Katzer, A.; Hartmann, S.; Krohne, G.; Zimmermann, H.; Scholz, C.J.; Polat, B.; Flentje, M.; et al. Actin cytoskeleton organization, cell surface modification and invasion rate of 5 glioblastoma cell lines differing in PTEN and p53 status. Exp. Cell Res. 2015, 330, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Park, C.M.; Park, M.J.; Kwak, H.J.; Moon, S.I.; Yoo, D.H.; Lee, H.C.; Park, I.C.; Rhee, C.H.; Hong, S.I. Induction of p53-mediated apoptosis and recovery of chemosensitivity through p53 transduction in human glioblastoma cells by cisplatin. Int. J. Oncol. 2006, 28, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Rives, S.A.; Casique-Aguirre, D.; Germán-Castelán, L.; Velasco-Velázquez, M.A.; González-Arenas, A. Apoptotic Signaling Pathways in Glioblastoma and Therapeutic Implications. Biomed. Res. Int. 2017, 2017, 7403747. [Google Scholar] [CrossRef]
- Mohr, A.; Deedigan, L.; Jencz, S.; Mehrabadi, Y.; Houlden, L.; Albarenque, S.M.; Zwacka, R.M. Caspase-10: A molecular switch from cell-autonomous apoptosis to communal cell death in response to chemotherapeutic drug treatment. Cell Death Differ. 2018, 25, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.M.; Stricker, S.H.; Halavach, H.; Poetsch, A.R.; Cresswell, G.; Kelly, G.; Kanu, N.; Marino, S.; Luscombe, N.M.; Pollard, S.M.; et al. Inactivation of the ATMIN/ATM pathway protects against glioblastoma formation. Elife 2016, 5, e08711. [Google Scholar] [CrossRef]
- Celardo, I.; Grespi, F.; Antonov, A.; Bernassola, F.; Garabadgiu, A.V.; Melino, G.; Amelio, I. Caspase-1 is a novel target of p63 in tumor suppression. Cell Death Dis. 2013, 4, e645. [Google Scholar] [CrossRef]
- Xu, H.; Zong, H.; Ma, C.; Ming, X.; Shang, M.; Li, K.; He, X.; Du, H.; Cao, L. Epidermal growth factor receptor in glioblastoma. Oncol. Lett. 2017, 14, 512–516. [Google Scholar] [CrossRef]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef]
- Manfred, W.M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735. [Google Scholar]
- Škovierová, H.; Vidomanová, E.; Mahmood, S.; Sopková, J.; Drgová, A.; Červeňová, T.; Halašová, E.; Lehotský, J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int. J. Mol. Sci. 2016, 17, 1733. [Google Scholar] [CrossRef] [PubMed]
- Hasan, T.; Arora, R.; Bansal, A.K.; Bhattacharya, R.; Sharma, G.S.; Singh, L.R. Disturbed homocysteine metabolism is associated with cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Linnebank, M.; Semmler, A.; Moskau, S.; Smulders, Y.; Blom, H.; Simon, M. The methylenetetrahydrofolate reductase (MTHFR) variant c.677C>T (A222V) influences overall survival of patients with glioblastoma multiforme. Neuro Oncol. 2008, 10, 548–552. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway Database | Pathways | NCNA1 | NEXP2 | IAS.S 3 | IAS.NS 4 | diffIAS 5 |
|---|---|---|---|---|---|---|
| PID | IL3_PATHWAY | 10 | 10 | 0.023 | 0.407 | −52.037 |
| REACTOME | PROTEIN_METHYLATION | 14 | 14 | 0.197 | 0.524 | −48.578 |
| REACTOME | DUAL_INCISION_IN_GG_NER | 14 | 14 | 0.081 | 0.430 | −48.002 |
| BIOCARTA | FORMATION_OF_INCISION_COMPLEX_IN_GG_NER | 26 | 26 | 0.176 | 0.501 | −47.231 |
| REACTOME | MICRORNA_MIRNA_BIOGENESIS | 10 | 10 | 0.146 | 0.476 | −47.219 |
| REACTOME | TRIGLYCERIDE_CATABOLISM | 15 | 11 | 0.148 | −0.179 | 41.893 |
| REACTOME | DEGRADATION_OF_CYSTEINE_AND_HOMOCYSTEINE | 11 | 10 | 0.168 | −0.159 | 41.940 |
| BIOCARTA | EGF_PATHWAY | 14 | 14 | 0.200 | −0.145 | 44.284 |
| KEGG | CYTOSOLIC_DNA_SENSING_PATHWAY | 16 | 15 | 0.240 | −0.127 | 47.355 |
| REACTOME | GLYCOSPHINGOLIPID_METABOLISM | 31 | 28 | 0.256 | −0.136 | 50.660 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, S.B. Set-Wise Differential Interaction between Copy Number Alterations and Gene Expressions of Lower-Grade Glioma Reveals Prognosis-Associated Pathways. Entropy 2020, 22, 1434. https://doi.org/10.3390/e22121434
Cho SB. Set-Wise Differential Interaction between Copy Number Alterations and Gene Expressions of Lower-Grade Glioma Reveals Prognosis-Associated Pathways. Entropy. 2020; 22(12):1434. https://doi.org/10.3390/e22121434
Chicago/Turabian StyleCho, Seong Beom. 2020. "Set-Wise Differential Interaction between Copy Number Alterations and Gene Expressions of Lower-Grade Glioma Reveals Prognosis-Associated Pathways" Entropy 22, no. 12: 1434. https://doi.org/10.3390/e22121434
APA StyleCho, S. B. (2020). Set-Wise Differential Interaction between Copy Number Alterations and Gene Expressions of Lower-Grade Glioma Reveals Prognosis-Associated Pathways. Entropy, 22(12), 1434. https://doi.org/10.3390/e22121434