
The Cellular Senescence Stress Response in Post-Mitotic Brain Cells: Cell Survival at the Expense of Tissue Degeneration


 ,
,  ,
,
Abstract
:1. Introduction
2. Identifying Senescent Brain Cells
2.1. Absence of Proliferation/Stable Cell Cycle Arrest
2.2. Cell Death Resistance
2.3. Secretory Phenotype
2.4. Senescence Associated β-Galactosidase
2.5. Concluding Remarks on Identifying Senescent Cells
3. Neurons
3.1. Neuronal Senescence in Tauopathies and Peripheral Neuropathies
3.2. Neuronal Senescence in Parkinson’s Disease
3.3. Neuronal Senescence in Aging
3.4. General Considerations for Evaluating Neuronal Senescence
3.5. Concluding Remarks
4. Astrocytes
Concluding Remarks
5. Endothelial Cells
Concluding Remarks
6. Oligodendrocytes
Concluding Remarks
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levi-Montalcini, R.; Booker, B. Destruction of the Sympathetic Ganglia in Mammals by an Antiserum to a Nerve-Growth Protein. Proc. Natl. Acad. Sci. USA 1960, 46, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Oppenheim, R.; Milligan, C. Programmed Cell Death. In Neuroscience and Biobehavioral Psychology; Elsevier: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.A.; Liu, D.X.; Troy, C.M.; Biswas, S.C. Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim. Biophys. Acta 2007, 1772, 392–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Wang, Z.; Liu, J.P. Roles of Telomere Biology in Cell Senescence, Replicative and Chronological Ageing. Cells 2019, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Matjusaitis, M.; Chin, G.; Sarnoski, E.A.; Stolzing, A. Biomarkers to identify and isolate senescent cells. Ageing Res. Rev. 2016, 29, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dodig, S.; Cepelak, I.; Pavic, I. Hallmarks of senescence and aging. Biochem. Med. 2019, 29, 030501. [Google Scholar] [CrossRef]
- Roy, A.L.; Sierra, F.; Howcroft, K.; Singer, D.S.; Sharpless, N.; Hodes, R.J.; Wilder, E.L.; Anderson, J.M. A Blueprint for Characterizing Senescence. Cell 2020, 183, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660 e2654. [Google Scholar] [CrossRef] [Green Version]
- Casella, G.; Munk, R.; Kim, K.M.; Piao, Y.; De, S.; Abdelmohsen, K.; Gorospe, M. Transcriptome signature of cellular senescence. Nucleic Acids Res. 2019, 47, 7294–7305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Gillispie, G.J.; Sah, E.; Krishnamurthy, S.; Ahmidouch, M.Y.; Zhang, B.; Orr, M.E. Evidence of the Cellular Senescence Stress Response in Mitotically Active Brain Cells-Implications for Cancer and Neurodegeneration. Life 2021, 11, 153. [Google Scholar] [CrossRef]
- Barrio-Alonso, E.; Fontana, B.; Valero, M.; Frade, J.M. Pathological Aspects of Neuronal Hyperploidization in Alzheimer’s Disease Evidenced by Computer Simulation. Front. Genet. 2020, 11, 287. [Google Scholar] [CrossRef] [PubMed]
- Ain, Q.; Schmeer, C.; Penndorf, D.; Fischer, M.; Bondeva, T.; Forster, M.; Haenold, R.; Witte, O.W.; Kretz, A. Cell cycle-dependent and -independent telomere shortening accompanies murine brain aging. Aging 2018, 10, 3397–3420. [Google Scholar] [CrossRef] [PubMed]
- Jacome Burbano, M.S.; Gilson, E. Long-lived post-mitotic cell aging: Is a telomere clock at play? Mech. Ageing Dev. 2020, 189, 111256. [Google Scholar] [CrossRef]
- Mosch, B.; Morawski, M.; Mittag, A.; Lenz, D.; Tarnok, A.; Arendt, T. Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. J. Neurosci. 2007, 27, 6859–6867. [Google Scholar] [CrossRef]
- Fischer, H.G.; Morawski, M.; Bruckner, M.K.; Mittag, A.; Tarnok, A.; Arendt, T. Changes in neuronal DNA content variation in the human brain during aging. Aging Cell 2012, 11, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Varvel, N.H.; Bhaskar, K.; Patil, A.R.; Pimplikar, S.W.; Herrup, K.; Lamb, B.T. Abeta oligomers induce neuronal cell cycle events in Alzheimer’s disease. J. Neurosci. 2008, 28, 10786–10793. [Google Scholar] [CrossRef]
- Modi, P.K.; Jaiswal, S.; Sharma, P. Regulation of Neuronal Cell Cycle and Apoptosis by MicroRNA 34a. Mol. Cell. Biol. 2016, 36, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, M.; Modi, P.K.; Sharma, P. Aberrant activation of neuronal cell cycle caused by dysregulation of ubiquitin ligase Itch results in neurodegeneration. Cell Death Dis. 2020, 11, 441. [Google Scholar] [CrossRef] [PubMed]
- Hradek, A.C.; Lee, H.P.; Siedlak, S.L.; Torres, S.L.; Jung, W.; Han, A.H.; Lee, H.G. Distinct chronology of neuronal cell cycle re-entry and tau pathology in the 3xTg-AD mouse model and Alzheimer’s disease patients. J. Alzheimers Dis. 2015, 43, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andorfer, C.; Acker, C.M.; Kress, Y.; Hof, P.R.; Duff, K.; Davies, P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 2005, 25, 5446–5454. [Google Scholar] [CrossRef] [Green Version]
- Delobel, P.; Lavenir, I.; Ghetti, B.; Holzer, M.; Goedert, M. Cell-cycle markers in a transgenic mouse model of human tauopathy: Increased levels of cyclin-dependent kinase inhibitors p21Cip1 and p27Kip1. Am. J. Pathol. 2006, 168, 878–887. [Google Scholar] [CrossRef] [Green Version]
- Barrio-Alonso, E.; Hernandez-Vivanco, A.; Walton, C.C.; Perea, G.; Frade, J.M. Cell cycle reentry triggers hyperploidization and synaptic dysfunction followed by delayed cell death in differentiated cortical neurons. Sci. Rep. 2018, 8, 14316. [Google Scholar] [CrossRef] [PubMed]
- Marlier, Q.; D’Aes, T.; Verteneuil, S.; Vandenbosch, R.; Malgrange, B. Core cell cycle machinery is crucially involved in both life and death of post-mitotic neurons. Cell. Mol. Life Sci. 2020, 77, 4553–4571. [Google Scholar] [CrossRef]
- Fujimaki, K.; Li, R.; Chen, H.; Della Croce, K.; Zhang, H.H.; Xing, J.; Bai, F.; Yao, G. Graded regulation of cellular quiescence depth between proliferation and senescence by a lysosomal dimmer switch. Proc. Natl. Acad. Sci. USA 2019, 116, 22624–22634. [Google Scholar] [CrossRef]
- Zhu, Y.I.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef]
- Kole, A.J.; Annis, R.P.; Deshmukh, M. Mature neurons: Equipped for survival. Cell Death Dis. 2013, 4, e689. [Google Scholar] [CrossRef] [Green Version]
- Oppenheim, R.W. Cell death during development of the nervous system. Annu. Rev. Neurosci. 1991, 14, 453–501. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.M.; Smith, M.I.; Farrag, L.; Deshmukh, M. Chromatin modification of Apaf-1 restricts the apoptotic pathway in mature neurons. J. Cell Biol. 2007, 179, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Donovan, M.; Cotter, T.G. Caspase-independent photoreceptor apoptosis in vivo and differential expression of apoptotic protease activating factor-1 and caspase-3 during retinal development. Cell Death Differ. 2002, 9, 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, A.G.; Ota, K.; Wang, G.; Movsesyan, V.; Bao, W.L.; Yoshihara, K.; Faden, A.I. Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J. Neurosci. 2001, 21, 7439–7446. [Google Scholar] [CrossRef] [Green Version]
- Annis, R.P.; Swahari, V.; Nakamura, A.; Xie, A.X.; Hammond, S.M.; Deshmukh, M. Mature neurons dynamically restrict apoptosis via redundant premitochondrial brakes. FEBS J. 2016, 283, 4569–4582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, P.; Liu, Y.; Chen, J.; Cheng, Z. Cell Cycle Proteins as Key Regulators of Postmitotic Cell Death. Yale J. Biol. Med. 2019, 92, 641–650. [Google Scholar] [PubMed]
- Sapieha, P.; Mallette, F.A. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol. 2018, 28, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Woods, G.; Rane, A.; Demaria, M.; Campisi, J.; Andersen, J.K. Cellular senescence and the aging brain. Exp. Gerontol. 2015, 68, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Ruben, L.N. Recombinant DNA produced human IL-2, injected in vivo, will substitute for carrier priming of helper function in the South African clawed toad, Xenopus laevis. Immunol. Lett. 1986, 13, 227–230. [Google Scholar] [CrossRef]
- Walker, L.; McAleese, K.E.; Thomas, A.J.; Johnson, M.; Martin-Ruiz, C.; Parker, C.; Colloby, S.J.; Jellinger, K.; Attems, J. Neuropathologically mixed Alzheimer’s and Lewy body disease: Burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol. 2015, 129, 729–748. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Koss, D.J.; Erskine, D.; Walker, L.; Kurzawa-Akanbi, M.; Burn, D.; Donaghy, P.; Morris, C.; Taylor, J.P.; Thomas, A.; et al. Dementia with Lewy bodies: An update and outlook. Mol. Neurodegener. 2019, 14, 5. [Google Scholar] [CrossRef]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic Interactions between Abeta, tau, and alpha-synuclein: Acceleration of neuropathology and cognitive decline. J. Neurosci. 2010, 30, 7281–7289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Piechota, M.; Sunderland, P.; Wysocka, A.; Nalberczak, M.; Sliwinska, M.A.; Radwanska, K.; Sikora, E. Is senescence-associated beta-galactosidase a marker of neuronal senescence? Oncotarget 2016, 7, 81099–81109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herculano-Houzel, S. The remarkable, yet not extraordinary, human brain as a scaled-up primate brain and its associated cost. Proc. Natl. Acad. Sci. USA 2012, 109 (Suppl. 1), 10661–10668. [Google Scholar] [CrossRef] [Green Version]
- von Bartheld, C.S. Myths and truths about the cellular composition of the human brain: A review of influential concepts. J. Chem. Neuroanat. 2018, 93, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [Green Version]
- Chow, H.M.; Shi, M.; Cheng, A.; Gao, Y.; Chen, G.; Song, X.; So, R.W.L.; Zhang, J.; Herrup, K. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat. Neurosci. 2019, 22, 1806–1819. [Google Scholar] [CrossRef] [PubMed]
- Riessland, M.; Kolisnyk, B.; Kim, T.W.; Cheng, J.; Ni, J.; Pearson, J.A.; Park, E.J.; Dam, K.; Acehan, D.; Ramos-Espiritu, L.S.; et al. Loss of SATB1 Induces p21-Dependent Cellular Senescence in Post-mitotic Dopaminergic Neurons. Cell Stem Cell 2019, 25, 514–530.e8. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Villasenor, I.; Garwood, C.J.; Heath, P.R.; Simpson, J.E.; Ince, P.G.; Wharton, S.B. Expression of p16 and p21 in the frontal association cortex of ALS/MND brains suggests neuronal cell cycle dysregulation and astrocyte senescence in early stages of the disease. Neuropathol. Appl. Neurobiol. 2020, 46, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Trias, E.; Beilby, P.R.; Kovacs, M.; Ibarburu, S.; Varela, V.; Barreto-Nunez, R.; Bradford, S.C.; Beckman, J.S.; Barbeito, L. Emergence of Microglia Bearing Senescence Markers During Paralysis Progression in a Rat Model of Inherited ALS. Front. Aging Neurosci. 2019, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.B.; Ai, R.; Kaeser, G.E.; Salathia, N.S.; Yung, Y.C.; Liu, R.; Wildberg, A.; Gao, D.; Fung, H.L.; Chen, S.; et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 2016, 352, 1586–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreadis, A.; Brown, W.M.; Kosik, K.S. Structure and novel exons of the human tau gene. Biochemistry 1992, 31, 10626–10633. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Orr, M.E.; Sullivan, A.C.; Frost, B. A Brief Overview of Tauopathy: Causes, Consequences, and Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 637–648. [Google Scholar] [CrossRef]
- de Calignon, A.; Spires-Jones, T.L.; Pitstick, R.; Carlson, G.A.; Hyman, B.T. Tangle-bearing neurons survive despite disruption of membrane integrity in a mouse model of tauopathy. J. Neuropathol. Exp. Neurol. 2009, 68, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Marquez, A.; Guernsey, L.S.; Frizzi, K.E.; Cundiff, M.; Constantino, I.; Muttalib, N.; Arenas, F.; Zhou, X.; Lim, S.H.; Ferdousi, M.; et al. Tau associated peripheral and central neurodegeneration: Identification of an early imaging marker for tauopathy. Neurobiol. Dis. 2021, 151, 105273. [Google Scholar] [CrossRef]
- Acklin, S.; Zhang, M.; Du, W.; Zhao, X.; Plotkin, M.; Chang, J.; Campisi, J.; Zhou, D.; Xia, F. Depletion of senescent-like neuronal cells alleviates cisplatin-induced peripheral neuropathy in mice. Sci. Rep. 2020, 10, 14170. [Google Scholar] [CrossRef] [PubMed]
- Calls, A.; Torres-Espin, A.; Navarro, X.; Yuste, V.J.; Udina, E.; Bruna, J. Cisplatin-induced peripheral neuropathy is associated with neuronal senescence-like response. Neuro Oncol. 2021, 23, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Ezquerra, M.; Pastor, P.; Gaig, C.; Vidal-Taboada, J.M.; Cruchaga, C.; Munoz, E.; Marti, M.J.; Valldeoriola, F.; Aguilar, M.; Calopa, M.; et al. Different MAPT haplotypes are associated with Parkinson’s disease and progressive supranuclear palsy. Neurobiol. Aging 2011, 32, 547.e11–547.e16. [Google Scholar] [CrossRef]
- Davis, A.A.; Andruska, K.M.; Benitez, B.A.; Racette, B.A.; Perlmutter, J.S.; Cruchaga, C. Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiol. Aging 2016, 37, 209.e201–209.e207. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gao, F.; Wang, D.; Li, C.; Fu, Y.; He, W.; Zhang, J. Tau Pathology in Parkinson’s Disease. Front. Neurol. 2018, 9, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. First of two parts. N. Engl. J. Med. 1998, 339, 1044–1053. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Santa-Maria, I.; Hernandez, F.; Smith, M.A.; Perry, G.; Avila, J.; Moreno, F.J. Neurotoxic dopamine quinone facilitates the assembly of tau into fibrillar polymers. Mol. Cell. Biochem. 2005, 278, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Losada, N.; de la Rosa, J.; Larriva, M.; Wendelbo, R.; Aguirre, J.A.; Castresana, J.S.; Ballaz, S.J. Overexpression of alpha-synuclein promotes both cell proliferation and cell toxicity in human SH-SY5Y neuroblastoma cells. J. Adv. Res. 2020, 23, 37–45. [Google Scholar] [CrossRef]
- Yasui, D.; Miyano, M.; Cai, S.; Varga-Weisz, P.; Kohwi-Shigematsu, T. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature 2002, 419, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Nalls, M.A.; Hallgrimsdottir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics, C.; andMe Research, T.; Kerchner, G.A.; Ayalon, G.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Brichta, L.; Shin, W.; Jackson-Lewis, V.; Blesa, J.; Yap, E.L.; Walker, Z.; Zhang, J.; Roussarie, J.P.; Alvarez, M.J.; Califano, A.; et al. Identification of neurodegenerative factors using translatome-regulatory network analysis. Nat. Neurosci. 2015, 18, 1325–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, D.H.; Seol, W.; Son, I. Upregulation of the p53–p21 pathway by G2019S LRRK2 contributes to the cellular senescence and accumulation of alpha-synuclein. Cell Cycle 2019, 18, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Levy, O.A.; Malagelada, C.; Greene, L.A. Cell death pathways in Parkinson’s disease: Proximal triggers, distal effectors, and final steps. Apoptosis 2009, 14, 478–500. [Google Scholar] [CrossRef]
- Golde, T.E.; Miller, V.M. Proteinopathy-induced neuronal senescence: A hypothesis for brain failure in Alzheimer’s and other neurodegenerative diseases. Alzheimers Res. Ther. 2009, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Gralle, M. The neuronal insulin receptor in its environment. J. Neurochem. 2017, 140, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Nie, J.; Van Skike, C.E.; Valentine, J.M.; Orr, M.E. Mammalian Target of Rapamycin at the Crossroad Between Alzheimer’s Disease and Diabetes. Adv. Exp. Med. Biol. 2019, 1128, 185–225. [Google Scholar] [CrossRef]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Haring, H.U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef] [Green Version]
- Orr, M.E.; Salinas, A.; Buffenstein, R.; Oddo, S. Mammalian target of rapamycin hyperactivity mediates the detrimental effects of a high sucrose diet on Alzheimer’s disease pathology. Neurobiol. Aging 2014, 35, 1233–1242. [Google Scholar] [CrossRef] [Green Version]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.P.; Tchkonia, T.; Kruger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1061–1077 e1068. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Herrup, K. Loss of neuronal cell cycle control in ataxia-telangiectasia: A unified disease mechanism. J. Neurosci. 2005, 25, 2522–2529. [Google Scholar] [CrossRef] [Green Version]
- Arendt, T.; Bruckner, M.K.; Mosch, B.; Losche, A. Selective cell death of hyperploid neurons in Alzheimer’s disease. Am. J. Pathol. 2010, 177, 15–20. [Google Scholar] [CrossRef]
- Toots, A.; Rosendahl, E.; Lundin-Olsson, L.; Nordstrom, P.; Gustafson, Y.; Littbrand, H. Usual gait speed independently predicts mortality in very old people: A population-based study. J. Am. Med. Dir. Assoc. 2013, 14, 529.e1–529.e6. [Google Scholar] [CrossRef]
- Blain, H.; Carriere, I.; Sourial, N.; Berard, C.; Favier, F.; Colvez, A.; Bergman, H. Balance and walking speed predict subsequent 8-year mortality independently of current and intermediate events in well-functioning women aged 75 years and older. J. Nutr. Health Aging 2010, 14, 595–600. [Google Scholar] [CrossRef]
- Moreno-Blas, D.; Gorostieta-Salas, E.; Pommer-Alba, A.; Mucino-Hernandez, G.; Geronimo-Olvera, C.; Maciel-Baron, L.A.; Konigsberg, M.; Massieu, L.; Castro-Obregon, S. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging 2019, 11, 6175–6198. [Google Scholar] [CrossRef]
- Goyal, V.K. Lipofuscin pigment accumulation in human brain during aging. Exp. Gerontol. 1982, 17, 481–487. [Google Scholar] [CrossRef]
- Sitte, N.; Merker, K.; Grune, T.; von Zglinicki, T. Lipofuscin accumulation in proliferating fibroblasts in vitro: An indicator of oxidative stress. Exp. Gerontol. 2001, 36, 475–486. [Google Scholar] [CrossRef]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2013, 5, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Munoz-Espin, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef]
- Calvo, M.; Sanz-Blasco, S.; Caballero, E.; Villalobos, C.; Nunez, L. Susceptibility to excitotoxicity in aged hippocampal cultures and neuroprotection by non-steroidal anti-inflammatory drugs: Role of mitochondrial calcium. J. Neurochem. 2015, 132, 403–417. [Google Scholar] [CrossRef] [PubMed]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Herculano-Houzel, S. The glia/neuron ratio: How it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 2014, 62, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Stiles, J.; Jernigan, T.L. The basics of brain development. Neuropsychol. Rev. 2010, 20, 327–348. [Google Scholar] [CrossRef] [Green Version]
- Cayre, M.; Canoll, P.; Goldman, J.E. Cell migration in the normal and pathological postnatal mammalian brain. Prog. Neurobiol. 2009, 88, 41–63. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Contet, C.; Goulding, S.P.; Kuljis, D.A.; Barth, A.L. BK Channels in the Central Nervous System. Int. Rev. Neurobiol. 2016, 128, 281–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabata, H. Diverse subtypes of astrocytes and their development during corticogenesis. Front. Neurosci. 2015, 9, 114. [Google Scholar] [CrossRef] [Green Version]
- de Majo, M.; Koontz, M.; Rowitch, D.; Ullian, E.M. An update on human astrocytes and their role in development and disease. Glia 2020, 68, 685–704. [Google Scholar] [CrossRef]
- Jung, Y.J.; Chung, W.S. Phagocytic Roles of Glial Cells in Healthy and Diseased Brains. Biomol. Ther. 2018, 26, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arboledas, A.; Davila, J.C.; Sanchez-Mejias, E.; Navarro, V.; Nunez-Diaz, C.; Sanchez-Varo, R.; Sanchez-Mico, M.V.; Trujillo-Estrada, L.; Fernandez-Valenzuela, J.J.; Vizuete, M.; et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 2018, 66, 637–653. [Google Scholar] [CrossRef] [Green Version]
- Limbad, C.; Oron, T.R.; Alimirah, F.; Davalos, A.R.; Tracy, T.E.; Gan, L.; Desprez, P.Y.; Campisi, J. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 2020, 15, e0227887. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [Google Scholar] [CrossRef] [Green Version]
- Barres, B.A. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Phatnani, H.; Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munger, E.L.; Edler, M.K.; Hopkins, W.D.; Ely, J.J.; Erwin, J.M.; Perl, D.P.; Mufson, E.J.; Hof, P.R.; Sherwood, C.C.; Raghanti, M.A. Astrocytic changes with aging and Alzheimer’s disease-type pathology in chimpanzees. J. Comp. Neurol. 2019, 527, 1179–1195. [Google Scholar] [CrossRef]
- Boisvert, M.M.; Erikson, G.A.; Shokhirev, M.N.; Allen, N.J. The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 2018, 22, 269–285. [Google Scholar] [CrossRef] [Green Version]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnquist, C.; Beck, J.A.; Horikawa, I.; Obiorah, I.E.; Von Muhlinen, N.; Vojtesek, B.; Lane, D.P.; Grunseich, C.; Chahine, J.J.; Ames, H.M.; et al. Radiation-induced astrocyte senescence is rescued by Delta133p53. Neuro Oncol. 2019, 21, 474–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.; Torres, C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18, e12937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Zhang, T.; Liu, H.; Mi, Y.; Gou, X. Astrocyte Senescence and Alzheimer’s Disease: A Review. Front. Aging Neurosci. 2020, 12, 148. [Google Scholar] [CrossRef]
- Evans, R.J.; Wyllie, F.S.; Wynford-Thomas, D.; Kipling, D.; Jones, C.J. A P53-dependent, telomere-independent proliferative life span barrier in human astrocytes consistent with the molecular genetics of glioma development. Cancer Res. 2003, 63, 4854–4861. [Google Scholar]
- Pertusa, M.; García-Matas, S.; Rodríguez-Farré, E.; Sanfeliu, C.; Cristòfol, R. Astrocytes aged in vitro show a decreased neuroprotective capacity. J. Neurochem. 2007, 101, 794–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spilsbury, A.; Miwa, S.; Attems, J.; Saretzki, G. The role of telomerase protein TERT in Alzheimer’s disease and in tau-related pathology in vitro. J. Neurosci. 2015, 35, 1659–1674. [Google Scholar] [CrossRef] [Green Version]
- Bitto, A.; Sell, C.; Crowe, E.; Lorenzini, A.; Malaguti, M.; Hrelia, S.; Torres, C. Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 2010, 316, 2961–2968. [Google Scholar] [CrossRef]
- Görg, B.; Karababa, A.; Shafigullina, A.; Bidmon, H.J.; Häussinger, D. Ammonia-induced senescence in cultured rat astrocytes and in human cerebral cortex in hepatic encephalopathy. Glia 2015, 63, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Stigbrand, T. Radiation-induced cell death mechanisms. Tumour. Biol. 2010, 31, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Turnquist, C.; Horikawa, I.; Foran, E.; Major, E.O.; Vojtesek, B.; Lane, D.P.; Lu, X.; Harris, B.T.; Harris, C.C. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016, 23, 1515–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmnacher, K.; Krach, F.; Schneider, Y.; Alecu, J.E.; Mautner, L.; Klein, P.; Roybon, L.; Prots, I.; Xiang, W.; Winner, B. Unique signatures of stress-induced senescent human astrocytes. Exp. Neurol. 2020, 334, 113466. [Google Scholar] [CrossRef]
- Lye, J.J.; Latorre, E.; Lee, B.P.; Bandinelli, S.; Holley, J.E.; Gutowski, N.J.; Ferrucci, L.; Harries, L.W. Astrocyte senescence may drive alterations in GFAPα, CDKN2A p14(ARF), and TAU3 transcript expression and contribute to cognitive decline. Geroscience 2019, 41, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 2011, 34, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R.; Ronisz, A.; Tousseyn, T.; Rijal Upadhaya, A.; Balakrishnan, K.; Vandenberghe, R.; Vandenbulcke, M.; von Arnim, C.A.F.; Otto, M.; Beach, T.G.; et al. Different aspects of Alzheimer’s disease-related amyloid beta-peptide pathology and their relationship to amyloid positron emission tomography imaging and dementia. Acta Neuropathol. Commun. 2019, 7, 178. [Google Scholar] [CrossRef]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Csipo, T.; Lipecz, A.; Ashpole, N.M.; Balasubramanian, P.; Tarantini, S. Astrocyte senescence contributes to cognitive decline. Geroscience 2020, 42, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, J.; Heasman, S.J.; Alvarez, J.I.; Prat, A.; Lyck, R.; Engelhardt, B. Review: Leucocyte-endothelial cell crosstalk at the blood-brain barrier: A prerequisite for successful immune cell entry to the brain. Neuropathol. Appl. Neurobiol. 2011, 37, 24–39. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [Green Version]
- Barinda, A.J.; Ikeda, K.; Nugroho, D.B.; Wardhana, D.A.; Sasaki, N.; Honda, S.; Urata, R.; Matoba, S.; Hirata, K.I.; Emoto, N. Endothelial progeria induces adipose tissue senescence and impairs insulin sensitivity through senescence associated secretory phenotype. Nat. Commun. 2020, 11, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Matsui-Hirai, H.; Miyazaki-Akita, A.; Fukatsu, A.; Funami, J.; Ding, Q.F.; Kamalanathan, S.; Hattori, Y.; Ignarro, L.J.; Iguchi, A. Endothelial cellular senescence is inhibited by nitric oxide: Implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. USA 2006, 103, 17018–17023. [Google Scholar] [CrossRef] [Green Version]
- Ekstrand, J.; Hellsten, J.; Tingstrom, A. Environmental enrichment, exercise and corticosterone affect endothelial cell proliferation in adult rat hippocampus and prefrontal cortex. Neurosci. Lett. 2008, 442, 203–207. [Google Scholar] [CrossRef]
- Hobson, B.; Denekamp, J. Endothelial proliferation in tumours and normal tissues: Continuous labelling studies. Br. J. Cancer 1984, 49, 405–413. [Google Scholar] [CrossRef]
- Wong, A.D.; Ye, M.; Levy, A.; Rothstein, J.; Bergles, D.; Searson, P.C. The blood-brain barrier: An engineering perspective. Front Neuroeng. 2013, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Brain endothelial cell-cell junctions: How to “open” the blood brain barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katt, M.E.; Xu, Z.S.; Gerecht, S.; Searson, P.C. Human Brain Microvascular Endothelial Cells Derived from the BC1 iPS Cell Line Exhibit a Blood-Brain Barrier Phenotype. PLoS ONE 2016, 11, e0152105. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Hernandez, H.; Cuevas, E.; Lantz, S.M.; Paule, M.G.; Ali, S.F. Isolation and Culture of Brain Microvascular Endothelial Cells for In Vitro Blood-Brain Barrier Studies. Methods Mol. Biol. 2018, 1727, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Song, H.W.; Foreman, K.L.; Gastfriend, B.D.; Kuo, J.S.; Palecek, S.P.; Shusta, E.V. Transcriptomic comparison of human and mouse brain microvessels. Sci. Rep. 2020, 10, 12358. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca(2+) Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. The Isolated Brain Microvessel: A Versatile Experimental Model of the Blood-Brain Barrier. Front. Physiol. 2020, 11, 398. [Google Scholar] [CrossRef]
- Bauer, H.C.; Krizbai, I.A.; Bauer, H.; Traweger, A. “You Shall Not Pass”—Tight junctions of the blood brain barrier. Front. Neurosci. 2014, 8, 392. [Google Scholar] [CrossRef]
- Soares, J.P.; Cortinhas, A.; Bento, T.; Leitao, J.C.; Collins, A.R.; Gaivao, I.; Mota, M.P. Aging and DNA damage in humans: A meta-analysis study. Aging 2014, 6, 432–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarantini, S.; Tran, C.H.T.; Gordon, G.R.; Ungvari, Z.; Csiszar, A. Impaired neurovascular coupling in aging and Alzheimer’s disease: Contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp. Gerontol. 2017, 94, 52–58. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Farrall, A.J.; Wardlaw, J.M. Blood-brain barrier: Ageing and microvascular disease-systematic review and meta-analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Machida, T.; Dohgu, S.; Takata, F.; Matsumoto, J.; Kimura, I.; Koga, M.; Nakamoto, K.; Yamauchi, A.; Kataoka, Y. Role of thrombin-PAR1-PKCtheta/delta axis in brain pericytes in thrombin-induced MMP-9 production and blood-brain barrier dysfunction in vitro. Neuroscience 2017, 350, 146–157. [Google Scholar] [CrossRef]
- Brouns, R.; Wauters, A.; De Surgeloose, D.; Marien, P.; De Deyn, P.P. Biochemical markers for blood-brain barrier dysfunction in acute ischemic stroke correlate with evolution and outcome. Eur. Neurol. 2011, 65, 23–31. [Google Scholar] [CrossRef]
- Zhang, S.; An, Q.; Wang, T.; Gao, S.; Zhou, G. Autophagy- and MMP-2/9-mediated Reduction and Redistribution of ZO-1 Contribute to Hyperglycemia-increased Blood-Brain Barrier Permeability During Early Reperfusion in Stroke. Neuroscience 2018, 377, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Guilfoyle, M.R.; Carpenter, K.L.; Helmy, A.; Pickard, J.D.; Menon, D.K.; Hutchinson, P.J. Matrix Metalloproteinase Expression in Contusional Traumatic Brain Injury: A Paired Microdialysis Study. J. Neurotrauma 2015, 32, 1553–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fainardi, E.; Castellazzi, M.; Bellini, T.; Manfrinato, M.C.; Baldi, E.; Casetta, I.; Paolino, E.; Granieri, E.; Dallocchio, F. Cerebrospinal fluid and serum levels and intrathecal production of active matrix metalloproteinase-9 (MMP-9) as markers of disease activity in patients with multiple sclerosis. Mult. Scler. 2006, 12, 294–301. [Google Scholar] [CrossRef]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef]
- Rao, J.S.; Bhoopathi, P.; Chetty, C.; Gujrati, M.; Lakka, S.S. MMP-9 short interfering RNA induced senescence resulting in inhibition of medulloblastoma growth via p16(INK4a) and mitogen-activated protein kinase pathway. Cancer Res. 2007, 67, 4956–4964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungvari, Z.; Podlutsky, A.; Sosnowska, D.; Tucsek, Z.; Toth, P.; Deak, F.; Gautam, T.; Csiszar, A.; Sonntag, W.E. Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells: Role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1443–1457. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.R. The comet assay for DNA damage and repair: Principles, applications, and limitations. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef]
- Kiss, T.; Nyul-Toth, A.; Balasubramanian, P.; Tarantini, S.; Ahire, C.; DelFavero, J.; Yabluchanskiy, A.; Csipo, T.; Farkas, E.; Wiley, G.; et al. Single-cell RNA sequencing identifies senescent cerebromicrovascular endothelial cells in the aged mouse brain. Geroscience 2020, 42, 429–444. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Baker, D.J.; Tachibana, M.; Liu, C.C.; van Deursen, J.M.; Brott, T.G.; Bu, G.; Kanekiyo, T. Vascular Cell Senescence Contributes to Blood-Brain Barrier Breakdown. Stroke 2016, 47, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Graves, S.I.; Baker, D.J. Implicating endothelial cell senescence to dysfunction in the ageing and diseased brain. Basic Clin. Pharmacol. Toxicol. 2020, 127, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.E.; Robbins, A.B.; Hu, M.; Cao, X.; Betensky, R.A.; Clark, T.; Das, S.; Hyman, B.T. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1289–E1298. [Google Scholar] [CrossRef] [Green Version]
- Bryant, A.G.; Hu, M.; Carlyle, B.C.; Arnold, S.E.; Frosch, M.P.; Das, S.; Hyman, B.T.; Bennett, R.E. Cerebrovascular Senescence Is Associated With Tau Pathology in Alzheimer’s Disease. Front. Neurol. 2020, 11, 575953. [Google Scholar] [CrossRef] [PubMed]
- Michalicova, A.; Majerova, P.; Kovac, A. Tau Protein and Its Role in Blood-Brain Barrier Dysfunction. Front. Mol. Neurosci. 2020, 13, 570045. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Tateno, K.; Kunieda, T.; Komuro, I. Vascular cell senescence and vascular aging. J. Mol. Cell. Cardiol. 2004, 36, 175–183. [Google Scholar] [CrossRef]
- Spazzafumo, L.; Mensa, E.; Matacchione, G.; Galeazzi, T.; Zampini, L.; Recchioni, R.; Marcheselli, F.; Prattichizzo, F.; Testa, R.; Antonicelli, R.; et al. Age-related modulation of plasmatic beta-Galactosidase activity in healthy subjects and in patients affected by T2DM. Oncotarget 2017, 8, 93338–93348. [Google Scholar] [CrossRef] [Green Version]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [Green Version]
- van Tilborg, E.; de Theije, C.G.M.; van Hal, M.; Wagenaar, N.; de Vries, L.S.; Benders, M.J.; Rowitch, D.H.; Nijboer, C.H. Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia 2018, 66, 221–238. [Google Scholar] [CrossRef]
- Moore, S.; Meschkat, M.; Ruhwedel, T.; Trevisiol, A.; Tzvetanova, I.D.; Battefeld, A.; Kusch, K.; Kole, M.H.P.; Strenzke, N.; Möbius, W.; et al. A role of oligodendrocytes in information processing. Nat. Commun. 2020, 11, 5497. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.A.; Ehrenreich, H. Myelination and oligodendrocyte functions in psychiatric diseases. JAMA Psychiatry 2014, 71, 582–584. [Google Scholar] [CrossRef]
- Tse, K.H.; Herrup, K. DNA damage in the oligodendrocyte lineage and its role in brain aging. Mech. Ageing Dev. 2017, 161, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Giacci, M.K.; Bartlett, C.A.; Smith, N.M.; Iyer, K.S.; Toomey, L.M.; Jiang, H.; Guagliardo, P.; Kilburn, M.R.; Fitzgerald, M. Oligodendroglia Are Particularly Vulnerable to Oxidative Damage after Neurotrauma In Vivo. J. Neurosci. 2018, 38, 6491–6504. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.H.; Hales, C.N.; Ozanne, S.E. DNA damage, cellular senescence and organismal ageing: Causal or correlative? Nucleic Acids Res. 2007, 35, 7417–7428. [Google Scholar] [CrossRef] [PubMed]
- Ettle, B.; Schlachetzki, J.C.M.; Winkler, J. Oligodendroglia and Myelin in Neurodegenerative Diseases: More Than Just Bystanders? Mol. Neurobiol. 2016, 53, 3046–3062. [Google Scholar] [CrossRef] [Green Version]
- Kritsilis, M.; Rizou, S.; Koutsoudaki, P.N.; Evangelou, K.; Gorgoulis, V.G.; Papadopoulos, D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int. J. Mol. Sci. 2018, 19, 2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, e13296. [Google Scholar] [CrossRef]
- Werkman, I.L.; Lentferink, D.H.; Baron, W. Macroglial diversity: White and grey areas and relevance to remyelination. Cell. Mol. Life Sci. 2021, 78, 143–171. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Al-Mashhadi, S.; Simpson, J.E.; Heath, P.R.; Dickman, M.; Forster, G.; Matthews, F.E.; Brayne, C.; Ince, P.G.; Wharton, S.B.; Medical Research Council Cognitive, F.; et al. Oxidative Glial Cell Damage Associated with White Matter Lesions in the Aging Human Brain. Brain Pathol. 2015, 25, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, S.C.; Freeland, A.; Rusiniak, M.E.; Kunnev, D.; Cady, G.K. Cdkn1b overexpression in adult mice alters the balance between genome and tissue ageing. Nat. Commun. 2013, 4, 2626. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Cue, C.; Rueda, N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef] [PubMed]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
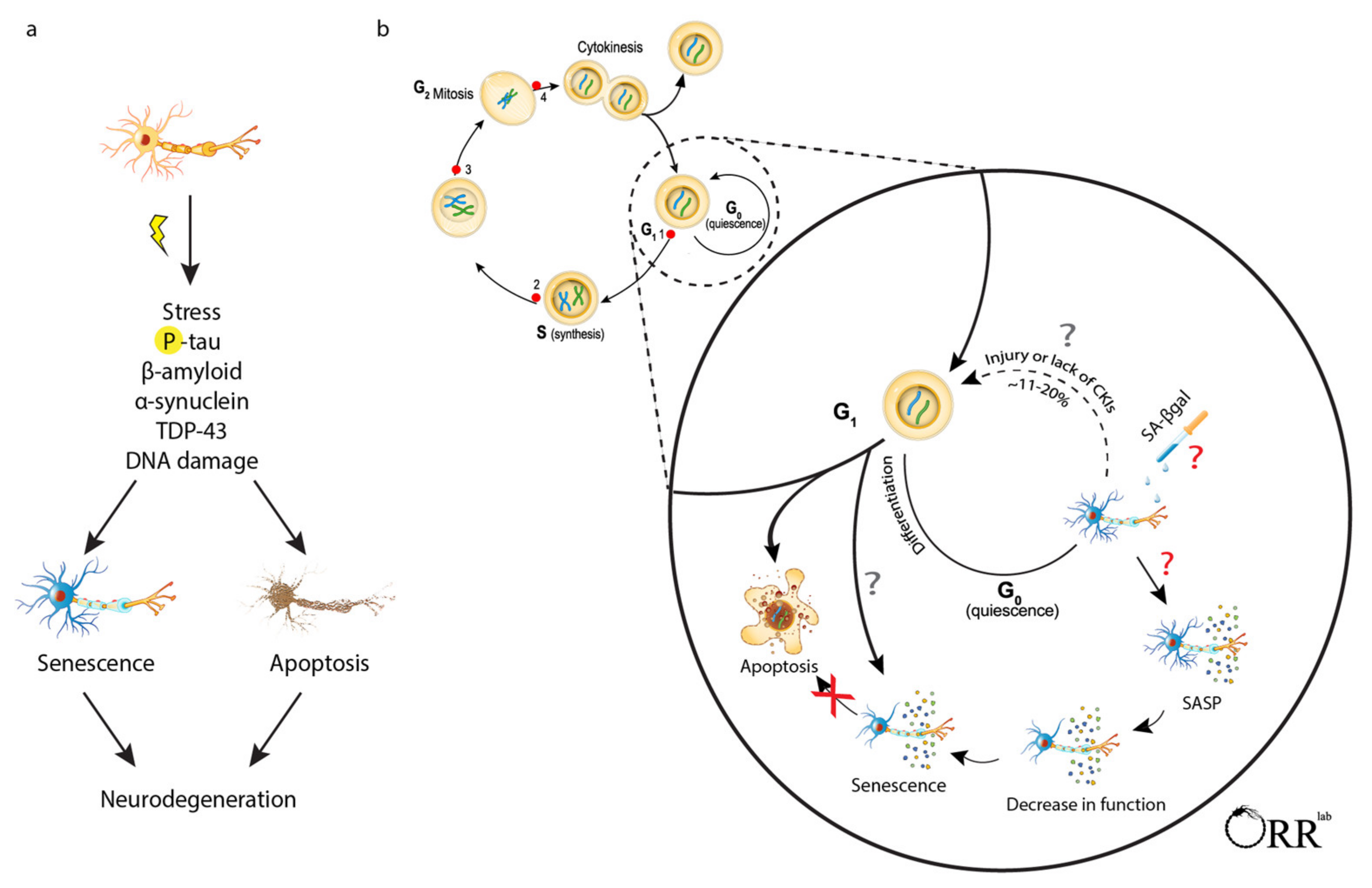

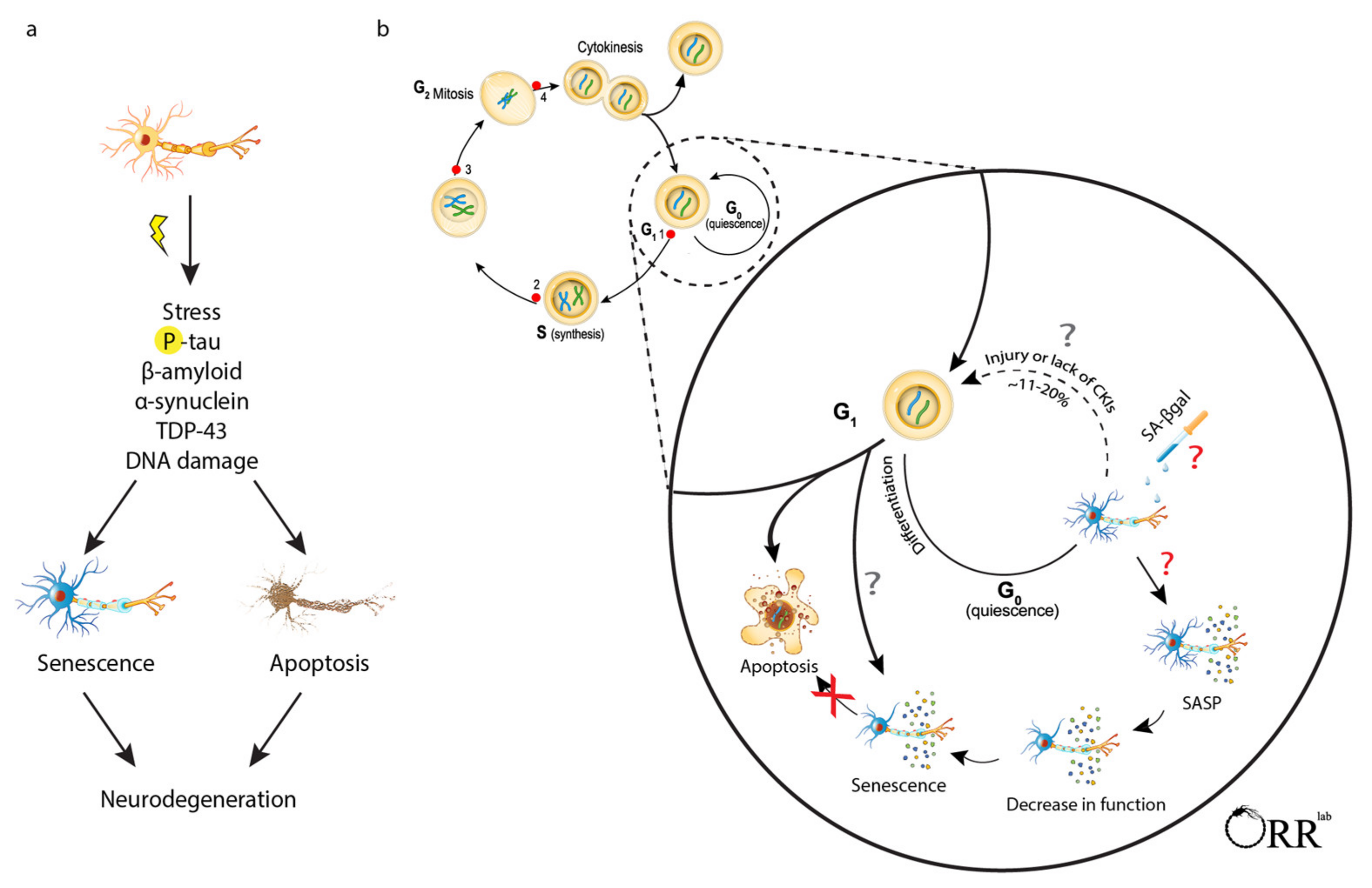{kind=link}
| Cell Type | Biomarkers | Reference |
|---|---|---|
| Neurons | SA-β-gal | [54], [55], [56], [65], [66], [77], [84], [89] |
| H2ax | [49], [66], [89] | |
| Comet Assay | [89] | |
| Nuclear morphology | [89] | |
| Cytosolic HMGB1 | [65] | |
| Telomere-associated DNA damage foci | [84] | |
| p53 | [77] | |
| p21 | [49], [54], [55], [56], [57], [65], [66], [77], [89] | |
| p16 | [49], [55], [65], [84] | |
| SASP | [49], [54], [56], [65], [66], [89] | |
| Lipofuscin | [54], [56], [66], [89] | |
| p38MAPK | [54] | |
| Astrocytes | SA-β-gal | [106], [113], [118], [119], [121], [122], [124], [125], [126], [127], [129] |
| Heterochromatin protein Hp1γ | [113] | |
| Senescence-associated heterochromatin foci | [121] | |
| BrdU | [118], [129] | |
| 53BP1 foci | [129] | |
| Lamins | [106], [122], [129] | |
| GADD45A | [122] | |
| p53 | [118], [121], [122], [124], [125] | |
| p21 | [118], [121], [122], [124], [125] | |
| p16 | [106], [113], [118], [121], [126], [127], [129] | |
| SASP | [106], [113], [124], [125], [126], [127], [129] | |
| p38MAPK | [127] | |
| Brain Endothelial Cells | Senescence-associated genes | [161], [163], [167] |
| Comet Assay | [161] | |
| SASP | [161], [167] | |
| Oligodendrocytes | SA-β-gal | [184] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sah, E.; Krishnamurthy, S.; Ahmidouch, M.Y.; Gillispie, G.J.; Milligan, C.; Orr, M.E. The Cellular Senescence Stress Response in Post-Mitotic Brain Cells: Cell Survival at the Expense of Tissue Degeneration. Life 2021, 11, 229. https://doi.org/10.3390/life11030229
Sah E, Krishnamurthy S, Ahmidouch MY, Gillispie GJ, Milligan C, Orr ME. The Cellular Senescence Stress Response in Post-Mitotic Brain Cells: Cell Survival at the Expense of Tissue Degeneration. Life. 2021; 11(3):229. https://doi.org/10.3390/life11030229
Chicago/Turabian StyleSah, Eric, Sudarshan Krishnamurthy, Mohamed Y. Ahmidouch, Gregory J. Gillispie, Carol Milligan, and Miranda E. Orr. 2021. "The Cellular Senescence Stress Response in Post-Mitotic Brain Cells: Cell Survival at the Expense of Tissue Degeneration" Life 11, no. 3: 229. https://doi.org/10.3390/life11030229