
Hepatitis B Virus X Protein (HBx) Suppresses Transcription Factor EB (TFEB) Resulting in Stabilization of Integrin Beta 1 (ITGB1) in Hepatocellular Carcinoma Cells


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Downregulation of Transcription Factor EB (TFEB) and Lysosomal Components Are Related to Liver Cancer Incidence and Poor Prognosis
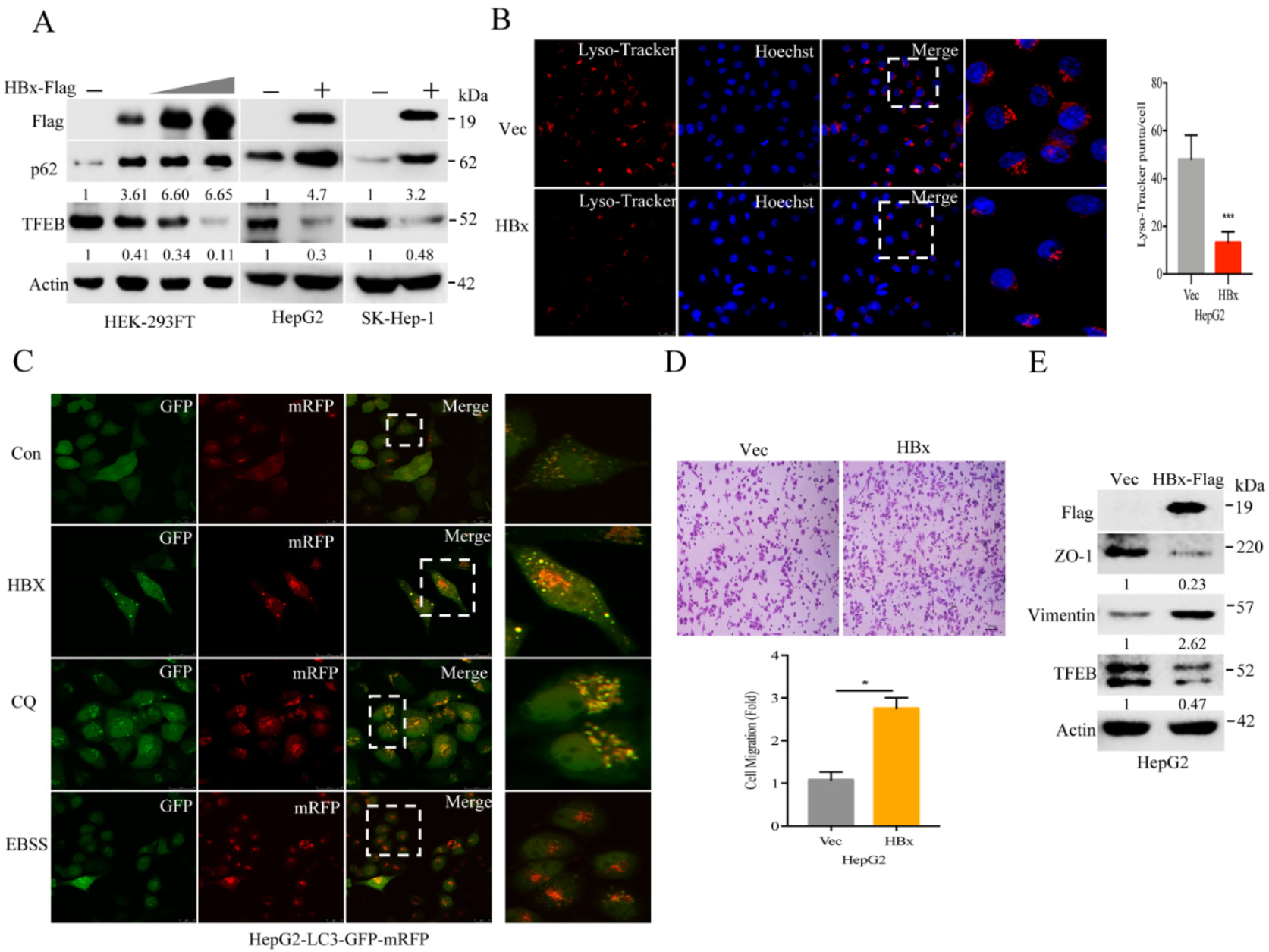
2.2. Hepatitis B Virus X Protein (HBx) Suppresses TFEB and Impairs Lysosomal Biogenesis to Promote Cancer Cell Migration
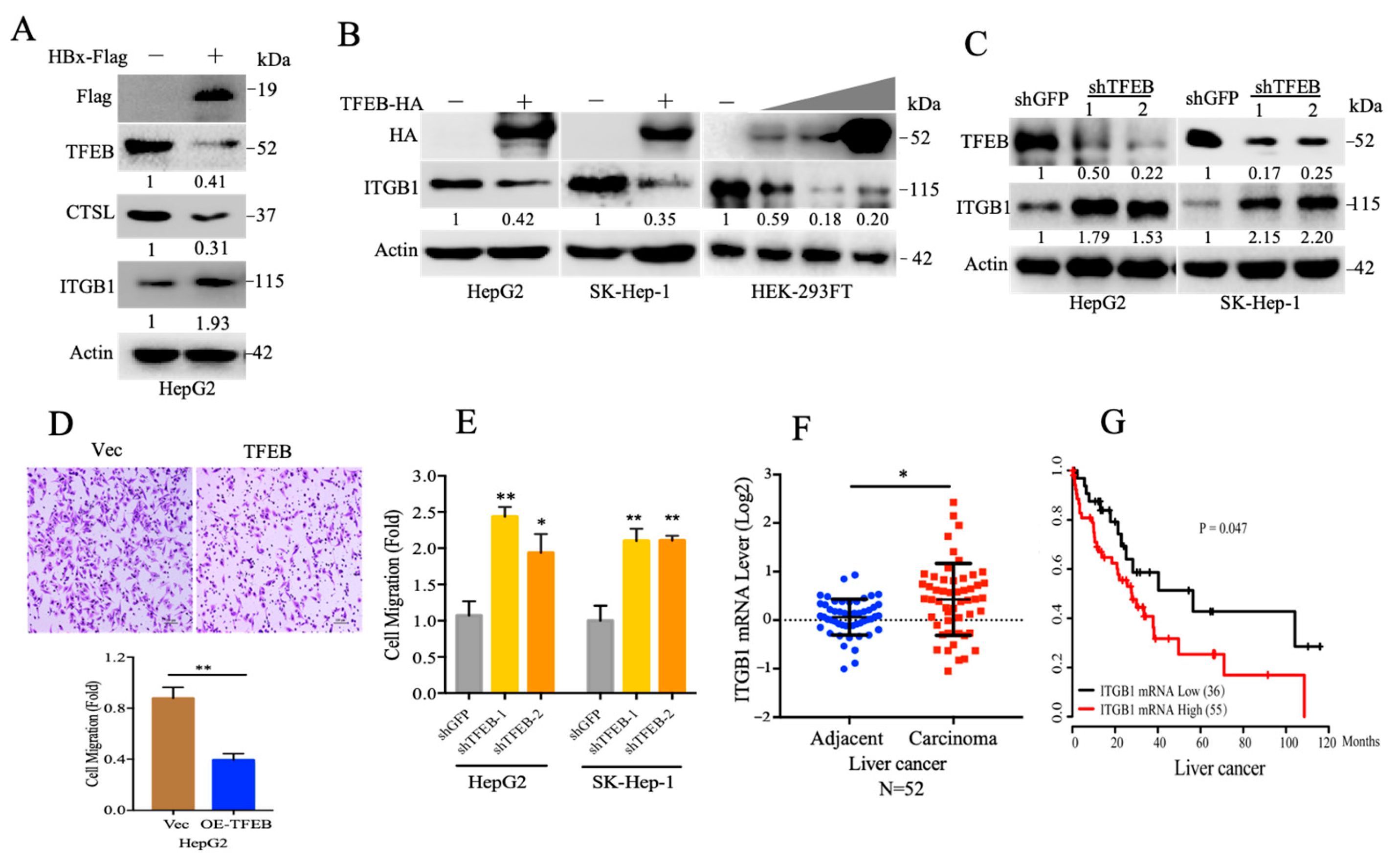
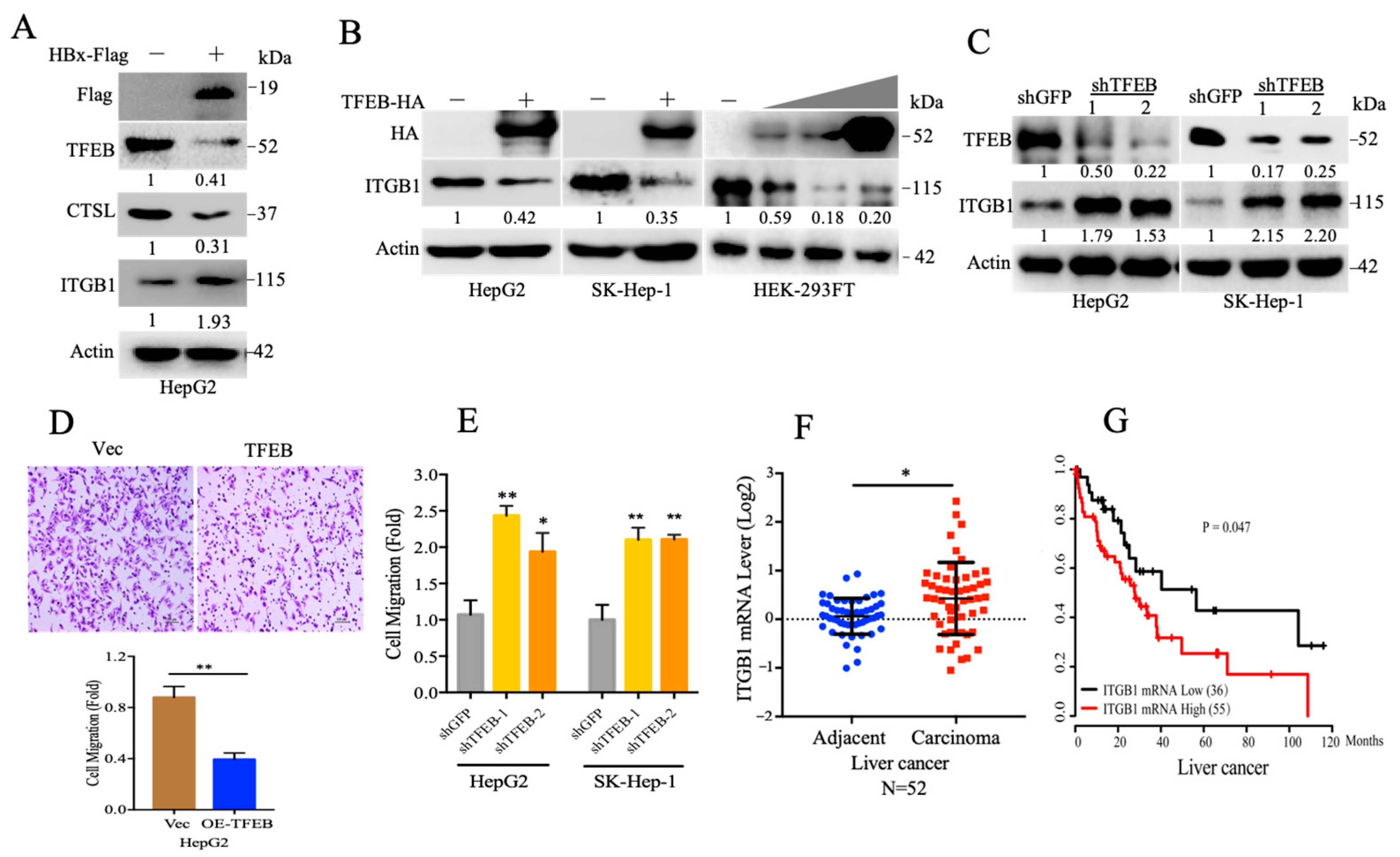
2.3. TFEB Inhibits Cell Migration through Downregulation of Integrin β1
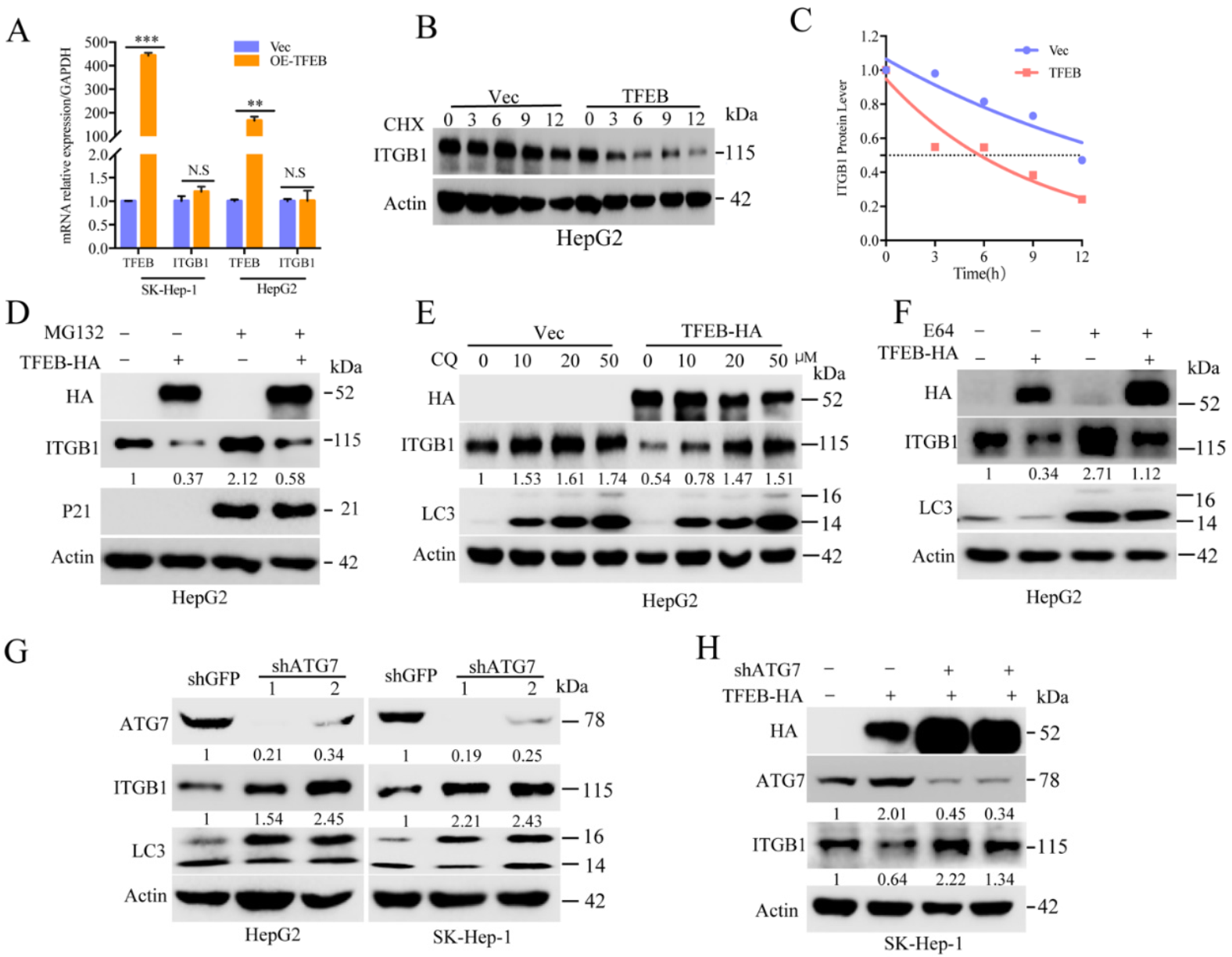
2.4. Cellular Turnover of ITGB1 Is Controlled by TFEB and Mediated by Autolysosomal Flux
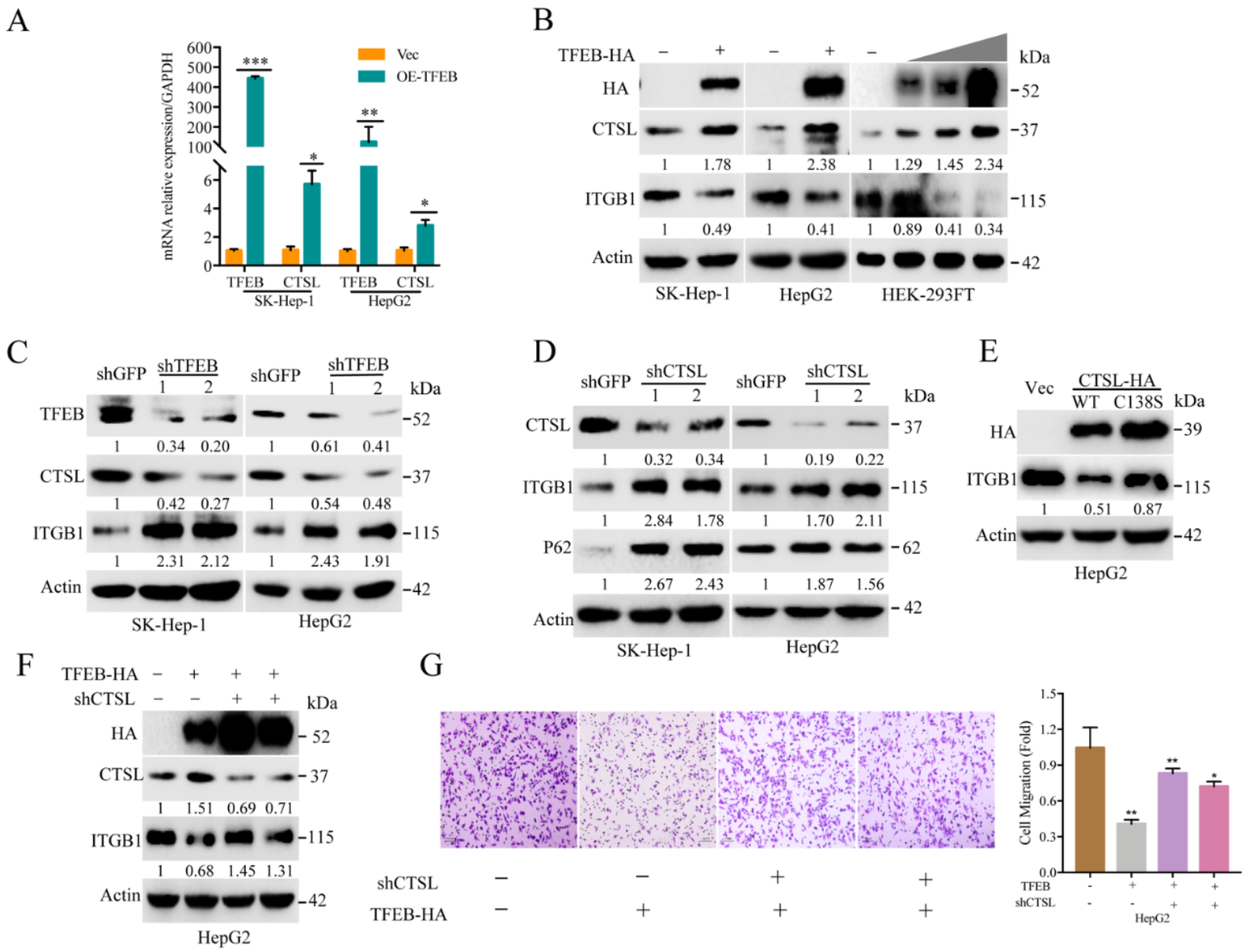
2.5. TFEB-Mediated Upregulation of Lysosomal CTSL Promotes ITGB1 Degradation
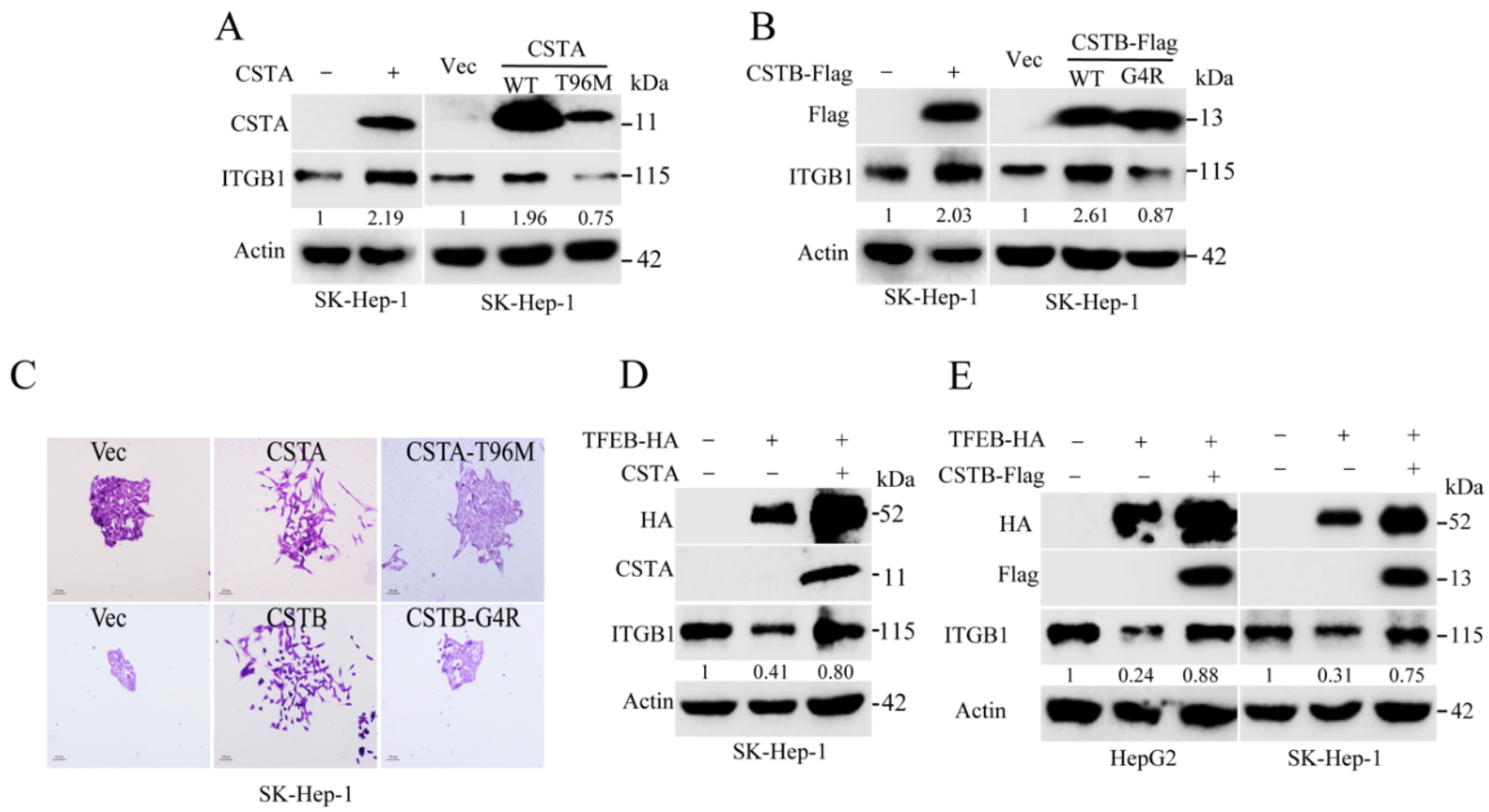
2.6. Endogenous Inhibitors of Lysosomal Cysteine Proteinases Control the Degradation Flux of ITGB1
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Plasmids
4.3. Cell Culture and Construction of Stable Cell Lines
4.4. Western Blotting Analysis
4.5. Reverse Transcription Quantitative PCR (RT-qPCR) Analysis
4.6. Transwell Cell Migration Assay
4.7. Cell Morphology
4.8. Cellular Protein Turnover Assay
4.9. Tandem Fluorescence-Tagged LC3 Probe
4.10. Lysosome Staining
4.11. Gene Expression in HCC, Retrieved from Public Data Bank
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Ahn, S.H.; Chen, D.S.; Chen, P.J.; Dusheiko, G.M.; Hou, J.L. Chronic Hepatitis B Virus Infection. J. Clin. Gastroenterol. 2016, 50, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Caines, A.; Selim, R.; Salgia, R. The Changing Global Epidemiology of Hepatocellular Carcinoma. Clin. Liver Dis. 2020, 24, 535–547. [Google Scholar] [CrossRef]
- Keasler, V.V.; Hodgson, A.J.; Madden, C.R.; Slagle, B.L. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J. Virol. 2007, 81, 2656–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremsdorf, D.; Soussan, P.; Paterlinibrechot, P.; Brechot, C. Hepatitis B virus-related hepatocellular carcinoma: Paradigms for viral-related human carcinogenesis. Oncogene 2006, 25, 3823–3833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Goel, A. Genetic and epigenetic signatures in human hepatocellular carcinoma: A systematic review. Curr. Genomics. 2011, 12, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Feitelson, M.A.; Zhu, M.; Duan, L.X.; London, W.T. Hepatitis B x antigen and p53 are associated in vitro and in liver tissues from patients with primary hepatocellular carcinoma. Oncogene 1993, 8, 1109–1117. [Google Scholar]
- Wang, X.W.; Forrester, K.; Yeh, H.; Feitelson, M.A.; Gu, J.R.; Harris, C.C. Hepatitis-B-Virus-X Protein Inhibits P53 Sequence-Specific DNA-Binding, Transcriptional Activity, and Association with Transcription Factor ERCC3. Natl. Acad. Sci. 1994, 91, 2230–2234. [Google Scholar] [CrossRef] [Green Version]
- Koike, K.; Moriya, K.; Iino, S.; Yotsuyanagi, H.; Endo, Y.; Miyamura, T.; Kurokawa, K. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology 1994, 19, 810–819. [Google Scholar] [CrossRef]
- Yu, D.-Y.; Moon, H.-B.; Son, J.-K.; Jeong, S.; Yu, S.-L.; Yoon, H.; Han, Y.-M.; Lee, C.-S.; Park, J.-S.; Hyun, B.-H.; et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J. Hepatol. 1999, 31, 123–132. [Google Scholar] [CrossRef]
- Saftig, P.; Puertollano, R. How Lysosomes Sense, Integrate, and Cope with Stress. Trends Biochem. Sci. 2021, 46, 97–112. [Google Scholar] [CrossRef]
- Piao, S.; Amaravadi, R.K.; New York Academic Science. Targeting the Lysosome in Cancer. Targeting the Lysosome; Annals of the New York Academy of Sciences: New York, NY, USA, 2016; pp. 45–54. [Google Scholar]
- Davidson, S.M.; Heiden, M.G.V. Critical Functions of the Lysosome in Cancer Biology. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 481–507. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, C.; Zhu, H.; Han, M.; Zhuang, Y.; Wu, X.; Xu, T. Disruption of Lysosome Function Promotes Tumor Growth and Metastasis in Drosophila. J. Biol. Chem. 2010, 285, 21817–21823. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Fang, M.; Hu, Y.; Huang, B.; Li, N.; Chang, C.; Huang, R.; Xu, X.; Yang, Z.; Chen, Z.; et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy 2013, 10, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Serkan, E.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Chao, X.; Wang, S.; Zhao, K.; Li, Y.; Williams, J.A.; Li, T.; Chavan, H.; Krishnamurthy, P.; He, X.C.; Li, L.; et al. Impaired TFEB-Mediated Lysosome Biogenesis and Autophagy Promote Chronic Ethanol-Induced Liver Injury and Steatosis in Mice. Gastroenterology 2018, 155, 865–879.e12. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nat. Cell Biol. 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Matsuda, K.; Jia, W.-H.; Chang, J.; Kweon, S.-S.; Xiang, Y.-B.; Shin, A.; Jee, S.H.; Kim, D.-H.; Zhang, B.; et al. Identification of Susceptibility Loci and Genes for Colorectal Cancer Risk. Gastroenterol. 2016, 150, 1633–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Hodge, J.; Saaoud, F.; Wang, J.; Iwanowycz, S.; Wang, Y.; Hui, Y.; Evans, T.D.; Razani, B.; Fan, D. Transcriptional factor EB regulates macrophage polarization in the tumor microenvironment. OncoImmunology 2017, 6, e1312042. [Google Scholar] [CrossRef] [Green Version]
- Dennemärker, J.; Lohmüller, T.; Mayerle, J.; Tacke, M.; Lerch, M.M.; Coussens, L.M.; Peters, C.; Reinheckel, T. Deficiency for the cysteine protease cathepsin L promotes tumor progression in mouse epidermis. Oncogene 2009, 29, 1611–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parigiani, M.A.; Ketscher, A.; Timme, S.; Bronsert, P.; Schlimpert, M.; Kammerer, B.; Jacquel, A.; Chaintreuil, P.; Reinheckel, T. Conditional Gene Targeting Reveals Cell Type-Specific Roles of the Lysosomal Protease Cathepsin L in Mammary Tumor Progression. Cancers 2020, 12, 2004. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-J.; Yu, G.-R.; Park, S.-H.; Cho, B.-H.; Ahn, J.-S.; Park, H.-J.; Song, E.-Y.; Kim, D.-G. Identification of Cystatin B as a Potential Serum Marker in Hepatocellular Carcinoma. Clin. Cancer Res. 2008, 14, 1080–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, N.Y.; Kang, Y.H.; Park, M.-Y.; Lee, C.I.; Kim, M.K.; Kim, D.G.; Kim, J.W.; Song, E.Y. Development of a fluorescent microsphere immunoassay for cystatin B (CSTB) in serum of patients with hepatocellular carcinoma. Clin. Chem. Lab. Med. 2011, 49, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, 11–25. [Google Scholar] [CrossRef]
- Aplin, A.E.; Howe, A.K.; Juliano, R.L. Cell adhesion molecules, signal transduction and cell growth. Curr. Opin. Cell Biol. 1999, 11, 737–744. [Google Scholar] [CrossRef]
- Burridge, K.; Chrzanowska-Wodnicka, M. Focal Adhesions, Contractility, and Signaling. Annu. Rev. Cell Dev. Biol. 1996, 12, 463–519. [Google Scholar] [CrossRef] [Green Version]
- Proux-Gillardeaux, V.; Gavard, J.; Irinopoulou, T.; Mège, R.-M.; Galli, T. Tetanus neurotoxin-mediated cleavage of cellubrevin impairs epithelial cell migration and integrin-dependent cell adhesion. Proc. Natl. Acad. Sci USA. 2005, 102, 6362–6367. [Google Scholar] [CrossRef] [Green Version]
- White, D.E.; Kurpios, N.A.; Zuo, D.; Hassell, J.A.; Blaess, S.; Mueller, U. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell 2004, 6, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Qiao, X.; Qiao, S.; Yu, L. Targeting integrins in hepatocellular carcinoma. Expert Opin. Ther. Targets 2011, 15, 421–437. [Google Scholar] [CrossRef]
- Ozaki, I.; Yamamoto, K.; Mizuta, T.; Kajihara, S.; Fukushima, N.; Setoguchi, Y.; Morito, F.; Sakai, T. Differential expression of laminin receptors in human hepatocellular carcinoma. Gut 1998, 43, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Masumoto, A.; Arao, S.; Otsuki, M. Role of beta1 integrins in adhesion and invasion of hepatocellular carcinoma cells. Hepatology 1999, 29, 68–74. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the Autophagosome Maturation Process by a Novel Reporter Protein, Tandem Fluorescent-Tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.K.; Man, K.; Poon, R.T.; Lo, C.M.; Yuen, A.P.; Ng, I.O.; Ng, K.T.; Leonard, W.; Fan, S.T. Signal Transducers and Activators of Transcription 5b Activation Enhances Hepatocellular Carcinoma Aggressiveness through Induction of Epithelial-Mesenchymal Transition. Cancer Res. 2006, 66, 9948–9956. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.Z.; Zhang, L.D.; Zhang, Y.; Xiong, Y.; Zhang, Y.J.; Li, H.L. HBx protein induces EMT through c-Src activation in SMMC-7721 hepatoma cell line. Biochem. Biophys. Res. Commun. 2009, 382, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.-Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of Metastatic Outgrowth from Single Dormant Tumor Cells by Targeting the Cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef] [Green Version]
- Singal, A.G.; El-Serag, H.B. Hepatocellular Carcinoma From Epidemiology to Prevention: Translating Knowledge into Practice. Clin. Gastroenterol. Hepatol. 2015, 13, 2140–2151. [Google Scholar] [CrossRef] [Green Version]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Masuzaki, R.; Ray, K.C.; Roland, J.; Zent, R.; Lee, Y.A.; Karp, S.J. Integrin β1 Establishes Liver Microstructure and Modulates Transforming Growth Factor β during Liver Development and Regeneration. Am. J. Pathol. 2021, 191, 309–319. [Google Scholar] [CrossRef]
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouzé, E.; Imbeaud, S.; Pilati, C.; Nault, J.-C.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-phenotype correlation ofCTNNB1mutations reveals different ß-catenin activity associated with liver tumor progression. Hepatology 2016, 64, 2047–2061. [Google Scholar] [CrossRef] [Green Version]
- Abitbol, S.; Dahmani, R.; Coulouarn, C.; Ragazzon, B.; Mlecnik, B.; Senni, N.; Savall, M.; Bossard, P.; Sohier, P.; Drouet, V.; et al. AXIN deficiency in human and mouse hepatocytes induces hepatocellular carcinoma in the absence of β-catenin activation. J. Hepatol. 2018, 68, 1203–1213. [Google Scholar] [CrossRef]
- Torimura, T.; Ueno, T.; Kin, M.; Inuzuka, S.; Sugawara, H.; Tamaki, S.; Tsuji, R.; Sujaku, K.; Sata, M.; Tanikawa, K. Coordinated expression of integrin alpha6beta1 and laminin in hepatocellular carcinoma. Hum. Pathol. 1997, 28, 1131–1138. [Google Scholar] [CrossRef]
- Volpes, R.; Oord, J.J.V.D.; Desmet, V.J. Integrins as differential cell lineage markers of primary liver tumors. Am. J. Pathol. 1993, 142, 1483–1492. [Google Scholar]
- Li, S.; Song, Y.; Quach, C.; Guo, H.; Jang, G.-B.; Maazi, H.; Zhao, S.; Sands, N.A.; Liu, Q.; In, G.K.; et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [Green Version]
- Zada, S.; Hwang, J.S.; Ahmed, M.; Lai, T.H.; Pham, T.M.; Kim, D.R. Control of the Epithelial-to-Mesenchymal Transition and Cancer Metastasis by Autophagy-Dependent SNAI1 Degradation. Cells 2019, 8, 129. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Liu, Y.; Qin, L.; Wu, P.; Xia, Z.; Luo, M. Cathepsin H–Mediated Degradation of HDAC4 for Matrix Metalloproteinase Expression in Hepatic Stellate Cells: Implications of Epigenetic Suppression of Matrix Metalloproteinases in Fibrosis through Stabilization of Class IIa Histone Deacetylases. Am. J. Pathol. 2017, 187, 781–797. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Weng, J.; Wu, K.; Xu, X.; Wang, H.; Zhang, J.; Zhao, C.; Yang, J.; Zhang, Y.; Shen, W. Silencing of TAZ inhibits the motility of hepatocellular carcinoma cells through autophagy induction. Cancer Manag. Res. 2019, 11, 8743–8753. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; You, Y.; Hua, M.; Wu, P.; Liu, Y.; Chen, Z.; Zhang, L.; Wei, H.; Li, Y.; Luo, M.; et al. Chlorophyllin Modulates Gut Microbiota and Inhibits Intestinal Inflammation to Ameliorate Hepatic Fibrosis in Mice. Front. Physiol. 2018, 9, 1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, D.; Nie, Y.; Zhu, A.; Chen, Z.; Wu, P.; Zhang, L.; Luo, M.; Sun, Q.; Cai, L.; Lai, Y.; et al. Vitamin D Signaling through Induction of Paneth Cell Defensins Maintains Gut Microbiota and Improves Metabolic Disorders and Hepatic Steatosis in Animal Models. Front. Physiol. 2016, 7, 498. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Yang, H.; Pan, L.; Zhao, G.; Zhang, R.; Zhang, T.; Xiao, Z.; Tong, Y.; Zhang, Y.; Hu, R.; et al. Hepatitis B Virus X Protein (HBx) Suppresses Transcription Factor EB (TFEB) Resulting in Stabilization of Integrin Beta 1 (ITGB1) in Hepatocellular Carcinoma Cells. Cancers 2021, 13, 1181. https://doi.org/10.3390/cancers13051181
Zhang C, Yang H, Pan L, Zhao G, Zhang R, Zhang T, Xiao Z, Tong Y, Zhang Y, Hu R, et al. Hepatitis B Virus X Protein (HBx) Suppresses Transcription Factor EB (TFEB) Resulting in Stabilization of Integrin Beta 1 (ITGB1) in Hepatocellular Carcinoma Cells. Cancers. 2021; 13(5):1181. https://doi.org/10.3390/cancers13051181
Chicago/Turabian StyleZhang, Chunyan, Huan Yang, Liwei Pan, Guangfu Zhao, Ruofei Zhang, Tianci Zhang, Zhixiong Xiao, Ying Tong, Yi Zhang, Richard Hu, and et al. 2021. "Hepatitis B Virus X Protein (HBx) Suppresses Transcription Factor EB (TFEB) Resulting in Stabilization of Integrin Beta 1 (ITGB1) in Hepatocellular Carcinoma Cells" Cancers 13, no. 5: 1181. https://doi.org/10.3390/cancers13051181