Crystallization of Supercooled Liquids: Self-Consistency Correction of the Steady-State Nucleation Rate



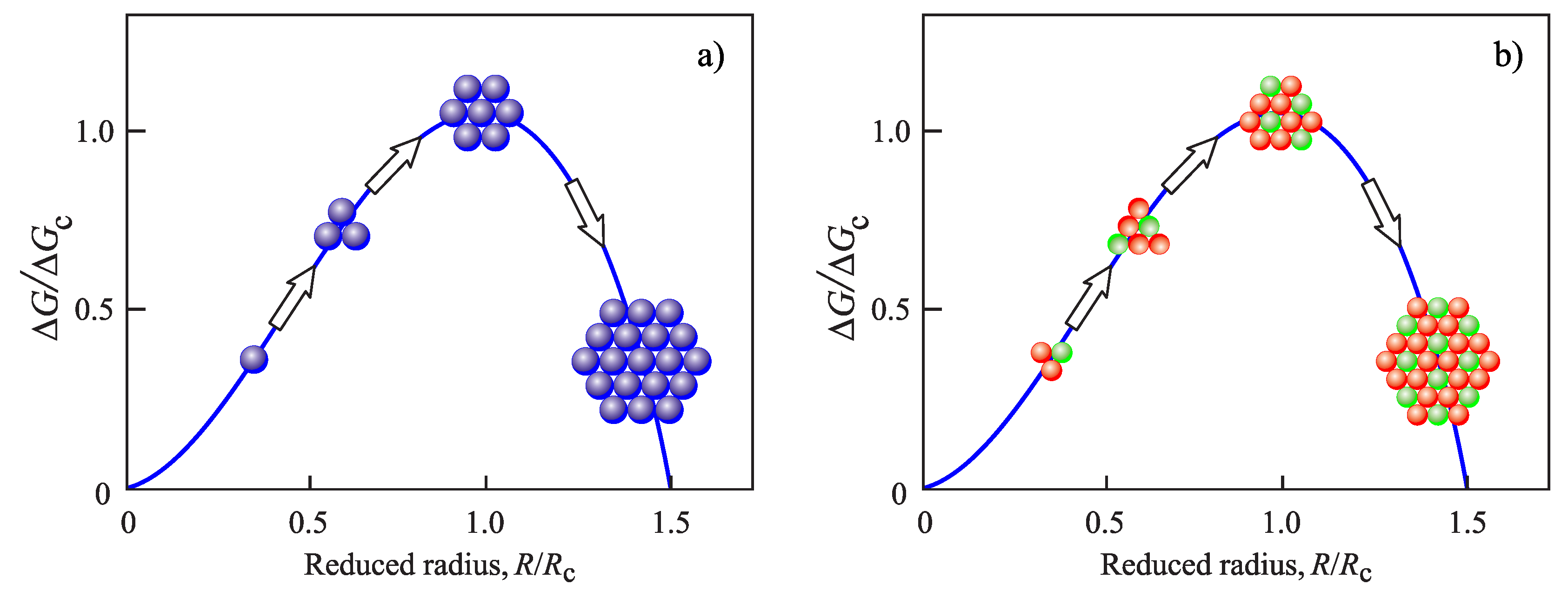{kind=link}
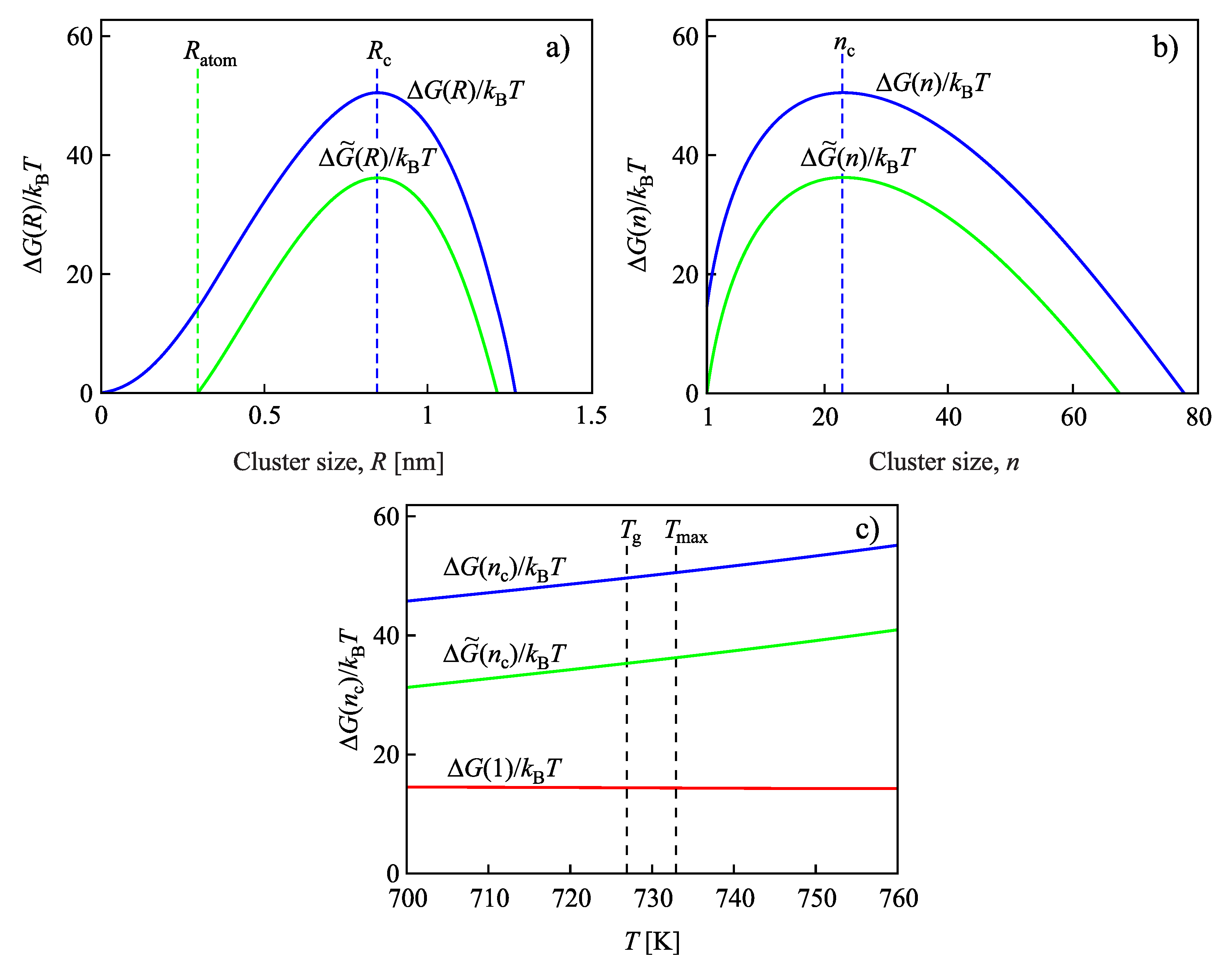{kind=link}
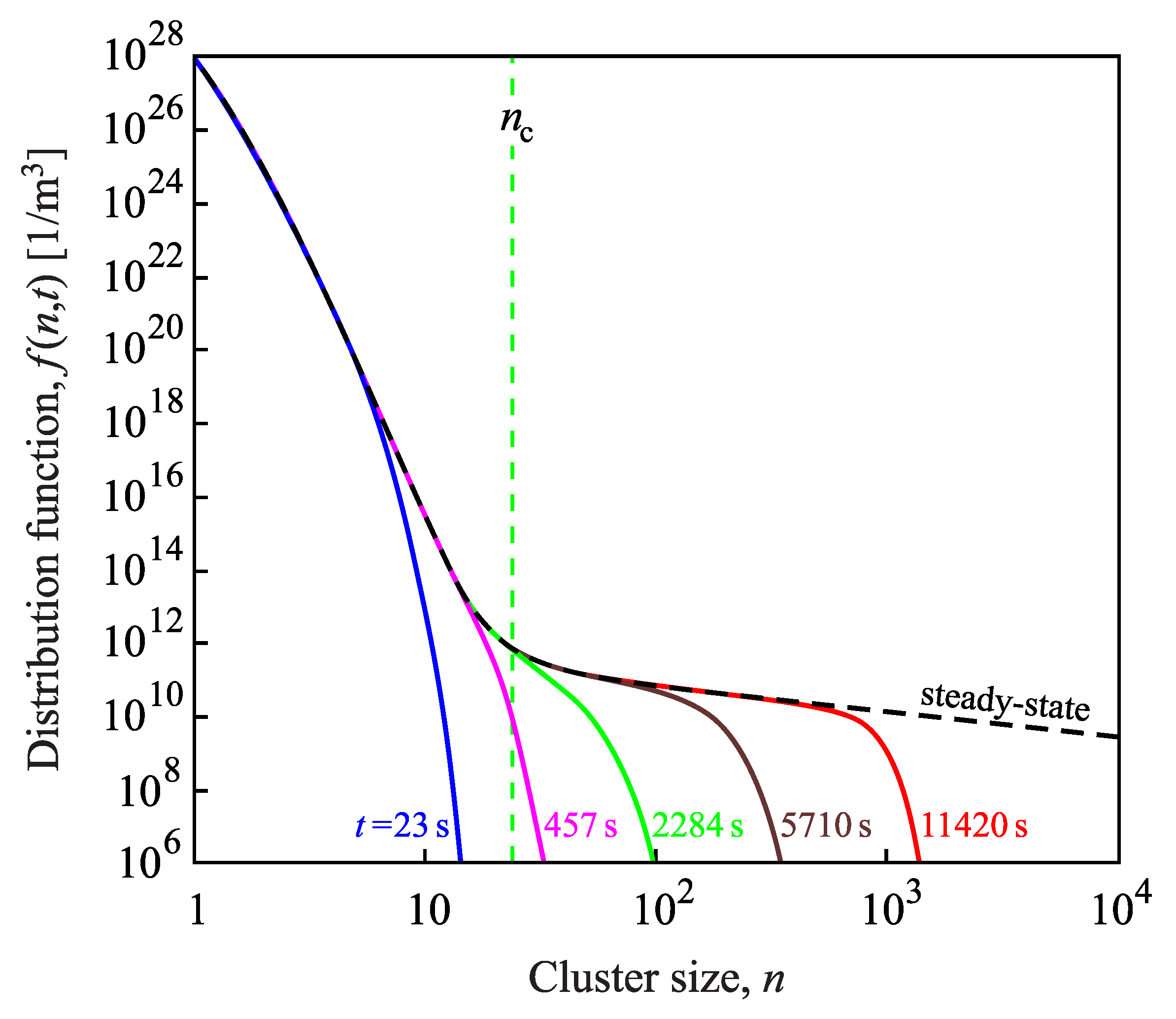{kind=link}
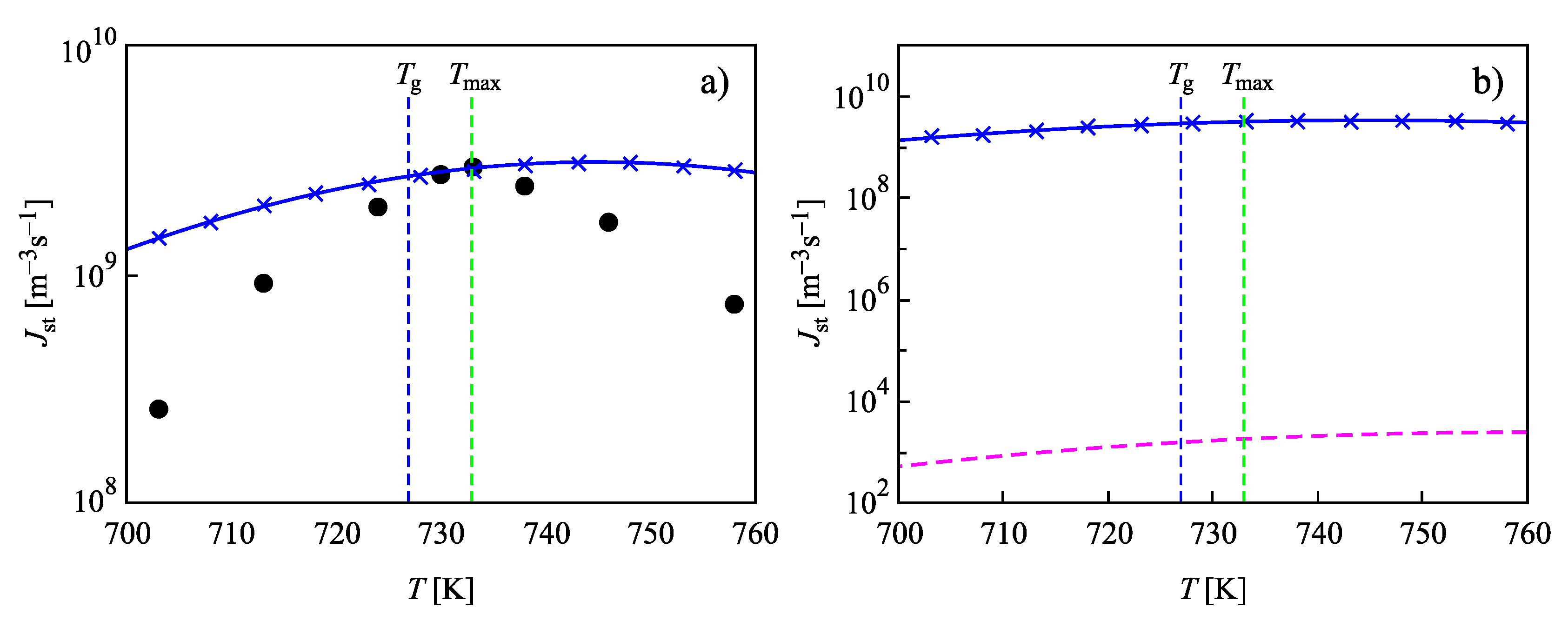{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Basic Equations of Classical Nucleation Theory
3. Comparison of the Different Theoretical Treatments
3.1. Application of the Capillarity Approximation
3.2. Account of a Temperature or Size Dependence of the Surface Tension
3.2.1. Some General Comments
3.2.2. Account of a Temperature Dependence of the Surface Tension for Sub- and Super-Critical Clusters
3.2.3. Account of a Curvature or Size Dependence of the Surface Tension for Sub- and Super-Critical Clusters
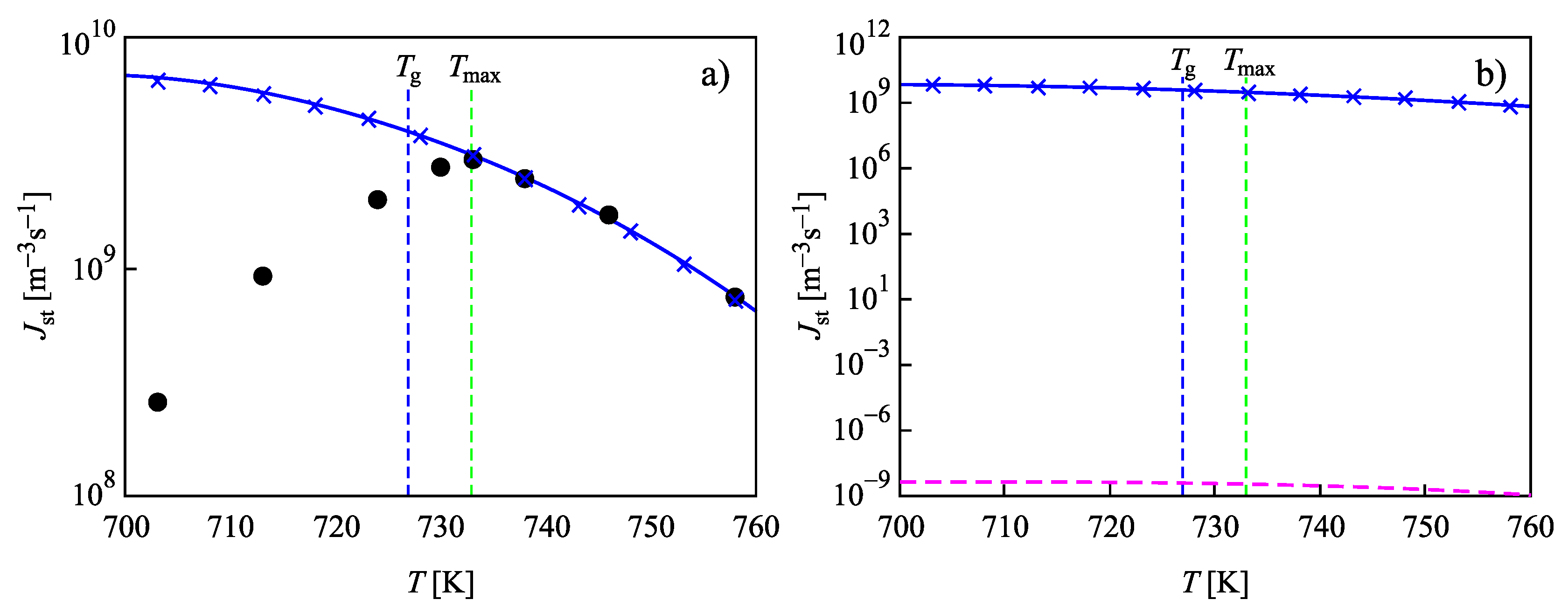
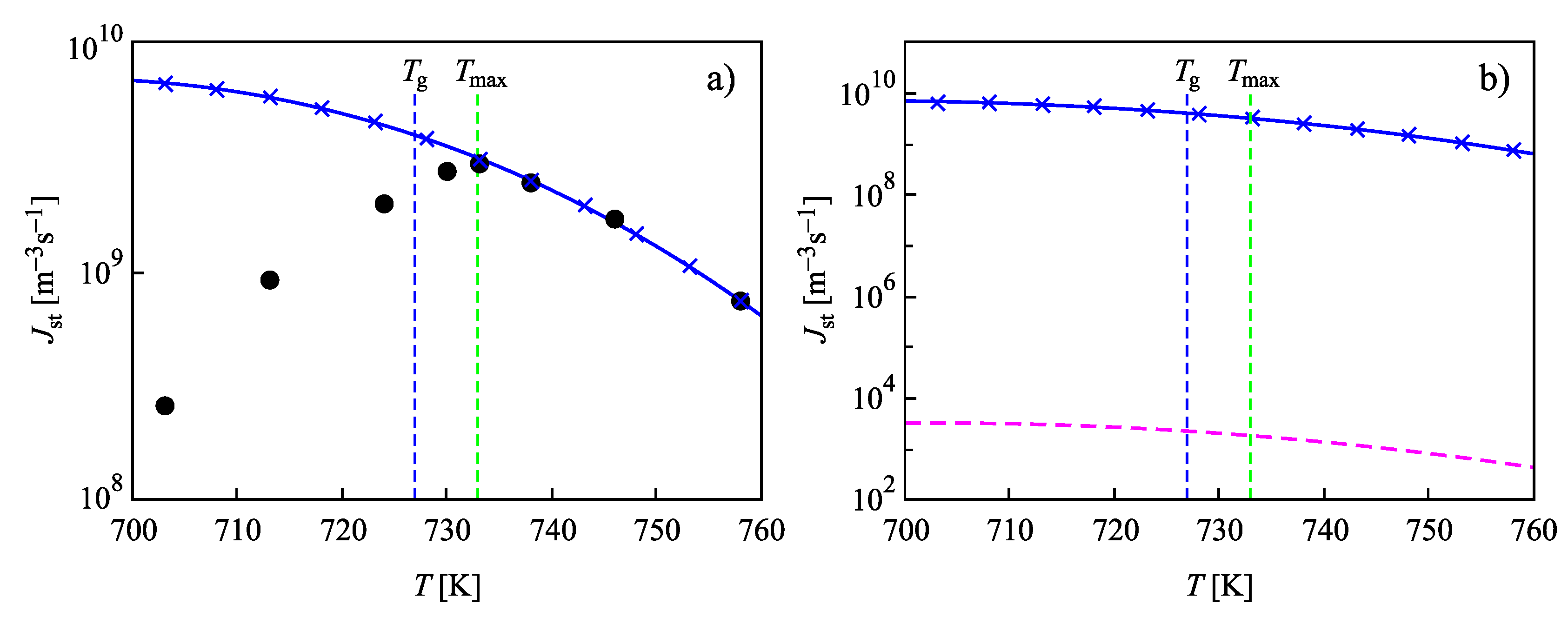
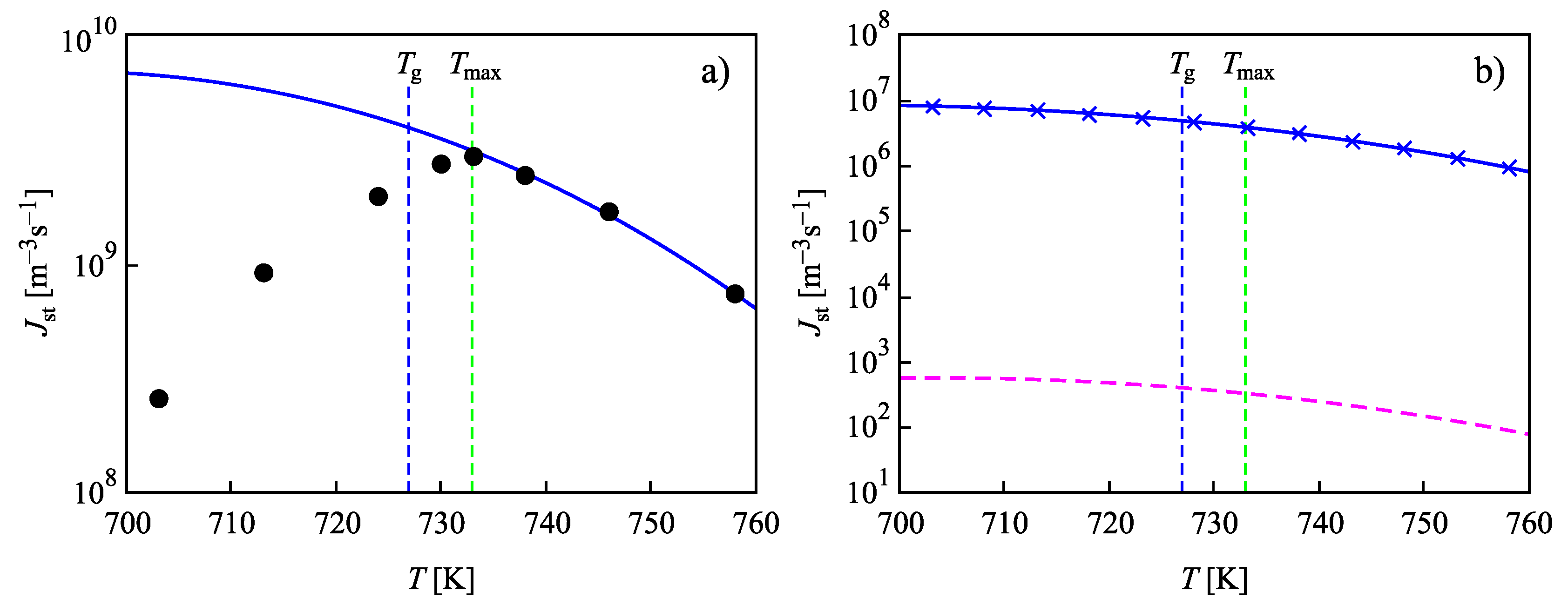
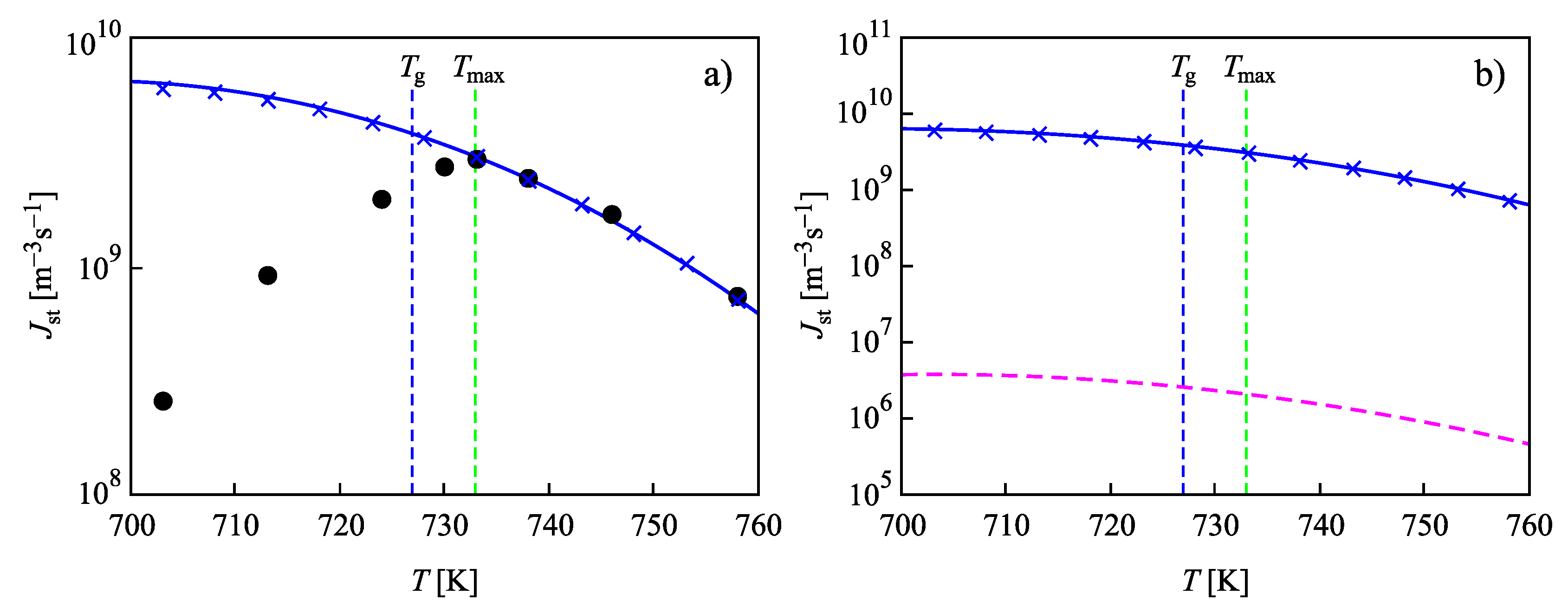
4. Summary of Results and Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Höland, W.; Beall, G.H. Glass-Ceramic Technology, 3rd ed.; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar]
- Kelton, K.F.; Greer, A.L. Nucleation in Condensed Matter: Applications in Materials and Biology; Pergamon: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Gusak, A.M. Diffusion-Controlled Solid State Reactions; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Gutzow, I.S.; Schmelzer, J.W.P. The Vitreous State: Thermodynamics, Structure, Rheology, and Crystallization, 1st ed.; Springer: Berlin, Germany, 1995. [Google Scholar]
- Gutzow, I.S.; Schmelzer, J.W.P. The Vitreous State: Thermodynamics, Structure, Rheology, and Crystallization, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Schick, C.; Zhuravlev, E.; Androsch, R.; Wurm, A.; Schmelzer, J.W.P. Influence of Thermal Prehistory on Crystal Nucleation and Growth in Polymers. In Glass: Selected Properties and Crystallization; Schmelzer, J.W.P., Ed.; De Gruyter: Berlin, Germany, 2014; pp. 1–93. [Google Scholar]
- Schick, C.; Androsch, R.; Schmelzer, J.W.P. Topical Review: Homogeneous crystal nucleation in polymers. J. Phys. Condens. Matter 2017, 29, 453002/1–453002/35. [Google Scholar] [CrossRef]
- Wilde, G. Early Stages of Crystal Formation in Glass-forming Metallic Alloys. In Glass: Selected Properties and Crystallization; Schmelzer, J.W.P., Ed.; De Gruyter: Berlin, Germany, 2014; pp. 95–136. [Google Scholar]
- Fokin, V.M.; Zanotto, E.D.; Yuritsyn, N.S.; Schmelzer, J.W.P. Homogeneous Crystal Nucleation in Silicate Glasses: A Forty Years Perspective. J. Non-Cryst. Solids 2006, 352, 2681–2714. [Google Scholar] [CrossRef]
- Skripov, V.P.; Koverda, V.P. Spontaneous Crystallization of Undercooled Liquids; Nauka: Moscow, Russia, 1984. (In Russian) [Google Scholar]
- Debenedetti, P. Metastable Liquids: Concepts and Principles; Princeton University Press: Princeton, NJ, USA, 1996. [Google Scholar]
- Skripov, V.P.; Faizullin, M.Z. Crystal-Liquid-Gas Phase Transitions and Thermodynamic Similarity; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Morey, G.W. The Properties of Glass; Reinhold Publishers: New York, NY, USA, 1938. [Google Scholar]
- Olovsson, I. Wonders of Water: The Hydrogen Bond in Action; World Scientific Publishers: Singapore, 2017. [Google Scholar]
- Markov, I. Crystal Growth for Beginners: Fundamentals of Nucleation, Crystal Growth, and Epitaxy; World Scientific: Singapore, 2002. [Google Scholar]
- Feigelson, R.S. (Ed.) 50 Years Progress in Crystal Growth: A Reprint Collection; Elsevier: Amsterdam, The Netherlands, 2004. [Google Scholar]
- Earle, S. Physical Geology, 2nd ed.; BCcampus: Victoria, BC, Canada, 2019; Available online: https://opentextbc.ca/physicalgeology2ed/ (accessed on 23 September 2019).
- Schmelzer, J.W.P. Micro- and Nanostructures: A Little Picture Book. In Nucleation Theory and Applications; Schmelzer, J.W.P., Röpke, G., Priezzhev, V.B., Eds.; Joint Institute for Nuclear Research Publishing House: Dubna, Russia, 2002; pp. 469–488. [Google Scholar]
- Zanotto, E.D. Crystal in Glass: A Hidden Beauty; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Gutzow, I.; Kashchiev, D.; Avramov, I. Nucleation and Crystallization in Glass-forming melts: Old problems and new questions. J. Non-Cryst. Solids 1985, 73, 477–499. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Abyzov, A.S. Generalized Gibbs’ Approach to the Thermodynamics of Heterogeneous Systems and the Kinetics of First-Order Phase Transitions. J. Eng. Thermophys. 2007, 16, 119–129. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Fokin, V.M.; Abyzov, A.S.; Zanotto, E.D.; Gutzow, I. How Do Crystals Form and Grow in Glass-Forming Liquids: Ostwald’s Rule of Stages and Beyond. Int. J. Appl. Glass Sci. 2010, 1, 16–26. [Google Scholar] [CrossRef]
- Johari, G.; Schmelzer, J.W.P. Crystal Nucleation and Growth in Glass-forming Systems: Some New Results and Open Problems. In Glass: Selected Properties and Crystallization; Schmelzer, J.W.P., Ed.; De Gruyter: Berlin, Germany, 2014; pp. 521–585. [Google Scholar]
- Schmelzer, J.W.P.; Abyzov, A.S.; Fokin, V.M. Crystallization of glass: What we know, what we need to know. Int. J. Appl. Glass Sci. 2016, 7, 253–261. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Abyzov, A.S. Crystallization of glass-forming melts: New answers to old questions. J. Non-Cryst. Solids 2018, 501, 11–20. [Google Scholar] [CrossRef]
- Schottelius, A.; Mambretti, F.; Kalinin, A.; Beyersdorff, B.; Rothkirch, A.; Goy, C.; Müller, J.; Petridis, N.; Ritzer, M.; Trinter, F.; et al. Crystal growth rates in supercooled atomic liquid mixtures. Nat. Mater. 2020, 19, 512–516. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Schick, C. General concepts of crystallization: Some recent results and possible future developments. In Dielectrics and Crystallization; Ezquerra, T.A., Nogales, A., Eds.; Advances in Dielectrics Series (Kremer, F., series editor); Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Band, W. Dissociation Treatment of Condensing Systems. J. Chem. Phys. 1939, 7, 324–326. [Google Scholar] [CrossRef]
- Oxtoby, D.W. Crystal nucleation in simple and complex fluids. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2003, 361, 419–428. [Google Scholar] [CrossRef]
- Oxtoby, D.W. Nucleation of Crystals from the Melt. Ann. N. Y. Acad. Sci. 2006, 484, 26–38. [Google Scholar] [CrossRef]
- Asta, M.; Beckermann, C.; Karma, A.; Kurz, W.; Napolitano, R.; Plapp, M.; Purdy, G.; Rappaz, M.; Trivedi, R. Solidification microstructures and solid-state parallels: Recent developments, future directions. Acta Mater. 2009, 57, 941–971. [Google Scholar] [CrossRef]
- Granasy, L.; Toth, G.I.; Warren, J.A.; Podmaniczky, F.; Tegze, G.; Ratkai, L.; Pusztai, T. Phase-field modeling of crystal nucleation in undercooled liquids—A review. Prog. Mater. Sci. 2019, 106, 100569/1–100569/51. [Google Scholar] [CrossRef]
- Baidakov, V.G. Crystallization of Undercooled Liquids: Results of Molecular Dynamics Simulations. In Glass: Selected Properties and Crystallization; Schmelzer, J.W.P., Ed.; De Gruyter: Berlin, Germany, 2014; pp. 481–520. [Google Scholar]
- Schmelzer, J.W.P.; Abyzov, A.S.; Ferreira, E.B.; Fokin, V.M. Curvature dependence of the surface tension and crystal nucleation in liquids. Int. J. Appl. Glass Sci. 2019, 10, 57–60. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Abyzov, A.S.; Baidakov, V.G. Entropy and the Tolman Parameter in Nucleation Theory. Entropy 2019, 21, 670. [Google Scholar] [CrossRef]
- Gibbs, J.W. On the Equilibrium of Heterogeneous Substances. Trans. Conn. Acad. Sci. 1875–1878, 3, 108–248, 343–521. [Google Scholar] [CrossRef]
- Gibbs, J.W. On the Equilibrium of Heterogeneous Substances. In Thermodynamics; The Collected Works; Longmans & Green: New York, NY, USA; London, UK; Toronto, ON, Canada, 1928; Volume 1. [Google Scholar]
- Van der Waals, J.D.; Kohnstamm, P. Lehrbuch der Thermodynamik (English: Textbook on Thermodynamics); Johann-Ambrosius-Barth Verlag: Leipzig, Germany; Amsterdam, The Netherlands, 1908. [Google Scholar]
- Rowlinson, J.S. Translation of J. D. van der Waals’ “The Thermodynamic Theory of Capillarity Under the Hypothesis of a Continuous Variation of Density”. J. Stat. Phys. 1979, 20, 197–244. [Google Scholar] [CrossRef]
- Einstein, A. Zur Theorie der Brownschen Bewegung (Engl: On the theory of Brownian motion). Ann. Der Phys. 1906, 20, 199–206. [Google Scholar] [CrossRef]
- Einstein, A. Theorie der Opaleszenz von homogenen Flüssigkeiten und Flüssigkeitsgemischen in der Nähe des kritischen Zustandes (Engl: The Theory of the Opalescence of Homogeneous Fluids and Liquid Mixtures near the Critical State). Ann. Der Phys. 1910, 33, 1275–1298. [Google Scholar] [CrossRef]
- Landau, L.D.; Lifschitz, E.M. Statistische Physik (Engl.: Statistical Physics); Akademie-Verlag: Berlin, Germany, 1969. [Google Scholar]
- Volmer, M.; Weber, A. Keimbildung in übersättigten Gebilden (Engl.: Nucleation in supersaturated samples). Z. Phys. Chem. 1926, 119, 227–301. [Google Scholar]
- Schmelzer, J.W.P.; Abyzov, A.S. Crystallization of glass-forming liquids: Thermodynamic driving force. J. Non-Cryst. Solids 2016, 449, 41–49. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Pascova, R.; Möller, J.; Gutzow, I. Surface Induced Devitrification of Glasses: The Influence of Elastic Strains. J. Non-Cryst. Solids 1993, 162, 26–39. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Möller, J.; Gutzow, I.; Pascova, R.; Müller, R.; Pannhorst, W. Surface-energy and Structure Effects on Surface Crystallization. J. Non-Cryst. Solids 1995, 183, 215–233. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Müller, R.; Möller, J.; Gutzow, I.S. Elastic Stresses, Stress Relaxation, and Crystallization: Theory. Phys. Chem. Glas. 2002, 43 C, 291–298. [Google Scholar]
- Schmelzer, J.W.P.; Müller, R.; Möller, J.; Gutzow, I.S. Theory of Nucleation in Viscoelastic Media: Application to Phase Formation in Glassforming Melts. J. Non-Cryst. Solids 2003, 315, 144–160. [Google Scholar] [CrossRef]
- Fokin, V.M.; Zanotto, E.D.; Schmelzer, J.W.P.; Potapov, O.V. New Insights on the Thermodynamic Barrier for Nucleation in Lithium Disilicate Glass. J. Non-Cryst. Solids 2005, 351, 1491–1499. [Google Scholar] [CrossRef]
- Abyzov, A.S.; Fokin, V.M.; Rodrigues, A.M.; Zanotto, E.D.; Schmelzer, J.W.P. The effect of elastic stresses on the thermodynamic barrier for crystal nucleation. J. Non-Cryst. Solids 2016, 432, 325–333. [Google Scholar] [CrossRef]
- Slezov, V.V.; Schmelzer, J.W.P. Kinetics of formation of a phase with a definite stoichiometric composition. J. Phys. Chem. Solids 1994, 55, 243–251. [Google Scholar] [CrossRef]
- Slezov, V.V.; Schmelzer, J.W.P. Comments on Nucleation Theory. J. Phys. Chem. Solids 1998, 59, 1507–1519. [Google Scholar] [CrossRef]
- Slezov, V.V.; Schmelzer, J.W.P. Kinetics of formation of a phase with an arbitrary stoichiometric composition in a multi-component solid solution. Phys. Rev. E 2002, 65, 031506/1–031506/13. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P. Crystal nucleation and growth in glass-forming melts: Experiment and theory. J. Non-Cryst. Solids 2008, 354, 269–278. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P. On the determination of the kinetic pre-factor in classical nucleation theory. J. Non-Cryst. Solids 2010, 356, 2901–2907. [Google Scholar] [CrossRef]
- Blanc, W.; Martin, I.; Francois-Saint-Cyr, H.; Bidault, X.; Chaussedent, S.; Hombourger, C.; Lacomme, S.; le Coustumer, P.; Neuville, D.R.; Larson, D.J.; et al. Compositional Changes at the Early Stages of Nanoparticles Growth in Glasses. J. Phys. Chem. C 2019, 123, 29008–29014. [Google Scholar] [CrossRef]
- Abyzov, A.S.; Schmelzer, J.W.P.; Kovalchuk, A.A.; Slezov, V.V. Evolution of Cluster Size-Distributions in Nucleation-Growth and Spinodal Decomposition Processes in a Regular Solution. J. Non-Cryst. Solids 2010, 356, 2915–2922. [Google Scholar] [CrossRef]
- Abyzov, A.S.; Schmelzer, J.W.P. Kinetics of segregation processes in solutions: Saddle point versus ridge crossing of the thermodynamic potential barrier. J. Non-Cryst. Solids 2014, 384, 8–14. [Google Scholar] [CrossRef]
- Farkas, L. Keimbildungsgeschwindigkeit in übersättigten Dämpfen (Engl.: Nucleation rate in supersaturated vapors). Z. Phys. Chem. 1927, 125, 236–242. [Google Scholar]
- Volmer, M. Kinetik der Phasenbildung (English: Kinetics of Phase Formation); Th. Steinkopff: Dresden, Germany, 1939. [Google Scholar]
- Kaischew, R.; Stranski, I.N. Zur Theorie der linearen Kristallisationsgeschwindigkeit (Engl: On the theory of the linear rate of crystallization). Z. Phys. Chem. A 1934, 170, 295–299. [Google Scholar]
- Becker, R.; Döring, W. Kinetische Behandlung der Keimbildung in übersättigten Dämpfen (Engl.: Kinetic treatment of nucleation in supersaturated vapors). Ann. Der Phys. 1935, 24, 719–752. [Google Scholar] [CrossRef]
- Frenkel, Y.I. The Kinetic Theory of Liquids; Oxford University Press: Oxford, UK, 1946. [Google Scholar]
- Zeldovich, Y.B. On the Theory of New Phase Formation: Cavitation. Sov. Phys. JETP 1942, 12, 525–538. [Google Scholar]
- Turnbull, D.; Fisher, J.C. Rate of Nucleation in Condensed Systems. J. Chem. Phys. 1949, 17, 71–73. [Google Scholar] [CrossRef]
- Ulbricht, H.; Schmelzer, J.W.P.; Mahnke, R.; Schweitzer, F. Thermodynamics of Finite Systems and the Kinetics of First-Order Phase Transitions; Teubner-Texte zur Physik; Teubner-Verlag: Leipzig, Germany, 1988; Volume 17. [Google Scholar]
- Schmelzer, J.W.P. Comments on Curvature Dependent Surface Tension and Nucleation Theory. In Nucleation Theory and Applications; Schmelzer, J.W.P., Röpke, G., Priezzhev, V.B., Eds.; Workshop Proceedings 1997–1999; Joint Institute for Nuclear Research Publishing Department: Dubna, Russia, 1999; pp. 268–289. [Google Scholar]
- Schmelzer, J.W.P.; Boltachev, G.S.; Baidakov, V.G. Classical and Generalized Gibbs’ Approaches and the Work of Critical Cluster Formation in Nucleation Theory. J. Chem. Phys. 2006, 124, 194503/1–194503/18. [Google Scholar] [CrossRef] [PubMed]
- Schmelzer, J.W.P. Application of the Nucleation Theorem to Crystallization of Liquids: Some General Theoretical Results. Entropy 2019, 21, 1147. [Google Scholar] [CrossRef]
- Wu, D.T. Nucleation Theory. In Solid State Physics; Ehrenreich, H., Spaepen, F., Eds.; Academic Press: New York, NY, USA, 1997; Volume 50, pp. 37–187. [Google Scholar]
- Ford, I.J. Statistical mechanics of nucleation: A review. Proc. Inst. Mech. Eng. Part C J. Mech. Eng. Sci. 2004, 218, 883–899. [Google Scholar] [CrossRef]
- Bartels, J.; Lembke, U.; Pascova, R.; Schmelzer, J.; Gutzow, I. Evolution of Cluster Size Distributions in Nucleation and Growth Processes. J. Non-Cryst. Solids 1991, 136, 181–197. [Google Scholar] [CrossRef]
- Fokin, V.M. Investigation of Stationary and Non-Stationary Crystal Nucleation Rates in Model Glasses of Stoichiometric Composition Li2O·2SiO2 and 2Na2O·CaO·3SiO2. Ph.D. Thesis, Grebenshchikov Institute of Silicate Chemistry of the Russian Academy of Sciences, Leningrad, Russia, 1980. [Google Scholar]
- Fokin, V.M.; Kalinina, A.M.; Filipovich, V.N. Nucleation in silicate glasses and effect of preliminary heat treatment on it. J. Cryst. Growth 1981, 52, 115–121. [Google Scholar] [CrossRef]
- Slezov, V.V.; Schmelzer, J.W.P. Kinetics of Nucleation-Growth Processes: The First Stages. In Nucleation Theory and Applications; Schmelzer, J.W.P., Röpke, G., Priezzhev, V.B., Eds.; Joint Institute for Nuclear Research Publishing House: Dubna, Russia, 1999; pp. 6–81. [Google Scholar]
- Schmelzer, J.W.P.; Slezov, V.V.; Röpke, G.; Schmelzer, J., Jr. Shapes of Cluster Size Distributions Evolving in Nucleation—Growth Processes. In Nucleation Theory and Applications; Schmelzer, J.W.P., Röpke, G., Priezzhev, V.B., Eds.; Joint Institute for Nuclear Research Publishing House: Dubna, Russia, 1999; pp. 82–129. [Google Scholar]
- Slezov, V.V. Kinetics of First-Order Phase Transitions; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Von Smoluchowski, M. Versuch einer mathematischen Theorie der Koagulationskinetik kolloider Lösungen (Engl.: Attempt of a mathematical theory of the coagulation kinetics of colloidal solutions). Z. Phys. Chem. 1917, 92, 129–168. [Google Scholar]
- Binder, K.; Stauffer, D. Statistical theory of nucleation, condensation, and coagulation. Adv. Phys. 1976, 25, 343–396. [Google Scholar] [CrossRef]
- Wyslouzil, B.E.; Wilemski, G. Binary nucleation kinetics. II. Numerical solution of the birth-death equations. J. Chem. Phys. 1995, 103, 1137–1151. [Google Scholar] [CrossRef]
- Clouet, E. Modeling of Nucleation Processes. In Fundamentals of Modeling for Metals Processing; Furrer, D.U., Semiatin, S.L., Eds.; ASM Handbook; ASM International Publishers: Novelty, OH, USA, 2009; Volume 22A, pp. 203–219. [Google Scholar]
- Schmelzer, J.W.P.; Ulbricht, H. Thermodynamics of Finite Systems and the Kinetics of First-Order Phase Transitions. J. Colloid Interface Sci. 1987, 117, 325–338. [Google Scholar] [CrossRef]
- Abyzov, A.S.; Schmelzer, J.W.P. Nucleation versus Spinodal Decomposition in Confined Binary Solutions. J. Chem. Phys. 2007, 127, 114504/1–114504/17. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Abyzov, A.S. Thermodynamic analysis of nucleation in confined space: Generalized Gibbs’ approach. J. Chem. Phys. 2011, 134, 054511/1–054511/10. [Google Scholar] [CrossRef] [PubMed]
- Slezov, V.V.; Schmelzer, J.W.P.; Abyzov, A.S. A New Method of Determination of the Coefficients of Emission in Nucleation Theory. In Nucleation Theory and Applications; Schmelzer, J.W.P., Ed.; Selected Lectures of the Workshops 1997–2005; Joint Institute for Nuclear Research: Dubna, Russia; Wiley-VCH Publishers: Weinheim, Germany, 2005; pp. 39–73. [Google Scholar]
- Gutzow, I.; Schmelzer, J.W.P.; Dobreva, A. Kinetics of Transient Nucleation in Glass-Forming Liquids: A Retrospective and Recent Results. J. Non-Cryst. Solids 1997, 219, 1–16. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Abyzov, A.S.; Baidakov, V.G. Time of formation of the first supercritical nucleus, time-lag, and the steady-state nucleation rate. Int. J. Appl. Glass Sci. 2017, 8, 48–60. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Abyzov, A.S. On the theoretical description of nucleation in confined space. Am. Insitute Phys. Adv. 2011, 1, 042160/1–042160/9. [Google Scholar] [CrossRef]
- Tammann, G. Der Glaszustand (Engl.: The Vitreous State); Leopold Voss Verlag: Leipzig, Germany, 1933. [Google Scholar]
- Kalinina, A.M.; Filipovich, V.N.; Fokin, V.M. Stationary and non-stationary crystal nucleation in a glass of 2Na20·CaO·3SiO2 stoichiometric composition. J. Non-Cryst. Solids 1980, 38–39, 723–728. [Google Scholar] [CrossRef]
- Fokin, V.M.; Abyzov, A.S.; Zanotto, E.D.; Cassar, D.R.; Rodrigues, A.M.; Schmelzer, J.W.P. Crystal nucleation in glass-forming liquids: Variation of the size of the “structural units” with temperature. J. Non-Cryst. Solids 2016, 447, 35–44. [Google Scholar] [CrossRef]
- Abyzov, A.S.; Fokin, V.M.; Yuritsyn, N.S.; Rodrigues, A.M.; Schmelzer, J.W.P. The effect of heterogeneous structure of glass-forming liquids on crystal nucleation. J. Non-Cryst. Solids 2017, 462, 32–40. [Google Scholar] [CrossRef]
- Ono, S.; Kondo, S. Molecular theory of surface tension in liquids. In Handbuch der Physik (Encyclopedia of Physics); Flügge, S., Ed.; Springer: Berlin/Heidelberg, Germany, 1960; Volume 10, pp. 134–280. [Google Scholar]
- Renninger, R.G.; Hiller, F.C.; Bone, R.C. Comment on “Self-nucleation in the sulfuric acid-water system”. J. Chem. Phys. 1981, 75, 1584–1585. [Google Scholar] [CrossRef][Green Version]
- Wilemski, G. Composition of the critical nucleus in multi-component vapor nucleation. J. Chem. Phys. 1984, 80, 1370–1372. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Mahnke, R. General Formulae for the Curvature Dependence of Droplets and Bubbles. J. Chem. Soc. Faraday Trans. I 1986, 82, 1413–1420. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P. The Curvature Dependence of Surface Tension of Small Droplets. J. Chem. Soc. Faraday Trans. I 1986, 82, 1421–1428. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Gutzow, I.; Schmelzer, J., Jr. Curvature Dependent Surface Tension and Nucleation Theory. J. Colloid Interface Sci. 1996, 178, 657–665. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Baidakov, V.G. Comment on “Simple improvements to classical nucleation models”. Phys. Rev. E 2016, 94, 026801/1–026801/4. [Google Scholar] [CrossRef]
- Baidakov, V.G. Explosive Boiling of Superheated Cryogenic Liquids; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Tolman, R. The Effect of Droplet Size on Surface Tension. J. Chem. Phys. 1949, 17, 333–337. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Abyzov, A.S.; Fokin, V.M.; Schick, C. Kauzmann paradox and the crystallization of glass-forming melts. J. Non-Cryst. Solids 2018, 501, 21–35. [Google Scholar] [CrossRef]
- Fokin, V.M.; Zanotto, E.D. Crystal nucleation in silicate glasses: The temperature and size dependence of crystal/liquid surface energy. J. Non-Cryst. Solids 2000, 265, 105–112. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Abyzov, A.S. Comments on the thermodynamic analysis of nucleation in confined space. J. Non-Cryst. Solids 2014, 384, 2–7. [Google Scholar] [CrossRef]
- Blander, M.; Katz, J.L. The Thermodynamics of Cluster Formation in Nucleation Theory. J. Stat. Phys. 1972, 4, 55–59. [Google Scholar] [CrossRef]
- Girshick, S.L.; Chiu, C.-P. Kinetic nucleation theory: A new expression for the rate of homogeneous nucleation from an ideal supersaturated vapor. J. Chem. Phys. 1990, 93, 1273–1277. [Google Scholar] [CrossRef]
- Girshick, S.L. Comment on: “Self-consistency correction to homogeneous nucleation theory”. J. Chem. Phys. 1991, 94, 826–827. [Google Scholar] [CrossRef]
- Fokin, V.M.; Zanotto, E.D.; Schmelzer, J.W.P. Homogeneous Nucleation versus Glass Transition Temperature. J. Non-Cryst. Solids 2003, 321, 52–65. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Abyzov, A.S.; Fokin, V.M.; Schick, C.; Zanotto, E.D. Crystallization in glass-forming liquids: Effects of fragility and glass transition temperature. J. Non-Cryst. Solids 2015, 428, 68–74. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Schick, C. Dependence of Crystallization Processes of Glass-forming Melts on Prehistory: A Theoretical Approach to a Quantitative Treatment. Phys. Chem. Glas. Eur. J. Glass Sci. Technol. B 2012, 53, 99–106. [Google Scholar]
- Zanotto, E.D.; Cassar, D.R. The race within supercooled liquids: Relaxation versus crystallization. J. Chem. Phys. 2018, 149, 024503/1–024503/9. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Tropin, T.V. Glass transition, crystallization of glass-forming melts, and entropy. Entropy 2018, 20, 103. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abyzov, A.S.; Schmelzer, J.W.P.; Fokin, V.M.; Zanotto, E.D. Crystallization of Supercooled Liquids: Self-Consistency Correction of the Steady-State Nucleation Rate. Entropy 2020, 22, 558. https://doi.org/10.3390/e22050558
Abyzov AS, Schmelzer JWP, Fokin VM, Zanotto ED. Crystallization of Supercooled Liquids: Self-Consistency Correction of the Steady-State Nucleation Rate. Entropy. 2020; 22(5):558. https://doi.org/10.3390/e22050558
Chicago/Turabian StyleAbyzov, Alexander S., Jürn W. P. Schmelzer, Vladimir M. Fokin, and Edgar D. Zanotto. 2020. "Crystallization of Supercooled Liquids: Self-Consistency Correction of the Steady-State Nucleation Rate" Entropy 22, no. 5: 558. https://doi.org/10.3390/e22050558
APA StyleAbyzov, A. S., Schmelzer, J. W. P., Fokin, V. M., & Zanotto, E. D. (2020). Crystallization of Supercooled Liquids: Self-Consistency Correction of the Steady-State Nucleation Rate. Entropy, 22(5), 558. https://doi.org/10.3390/e22050558