KStable: A Computational Method for Predicting Protein Thermal Stability Changes by K-Star with Regular-mRMR Feature Selection


Abstract
1. Introduction
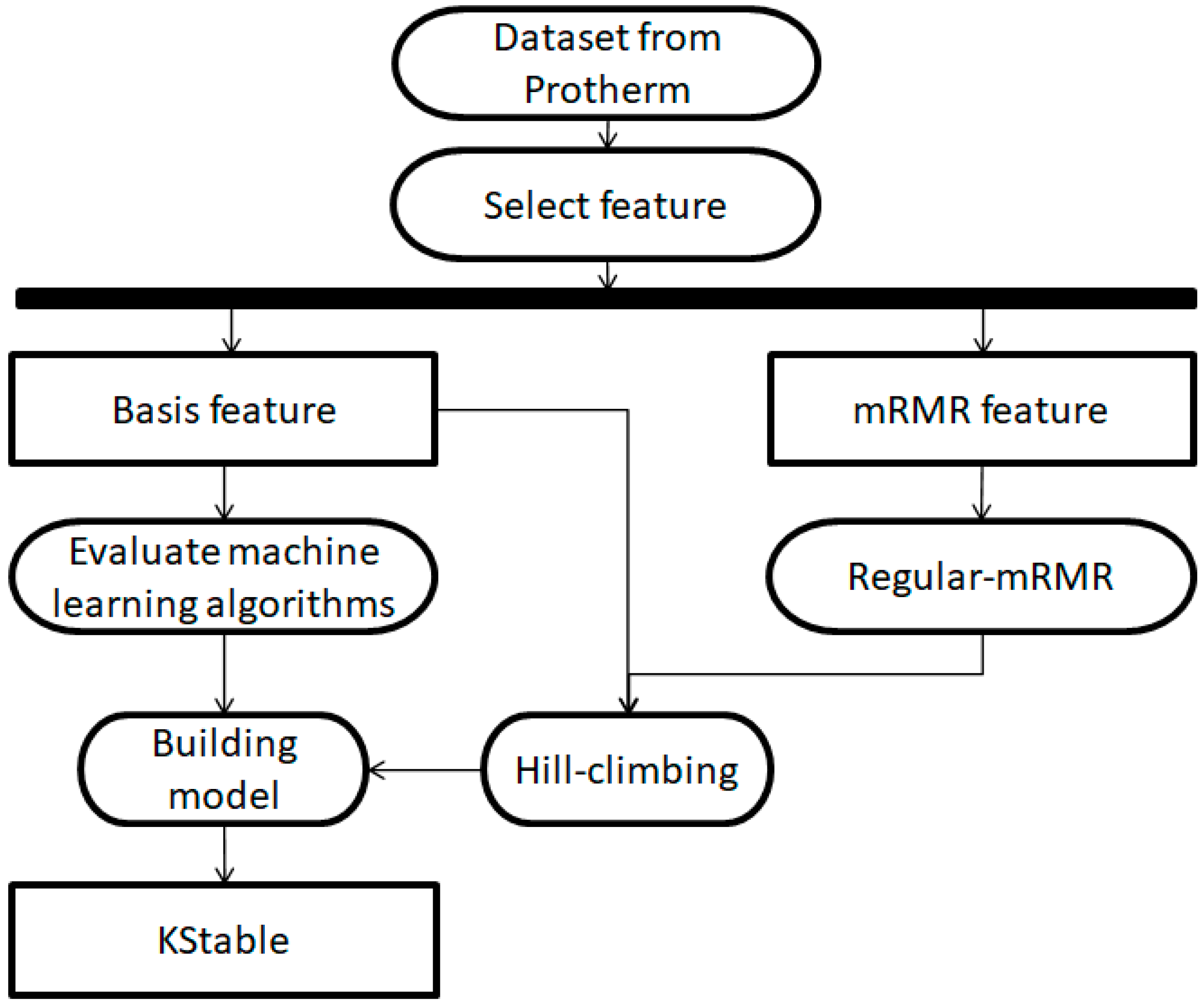
2. Materials and Methods
2.1. Dataset
2.2. Features
2.3. mRMR Feature Selection
2.4. Regular-mRMR Feature Selection
- Create N feature sets denoted as PT1~N by randomly selecting 868 samples N times from all data in S2864 (868 positive mutations and 1996 negative mutations).
- Perform mRMR-feature selection on each PT feature set.
- For each feature j, calculate the weight ki,j according to its mRMR score rank of each PTj, with n being the number of features to be chosen (Equation (4) below).
- The significance score Sj is defined by the average ki,j (Equation (5) below). The priority of each feature is determined by the significance score.
2.5. Hill-Climbing Algorithm
2.6. Evaluation
3. Results and Discussion
3.1. Ranking Features in the PT Files by mRMR
3.2. Classifier Selection
3.3. Basis Features Found Based on the Hill-Climbing Algorithm
3.4. Comparison of mRMR with Other Feature-Selection Methods
3.5. Comparison of KStable with Other Predictors
3.6. Feature Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Capriotti, E.; Fariselli, P.; Casadio, R. A neural-network-based method for predicting protein stability changes upon single point mutations. Bioinformatics 2004, 20, i63–i68. [Google Scholar] [CrossRef]
- Frenz, C.M. Neural network-based prediction of mutation-induced protein stability changes in staphylococcal nuclease at 20 residue positions. Proteins Struct. Funct. Bioinform. 2005, 59, 147–151. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Sarai, A. Thermodynamic database for proteins: Features and applications. Methods Mol. Biol. 2010, 609, 97–112. [Google Scholar]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. Cupsat: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 34, W239–W242. [Google Scholar] [CrossRef]
- Dehouck, Y.; Kwasigroch, J.M.; Gilis, D.; Rooman, M. Popmusic 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinform. 2011, 12, 151. [Google Scholar] [CrossRef]
- Masso, M.; Vaisman, I.I. Auto-mute: Web-based tools for predicting stability changes in proteins due to single amino acid replacements. Protein Eng. Des. Sel. 2010, 23, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Worth, C.L.; Preissner, R.; Blundell, T.L. SDM—A server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011, 39, W215–W222. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. INPS-MD: A web server to predict stability of protein variants from sequence and structure. Bioinformatics 2016, 32, 2542–2544. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. MCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2013, 30, 335–342. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Ochoa-Montaño, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.T.; Gromiha, M.M.; Ho, S.Y. IPTREE-STAB: Interpretable decision tree based method for predicting protein stability changes upon mutations. Bioinformatics 2007, 23, 1292–1293. [Google Scholar] [CrossRef] [PubMed]
- Folkman, L.; Stantic, B.; Sattar, A.; Zhou, Y. EASE-MM: Sequence-based prediction of mutation-induced stability changes with feature-based multiple models. J. Mol. Biol. 2016, 428, 1394–1405. [Google Scholar] [CrossRef] [PubMed]
- Folkman, L.; Stantic, B.; Sattar, A. Towards sequence-based prediction of mutation-induced stability changes in unseen non-homologous proteins. BMC Genom. 2014, 15, S4. [Google Scholar] [CrossRef]
- Folkman, L.; Stantic, B.; Sattar, A. Feature-based multiple models improve classification of mutation-induced stability changes. BMC Genom. 2014, 15, S6. [Google Scholar] [CrossRef] [PubMed]
- Fariselli, P.; Martelli, P.L.; Savojardo, C.; Casadio, R. INPS: Predicting the impact of non-synonymous variations on protein stability from sequence. Bioinformatics 2015, 31, 2816–2821. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Bioinform. 2006, 62, 1125–1132. [Google Scholar] [CrossRef]
- Chen, C.W.; Lin, J.; Chu, Y.W. iStable: Off-the-shelf predictor integration for predicting protein stability changes. BMC Bioinform. 2013, 14, S5. [Google Scholar]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. Duet: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef]
- Broom, A.; Jacobi, Z.; Trainor, K.; Meiering, E.M. Computational tools help improve protein stability but with a solubility tradeoff. J. Biol. Chem. 2017, 292, 14349–14361. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. Dynamut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W351. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.; Srivastava, A.K.; Wang, L. Sequence feature-based prediction of protein stability changes upon amino acid substitutions. BMC Genom. 2010, 11, S5. [Google Scholar] [CrossRef] [PubMed]
- Saeys, Y.; Inza, I.; Larrañaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef]
- Ding, C.; Peng, H. Minimum redundancy feature selection from microarray gene expression data. J. Bioinform. Comput. Biol. 2005, 3, 185–205. [Google Scholar] [CrossRef]
- Nunes, C.M.; Britto, A.D.S.; Kaestner, C.A.; Sabourin, R. Feature subset selection using an optimized hill climbing algorithm for handwritten character recognition. In Proceedings of the Joint IAPR International Workshops on Statistical Techniques in Pattern Recognition (SPR) and Structural and Syntactic Pattern Recognition (SSPR), Lisbon, Portugal, 18–20 August 2004; pp. 1018–1025. [Google Scholar]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The weka data mining software: An update. ACM SIGKDD Explor. Newslett. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Frank, E.; Hall, M.; Trigg, L.; Holmes, G.; Witten, I.H. Data mining in bioinformatics using weka. Bioinformatics 2004, 20, 2479–2481. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.G.; Trigg, L.E. K*: An instance-based learner using an entropic distance measure. In Proceedings of the Machine Learning Proceedings 1995, Tahoe City, CA, USA, 9–12 July 1995; Elsevier: Atlanta, GA, USA, 1995; pp. 108–114. [Google Scholar]
- Bava, K.A.; Gromiha, M.M.; Uedaira, H.; Kitajima, K.; Sarai, A. Protherm, version 4.0: Thermodynamic database for proteins and mutants. Nucleic Acids Res. 2004, 32, D120–D121. [Google Scholar] [CrossRef]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. Aaindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2007, 36, D202–D205. [Google Scholar] [CrossRef] [PubMed]
- Won, H.H.; Kim, M.J.; Kim, S.; Kim, J.W. Ensempro: An ensemble approach to predicting transcription start sites in human genomic DNA sequences. Genomics 2008, 91, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lu, L.; Feng, K.; Li, W.; Song, J.; Zheng, L.; Yuan, Y.; Zeng, Z.; Feng, K.; Lu, W. Multiple classifier integration for the prediction of protein structural classes. J. Comput. Chem. 2009, 30, 2248–2254. [Google Scholar] [CrossRef]
- Bontempi, G.; Meyer, P.E. Causal filter selection in microarray data. In Proceedings of the 27th International Conference on Machine Learning, Haifa, Israel, 21–24 June 2010; pp. 95–102. [Google Scholar]
- Chang, C.C.; Lin, C.J. Libsvm: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Quinlan, J.R. Induction of decision trees. Mach. Learn. 1986, 1, 81–106. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Kuncheva, L.I.; Alonso, C.J. Rotation forest: A new classifier ensemble method. IEEE Trans. Pattern Anal. Mach. Intell. 2006, 28, 1619–1630. [Google Scholar] [CrossRef]
- Popov, P.; Peng, Y.; Shen, L.; Stevens, R.C.; Cherezov, V.; Liu, Z.; Katritch, V. Computational design of thermostabilizing point mutations for G protein-coupled receptors. eLife 2018, 7, e34729. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.R.; de Araújo, G.C.; Zanphorlin, L.M.; Ruller, R.; Franco, F.C.; Torres, F.A.; Mertens, J.A.; Bowman, M.J.; Gomes, E.; Da Silva, R. Engineering increased thermostability in the GH-10 endo-1, 4-β-xylanase from Thermoascus aurantiacus CBMAI 756. Int. J. Biol. Macromol. 2016, 93, 20–26. [Google Scholar] [CrossRef]
- Magyar, C.; Gromiha, M.M.; Sávoly, Z.; Simon, I. The role of stabilization centers in protein thermal stability. Biochem. Biophys. Res. Commun. 2016, 471, 57–62. [Google Scholar] [CrossRef]


{kind=link}
| Category | Classifier | ACC | MCC |
|---|---|---|---|
| function | LIBSVM | 0.781 | 0.436 |
| lazy | KStar | 0.817 | 0.547 |
| meta | RotationForest | 0.805 | 0.513 |
| rules | PART 1 | 0.788 | 0.478 |
| trees | RandomForest | 0.784 | 0.474 |
| trees | RandomTree | 0.774 | 0.459 |
| Feature Combination | Sn | Sp | ACC | MCC |
|---|---|---|---|---|
| Basis | 0.641 | 0.906 | 0.818 | 0.578 |
| Basis + M324 | 0.656 | 0.887 | 0.810 | 0.562 |
| Basis + V651 | 0.677 | 0.894 | 0.822 | 0.590 |
| Basis + V651 + V453 | 0.687 | 0.888 | 0.821 | 0.590 |
| Basis + V651 + V453 + M149 | 0.708 | 0.887 | 0.827 | 0.607 |
| Method | Sn | Sp | ACC | MCC |
|---|---|---|---|---|
| Regular-mRMR | 0.708 | 0.887 | 0.827 | 0.607 |
| InfoGain | 0.683 | 0.896 | 0.825 | 0.597 |
| ChiSquared | 0.692 | 0.890 | 0.824 | 0.598 |
| Method | Sn | Sp | ACC | MCC |
|---|---|---|---|---|
| Sequence-based | ||||
| KStable | 0.411 | 0.856 | 0.708 | 0.298 |
| EASE-MM | 0.658 | 0.757 | 0.724 | 0.402 |
| I-Mutant2.0_seq | 0.185 | 0.918 | 0.674 | 0.151 |
| INPS_seq | 0.260 | 0.901 | 0.687 | 0.211 |
| iPTREE-STAB | 0.233 | 0.949 | 0.710 | 0.271 |
| MUpro | 0.267 | 0.901 | 0.689 | 0.218 |
| Structure-based | ||||
| AUTO-MUTE SVM | 0.075 | 0.969 | 0.671 | 0.101 |
| AUTO-MUTE RF | 0.137 | 0.976 | 0.696 | 0.222 |
| CUPSAT | 0.342 | 0.747 | 0.612 | 0.093 |
| DUET | 0.308 | 0.962 | 0.744 | 0.382 |
| I-Mutant2.0 | 0.233 | 0.918 | 0.689 | 0.210 |
| MAESTRO | 0.342 | 0.921 | 0.728 | 0.334 |
| mCSM | 0.212 | 0.979 | 0.724 | 0.325 |
| PoPMuSiC | 0.247 | 0.955 | 0.719 | 0.302 |
| SDM | 0.733 | 0.736 | 0.735 | 0.448 |
| SDM2 | 0.616 | 0.774 | 0.721 | 0.384 |
| Structure * | Method | Sn | Sp | ACC | MCC |
|---|---|---|---|---|---|
| Coil | KStable | 0.444 | 0.818 | 0.636 | 0.284 |
| EASE-MM | 0.571 | 0.682 | 0.628 | 0.255 | |
| Helix | KStable | 0.400 | 0.806 | 0.661 | 0.222 |
| EASE-MM | 0.750 | 0.676 | 0.702 | 0.409 | |
| Strand | KStable | 0.348 | 0.924 | 0.830 | 0.308 |
| EASE-MM | 0.652 | 0.873 | 0.837 | 0.474 | |
| Buried | KStable | 0.419 | 0.903 | 0.777 | 0.368 |
| EASE-MM | 0.500 | 0.835 | 0.748 | 0.339 | |
| Surface | KStable | 0.315 | 0.725 | 0.514 | 0.044 |
| EASE-MM | 0.796 | 0.549 | 0.676 | 0.357 | |
| Under surface | KStable | 0.567 | 0.831 | 0.747 | 0.405 |
| EASE-MM | 0.733 | 0.708 | 0.716 | 0.414 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-W.; Chang, K.-P.; Ho, C.-W.; Chang, H.-P.; Chu, Y.-W. KStable: A Computational Method for Predicting Protein Thermal Stability Changes by K-Star with Regular-mRMR Feature Selection. Entropy 2018, 20, 988. https://doi.org/10.3390/e20120988
Chen C-W, Chang K-P, Ho C-W, Chang H-P, Chu Y-W. KStable: A Computational Method for Predicting Protein Thermal Stability Changes by K-Star with Regular-mRMR Feature Selection. Entropy. 2018; 20(12):988. https://doi.org/10.3390/e20120988
Chicago/Turabian StyleChen, Chi-Wei, Kai-Po Chang, Cheng-Wei Ho, Hsung-Pin Chang, and Yen-Wei Chu. 2018. "KStable: A Computational Method for Predicting Protein Thermal Stability Changes by K-Star with Regular-mRMR Feature Selection" Entropy 20, no. 12: 988. https://doi.org/10.3390/e20120988
APA StyleChen, C.-W., Chang, K.-P., Ho, C.-W., Chang, H.-P., & Chu, Y.-W. (2018). KStable: A Computational Method for Predicting Protein Thermal Stability Changes by K-Star with Regular-mRMR Feature Selection. Entropy, 20(12), 988. https://doi.org/10.3390/e20120988