
High Density Lipoprotein Cholesterol Efflux Capacity and Atherosclerosis in Cardiovascular Disease: Pathophysiological Aspects and Pharmacological Perspectives


Abstract
1. Introduction
2. HDL and RCT
- Cellular cholesterol efflux from macrophages;
- HDL remodelling in plasma;
- Cholesterol hepatic uptake and excretion.
2.1. Cellular Cholesterol Efflux from Macrophages
2.2. HDL Remodeling in Plasma
2.3. Cholesterol Hepatic Uptake and Excretion
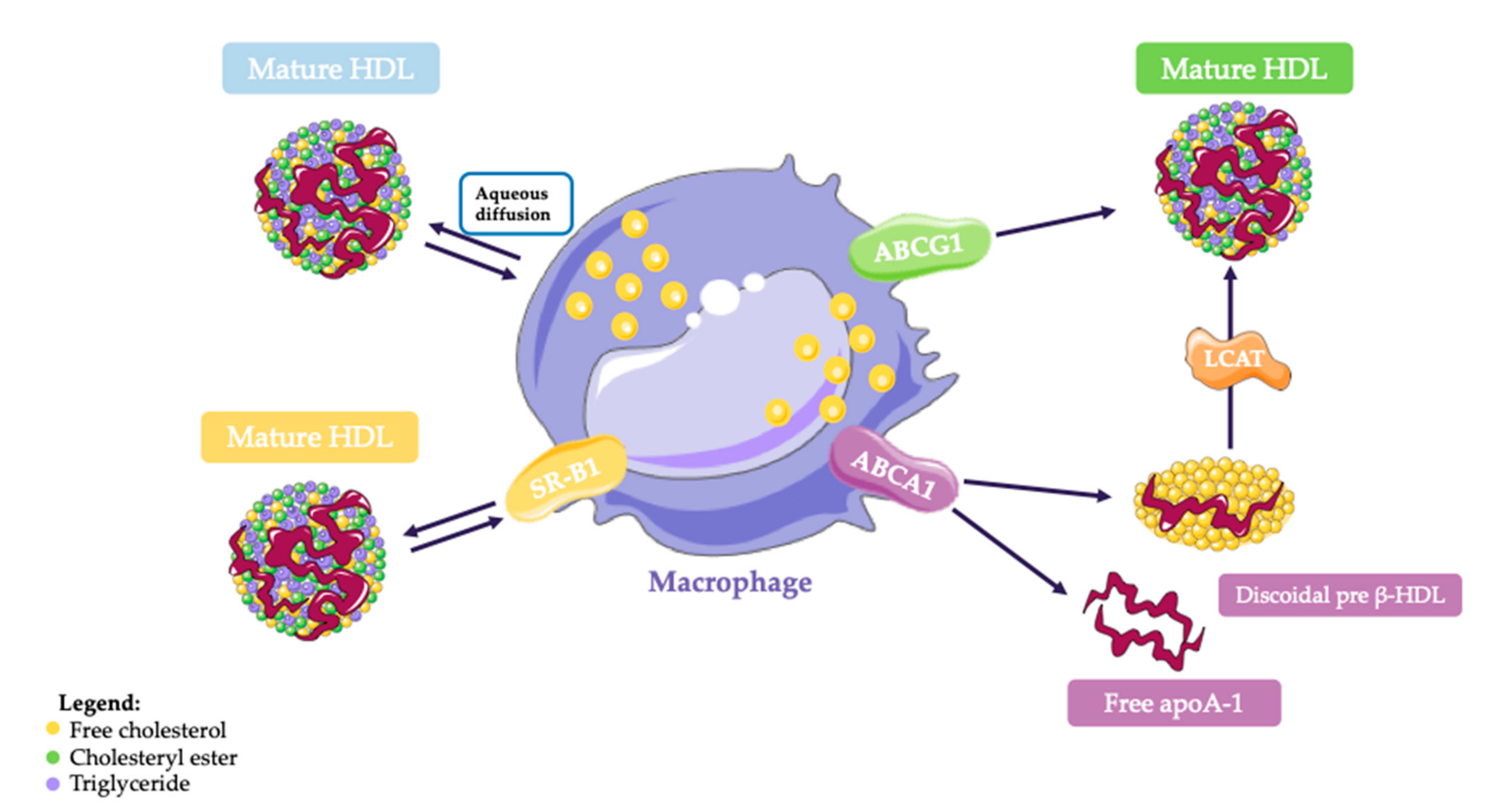
2.4. Mechanisms of Cell Cholesterol Efflux
- The aqueous diffusion (AD)-mediated efflux. It is a pathway that depends on the cholesterol gradient between cells and acceptors [9];
- The ABCA1-mediated efflux.
- The ABCG1-mediated efflux.
2.4.1. SR-BI
2.4.2. ABCA1
2.4.3. ABCG1
2.5. HDL CEC as an Index of RCT Efficiency
3. HDL CEC, Atherosclerosis and Cardiovascular Risk
3.1. Cross-Sectional Studies
3.2. Longitudinal Studies
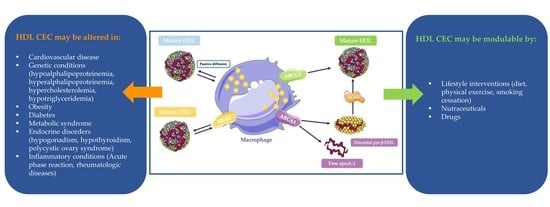
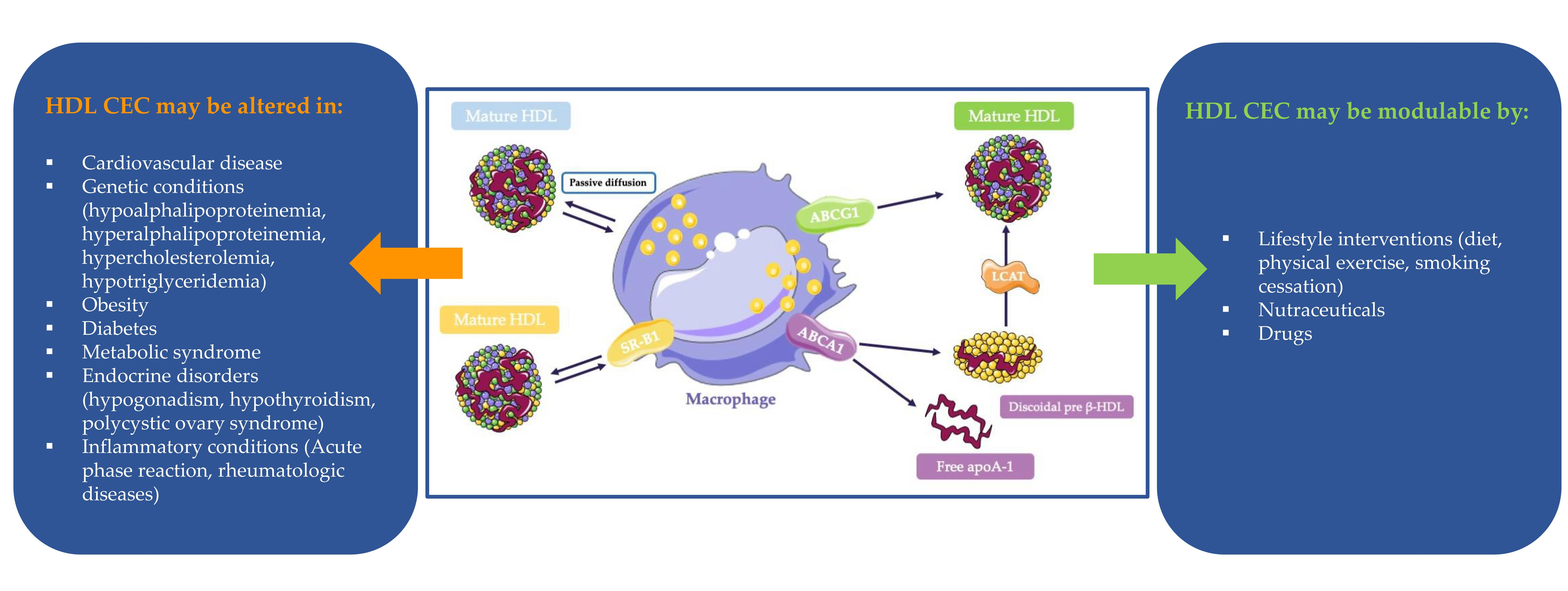
4. HDL CEC in Specific Conditions
4.1. HDL CEC in Genetic Disorders
4.1.1. Genic Alterations Leading to Hypoalphalipoproteinemia
4.1.2. Genic Alterations leading to Hyperalphalipoproteinaemia
4.1.3. Genic Alterations leading to Hypercholesterolemia
4.1.4. Genic Alterations leading to Hypotriglyceridemia
4.2. HDL CEC in Chronic Pathologies
4.2.1. Chronic Kidney Disease
4.2.2. Obesity
4.2.3. Diabetes
4.2.4. Metabolic Syndrome
4.2.5. Endocrine Disorders
Hypogonadism
Hypothyroidism
Polycystic Ovary Syndrome
4.3. HDL CEC and Inflammation
4.3.1. Inflammation and Cell Cholesterol Homeostasis
4.3.2. HDL CEC Impairment in Inflammatory Condition
Acute Phase Reaction
Rheumatologic Diseases
4.4. HDL CEC, Aging and Mortality
5. Strategies to Increase RCT in Humans
5.1. Lifestyle
5.2. Nutraceuticals
5.3. Drugs
6. Conclusions
Funding
Conflicts of Interest
References
- Wilson, P.W.; Garrison, R.J.; Castelli, W.P.; Feinleib, M.; McNamara, P.M.; Kannel, W.B. Prevalence of coronary heart disease in the Framingham Offspring Study: Role of lipoprotein cholesterols. Am. J. Cardiol. 1980, 46, 649–654. [Google Scholar] [CrossRef]
- Gordon, D.J.; Probstfield, J.L.; Garrison, R.J.; Neaton, J.D.; Castelli, W.P.; Knoke, J.D.; Jacobs, D.R.J.; Bangdiwala, S.; Tyroler, H.A. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 1989, 79, 8–15. [Google Scholar] [CrossRef]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: Two prospective cohort studies. Eur. Heart J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of Dalcetrapib in Patients with a Recent Acute Coronary Syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Hólm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef]
- Favari, E.; Chroni, A.; Tietge, U.J.F.; Zanotti, I.; Escola-Gil, J.C.; Bernini, F. Cholesterol efflux and reverse cholesterol transport. Handb. Exp. Pharmacol. 2015, 224, 181–206. [Google Scholar] [CrossRef]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Circ. Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C. Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 2014, 289, 24020–24029. [Google Scholar] [CrossRef]
- Cuchel, M.; Rohatgi, A.; Sacks, F.M.; Guyton, J.R. JCL roundtable: High-density lipoprotein function and reverse cholesterol transport. J. Clin. Lipidol. 2018, 12, 1086–1094. [Google Scholar] [CrossRef]
- Soria-Florido, M.T.; Schröder, H.; Grau, M.; Fitó, M.; Lassale, C. High density lipoprotein functionality and cardiovascular events and mortality: A systematic review and meta-analysis. Atherosclerosis 2020, 302, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Glomset, J.A.; Janssen, E.T.; Kennedy, R.; Dobbins, J. Role of plasma lecithin:cholesterol acyltransferase in the metabolism of high density lipoproteins. J. Lipid Res. 1966, 7, 638–648. [Google Scholar] [CrossRef]
- Cuchel, M. Macrophage Reverse Cholesterol Transport: Key to the Regression of Atherosclerosis? Circulation 2006, 113, 2548–2555. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, I.; Favari, E.; Bernini, F. Cellular Cholesterol Efflux Pathways: Impact on Intracellular Lipid Trafficking and Methodological Considerations. Curr. Pharm. Biotechnol. 2012, 13, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Talbot, C.P.J.; Plat, J.; Ritsch, A.; Mensink, R.P. Determinants of cholesterol efflux capacity in humans. Prog. Lipid Res. 2018, 69, 21–32. [Google Scholar] [CrossRef]
- Niisuke, K.; Kuklenyik, Z.; Horvath, K.V.; Gardner, M.S.; Toth, C.A.; Asztalos, B.F. Composition-function analysis of HDL subpopulations: Influence of lipid composition on particle functionality. J. Lipid Res. 2020, 61, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Ossoli, A.; Simonelli, S.; Vitali, C.; Franceschini, G.; Calabresi, L. Role of LCAT in Atherosclerosis. J. Atheroscler. Thromb. 2016, 23, 119–127. [Google Scholar] [CrossRef]
- Czarnecka, H.; Yokoyama, S. Regulation of cellular cholesterol efflux by lecithin:cholesterol acyltransferase reaction through nonspecific lipid exchange. J. Biol. Chem. 1996, 271, 2023–2028. [Google Scholar] [CrossRef]
- Tanigawa, H.; Billheimer, J.T.; Tohyama, J.; Fuki, I.V.; Ng, D.S.; Rothblat, G.H.; Rader, D.J. Lecithin: Cholesterol acyltransferase expression has minimal effects on macrophage reverse cholesterol transport in vivo. Circulation 2009, 120, 160–169. [Google Scholar] [CrossRef]
- Calabresi, L.; Favari, E.; Moleri, E.; Adorni, M.P.; Pedrelli, M.; Costa, S.; Jessup, W.; Gelissen, I.C.; Kovanen, P.T.; Bernini, F.; et al. Functional LCAT is not required for macrophage cholesterol efflux to human serum. Atherosclerosis 2009, 204, 141–146. [Google Scholar] [CrossRef]
- Tall, A.R. Plasma cholesteryl ester transfer protein. J. Lipid Res. 1993, 34, 1255–1274. [Google Scholar] [CrossRef]
- Barter, P.J.; Brewer, H.B.J.; Chapman, M.J.; Hennekens, C.H.; Rader, D.J.; Tall, A.R. Cholesteryl ester transfer protein: A novel target for raising HDL and inhibiting atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.P.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.-C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef]
- Rader, D.J.; Alexander, E.T.; Weibel, G.L.; Billheimer, J.; Rothblat, G.H. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J. Lipid Res. 2009, 50, S189–S194. [Google Scholar] [CrossRef]
- Bashore, A.C.; Liu, M.; Key, C.-C.C.; Boudyguina, E.; Wang, X.; Carroll, C.M.; Sawyer, J.K.; Mullick, A.E.; Lee, R.G.; Macauley, S.L.; et al. Targeted Deletion of Hepatocyte Abca1 Increases Plasma HDL (High-Density Lipoprotein) Reverse Cholesterol Transport via the LDL (Low-Density Lipoprotein) Receptor. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1747–1761. [Google Scholar] [CrossRef]
- Cedó, L.; Metso, J.; Santos, D.; García-León, A.; Plana, N.; Sabate-Soler, S.; Rotllan, N.; Rivas-Urbina, A.; Méndez-Lara, K.A.; Tondo, M.; et al. LDL Receptor Regulates the Reverse Transport of Macrophage-Derived Unesterified Cholesterol via Concerted Action of the HDL-LDL Axis: Insight From Mouse Models. Circ. Res. 2020, 127, 778–792. [Google Scholar] [CrossRef]
- Yu, L.; Hammer, R.E.; Li-Hawkins, J.; von Bergmann, K.; Lutjohann, D.; Cohen, J.C.; Hobbs, H.H. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc. Natl. Acad. Sci. USA 2002, 99, 16237–16242. [Google Scholar] [CrossRef]
- Tietge, U.J.F.; Groen, A.K. Role the TICE?: Advancing the concept of transintestinal cholesterol excretion. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1452–1453. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shen, W.-J.; Azhar, S.; Kraemer, F.B. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu. Rev. Physiol. 2018, 80, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; van Berkel, T.J.; van Eck, M. Relative roles of various efflux pathways in net cholesterol efflux from macrophage foam cells in atherosclerotic lesions. Curr. Opin. Lipidol. 2010, 21, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Jebari-Benslaiman, S.; Uribe, K.B.; Benito-Vicente, A.; Galicia-Garcia, U.; Larrea-Sebal, A.; Alloza, I.; Vandenbroeck, K.; Ostolaza, H.; Martín, C. Cholesterol Efflux Efficiency of Reconstituted HDL Is Affected by Nanoparticle Lipid Composition. Biomedicines 2020, 8, 373. [Google Scholar] [CrossRef]
- Favari, E.; Calabresi, L.; Adorni, M.P.; Jessup, W.; Simonelli, S.; Franceschini, G.; Bernini, F. Small discoidal pre-beta1 HDL particles are efficient acceptors of cell cholesterol via ABCA1 and ABCG1. Biochemistry 2009, 48, 11067–11074. [Google Scholar] [CrossRef]
- Adorni, M.P.; Zimetti, F.; Billheimer, J.T.; Wang, N.; Rader, D.J.; Phillips, M.C.; Rothblat, G.H. The roles of different pathways in the release of cholesterol from macrophages. J. Lipid Res. 2007, 48, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, M. SR-BI as target in atherosclerosis and cardiovascular disease—A comprehensive appraisal of the cellular functions of SR-BI in physiology and disease. Atherosclerosis 2017, 258, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.B.J.; Davidson, W.S.; Fayad, Z.A.; Fuster, V.; Goldstein, J.; Hellerstein, M.; Jiang, X.-C.; Phillips, M.C.; Rader, D.J.; et al. Cholesterol efflux and atheroprotection: Advancing the concept of reverse cholesterol transport. Circulation 2012, 125, 1905–1919. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; DerOhannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016, 351, 1166–1171. [Google Scholar] [CrossRef]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef]
- Qian, H.; Zhao, X.; Cao, P.; Lei, J.; Yan, N.; Gong, X. Structure of the Human Lipid Exporter ABCA1. Cell 2017, 169, 1228–1239. [Google Scholar] [CrossRef]
- Tall, A.R. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J. Intern. Med. 2008, 263, 256–273. [Google Scholar] [CrossRef]
- Favari, E.; Lee, M.; Calabresi, L.; Franceschini, G.; Zimetti, F.; Bernini, F.; Kovanen, P.T. Depletion of pre-beta-high density lipoprotein by human chymase impairs ATP-binding cassette transporter A1- but not scavenger receptor class B type I-mediated lipid efflux to high density lipoprotein. J. Biol. Chem. 2004, 279, 9930–9936. [Google Scholar] [CrossRef]
- Wang, N.; Westerterp, M. ABC Transporters, Cholesterol Efflux, and Implications for Cardiovascular Diseases. Adv. Exp. Med. Biol. 2020, 1276, 67–83. [Google Scholar] [CrossRef]
- Chiesa, G.; Parolini, C.; Canavesi, M.; Colombo, N.; Sirtori, C.R.; Fumagalli, R.; Franceschini, G.; Bernini, F. Human apolipoproteins A-I and A-II in cell cholesterol efflux: Studies with transgenic mice. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1417–1423. [Google Scholar] [CrossRef]
- Ayaori, M.; Ikewaki, K. Role of ATP-Binding Cassette Transporters A1 and G1 in Reverse Cholesterol Transport and Atherosclerosis, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2014; ISBN 9780124078673. [Google Scholar]
- Knight, B.L. ATP-binding cassette transporter A1: Regulation of cholesterol efflux. Biochem. Soc. Trans. 2004, 32, 124–127. [Google Scholar] [CrossRef]
- Frikke-Schmidt, R. Genetic variation in ABCA1 and risk of cardiovascular disease. Atherosclerosis 2011, 218, 281–282. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F. ATP-binding cassette transporter A1 and cholesterol trafficking. Curr. Opin. Lipidol. 2002, 13, 373–381. [Google Scholar] [CrossRef]
- Frambach, S.J.C.M.; de Haas, R.; Smeitink, J.A.M.; Rongen, G.A.; Russel, F.G.M.; Schirris, T.J.J. Brothers in Arms: ABCA1- and ABCG1-Mediated Cholesterol Efflux as Promising Targets in Cardiovascular Disease Treatment. Pharmacol. Rev. 2020, 72, 152–190. [Google Scholar] [CrossRef]
- Small, D.M. Role of ABC transporters in secretion of cholesterol from liver into bile. Proc. Natl. Acad. Sci. USA 2003, 100, 4–6. [Google Scholar] [CrossRef]
- Larrede, S.; Quinn, C.M.; Jessup, W.; Frisdal, E.; Olivier, M.; Hsieh, V.; Kim, M.-J.; van Eck, M.; Couvert, P.; Carrie, A.; et al. Stimulation of cholesterol efflux by LXR agonists in cholesterol-loaded human macrophages is ABCA1-dependent but ABCG1-independent. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1930–1936. [Google Scholar] [CrossRef]
- Hardy, L.M.; Frisdal, E.; le Goff, W. Critical Role of the Human ATP-Binding Cassette G1 Transporter in Cardiometabolic Diseases. Int. J. Mol. Sci. 2017, 18, 1892. [Google Scholar] [CrossRef] [PubMed]
- Assmann, G.; Cullen, P.; Schulte, H. Simple scoring scheme for calculating the risk of acute coronary events based on the 10-year follow-up of the Prospective Cardiovascular Münster (PROCAM) study. Circulation 2002, 105, 310–315. [Google Scholar] [CrossRef] [PubMed]
- AIM-HIGH Investigators, T.A.-H. The role of niacin in raising high-density lipoprotein cholesterol to reduce cardiovascular events in patients (AIM-HIGH) trial. Am. Heart J. 2011, 161, 538–543. [Google Scholar] [CrossRef]
- Frikke-Schmidt, R.; Nordestgaard, B.G.; Stene, M.C.A.; Sethi, A.A.; Remaley, A.T.; Schnohr, P.; Grande, P.; Tybjerg-Hansen, A. Association of Loss-of-Function Mutations in the ABCA1 Gene With High-Density. JAMA 2008, 299, 2524–2532. [Google Scholar] [CrossRef] [PubMed]
- Karathanasis, S.K.; Freeman, L.A.; Gordon, S.M.; Remaley, A.T. The changing face of HDL and the best way to measure it. Clin. Chem. 2017, 63, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Toh, R. Assessment of HDL cholesterol removal capacity: Toward clinical application. J. Atheroscler. Thromb. 2019, 26, 111–120. [Google Scholar] [CrossRef]
- de la Llera-Moya, M.; Drazul-Schrader, D.; Asztalos, B.; Cuchel, M.; Rader, D.; Rothblat, G. The Ability to Promote Efflux via ABCA1 Determines the Capacity of Serum Specimens with Similar HDL-C to Remove Cholesterol from Macrophages. Arter. Thromb. Vasc. Biol. 2010, 30, 796–801. [Google Scholar] [CrossRef]
- Favari, E.; Gomaraschi, M.; Zanotti, I.; Bernini, F.; Lee-Rueckert, M.; Kovanen, P.T.; Sirtori, C.R.; Franceschini, G.; Calabresi, L. A unique protease-sensitive high density lipoprotein particle containing the apolipoprotein A-IMilano dimer effectively promotes ATP-binding cassette A1-mediated cell cholesterol efflux. J. Biol. Chem. 2007, 282, 5125–5132. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL Cholesterol Efflux Capacity and Incident Cardiovascular Events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.; Luben, R.; Billheimer, J.; et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef]
- Ishikawa, T.; Ayaori, M.; Uto-Kondo, H.; Nakajima, T.; Mutoh, M.; Ikewaki, K. High-density lipoprotein cholesterol efflux capacity as a relevant predictor of atherosclerotic coronary disease. Atherosclerosis 2015, 242, 318–322. [Google Scholar] [CrossRef]
- Ogura, M.; Hori, M.; Harada-Shiba, M. Association between Cholesterol Efflux Capacity and Atherosclerotic Cardiovascular Disease in Patients with Familial Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.J.; Khera, A.V.; Wilensky, R.L.; Rader, D.J. Anti-oxidative and cholesterol efflux capacities of high-density lipoprotein are reduced in ischaemic cardiomyopathy. Eur. J. Heart Fail. 2013, 15, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Zhao, X.; Zhou, Q.; Zhang, Z. High-density lipoprotein cholesterol efflux capacity is inversely associated with cardiovascular risk: A systematic review and meta-analysis. Lipids Health Dis. 2017, 16, 212. [Google Scholar] [CrossRef]
- Thakkar, H.; Vincent, V.; Roy, A.; Singh, S.; Ramakrishnan, L.; Kalaivani, M.; Singh, A. HDL functions and their interaction in patients with ST elevation myocardial infarction: A case control study. Lipids Health Dis. 2020, 19, 75. [Google Scholar] [CrossRef] [PubMed]
- Favari, E.; Ronda, N.; Adorni, M.P.; Zimetti, F.; Salvi, P.; Manfredini, M.; Bernini, F.; Borghi, C.; Cicero, A.F.G. ABCA1-dependent serum cholesterol efflux capacity inversely correlates with pulse wave velocity in healthy subjects. J. Lipid Res. 2013, 54, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Vigna, G.B.; Satta, E.; Bernini, F.; Boarini, S.; Bosi, C.; Giusto, L.; Pinotti, E.; Tarugi, P.; Vanini, A.; Volpato, S.; et al. Flow-mediated dilation, carotid wall thickness and HDL function in subjects with hyperalphalipoproteinemia. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 777–783. [Google Scholar] [CrossRef]
- Agarwala, A.P.; Rodrigues, A.; Risman, M.; McCoy, M.; Trindade, K.; Qu, L.; Cuchel, M.; Billheimer, J.; Rader, D.J. High-Density Lipoprotein (HDL) Phospholipid Content and Cholesterol Efflux Capacity Are Reduced in Patients With Very High HDL Cholesterol and Coronary Disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1515–1519. [Google Scholar] [CrossRef]
- Ko, D.T.; Alter, D.A.; Guo, H.; Koh, M.; Lau, G.; Austin, P.C.; Booth, G.L.; Hogg, W.; Jackevicius, C.A.; Lee, D.S.; et al. High-Density Lipoprotein Cholesterol and Cause-Specific Mortality in Individuals Without Previous Cardiovascular Conditions: The CANHEART Study. J. Am. Coll. Cardiol. 2016, 68, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Josefs, T.; Wouters, K.; Tietge, U.J.F.; Annema, W.; Dullaart, R.P.F.; Vaisar, T.; Arts, I.C.W.; van der Kallen, C.J.H.; Stehouwer, C.D.A.; Schalkwijk, C.G.; et al. High-density lipoprotein cholesterol efflux capacity is not associated with atherosclerosis and prevalence of cardiovascular outcome: The CODAM study. J. Clin. Lipidol. 2020, 14, 122–132.e4. [Google Scholar] [CrossRef]
- Li, X.M.; Tang, W.H.W.; Mosior, M.K.; Huang, Y.; Wu, Y.; Matter, W.; Gao, V.; Schmitt, D.; di Donato, J.A.; Fisher, E.A.; et al. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1696–1705. [Google Scholar] [CrossRef] [PubMed]
- Ebtehaj, S.; Gruppen, E.G.; Bakker, S.J.L.; Dullaart, R.P.F.; Tietge, U.J.F. HDL (High-Density Lipoprotein) Cholesterol Efflux Capacity Is Associated With Incident Cardiovascular Disease in the General Population. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1874–1883. [Google Scholar] [CrossRef]
- Khera, A.V.; Demler, O.V.; Adelman, S.J.; Collins, H.L.; Glynn, R.J.; Ridker, P.M.; Rader, D.J.; Mora, S. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events: An Analysis From the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2017, 135, 2494–2504. [Google Scholar] [CrossRef] [PubMed]
- Ritsch, A.; Scharnagl, H.; März, W. HDL cholesterol efflux capacity and cardiovascular events. N. Engl. J. Med. 2015, 372, 1870–1871. [Google Scholar] [PubMed]
- Chindhy, S.; Joshi, P.; Khera, A.; Ayers, C.R.; Hedayati, S.; Rohatgi, A. Impaired Renal Function on Cholesterol Efflux Capacity, HDL particle Number and Cardiovascular Events. JAMA 2018, 72, 698–700. [Google Scholar] [CrossRef] [PubMed]
- Javaheri, A.; Molina, M.; Zamani, P.; Rodrigues, A.; Novak, E.; Chambers, S.; Stutman, P.; Maslanek, W.; Williams, M.; Lilly, S.M.; et al. Cholesterol efflux capacity of high-density lipoprotein correlates with survival and allograft vasculopathy in cardiac transplant recipients. J. Hear. Lung Transplant. Off. Publ. Int. Soc. Hear. Transplant. 2016, 35, 1295–1302. [Google Scholar] [CrossRef]
- Kopecky, C.; Ebtehaj, S.; Genser, B.; Drechsler, C.; Krane, V.; Antlanger, M.; Kovarik, J.J.; Kaltenecker, C.C.; Parvizi, M.; Wanner, C.; et al. HDL Cholesterol Efflux Does Not Predict Cardiovascular Risk in Hemodialysis Patients. J. Am. Soc. Nephrol. 2017, 28, 769–775. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Y.; Ding, D.; Li, X.; Yang, Y.; Li, Q.; Zheng, Y.; Wang, D.; Ling, W. Cholesterol efflux capacity is an independent predictor of all-cause and cardiovascular mortality in patients with coronary artery disease: A prospective cohort study. Atherosclerosis 2016, 249, 116–124. [Google Scholar] [CrossRef]
- Mody, P.; Joshi, P.H.; Khera, A.; Ayers, C.R.; Rohatgi, A. Beyond Coronary Calcification, Family History, and C-Reactive Protein: Cholesterol Efflux Capacity and Cardiovascular Risk Prediction. J. Am. Coll. Cardiol. 2016, 67, 2480–2487. [Google Scholar] [CrossRef]
- Soria-Florido, M.T.; Castañer, O.; Lassale, C.; Estruch, R.; Salas-Salvadó, J.; Martínez-González, M.Á.; Corella, D.; Ros, E.; Arós, F.; Elosua, R.; et al. Dysfunctional High-Density Lipoproteins Are Associated with a Greater Incidence of Acute Coronary Syndrome in a Population at High Cardiovascular Risk: A Nested Case-Control Study. Circulation 2020, 444–453. [Google Scholar] [CrossRef]
- Riggs, K.A.; Joshi, P.H.; Khera, A.; Singh, K.; Akinmolayemi, O.; Ayers, C.R.; Rohatgi, A. Impaired HDL Metabolism Links GlycA, A Novel Inflammatory Marker, with Incident Cardiovascular Events. J. Clin. Med. 2019, 8, 173. [Google Scholar] [CrossRef] [PubMed]
- Nazir, S.; Jankowski, V.; Bender, G.; Zewinger, S.; Rye, K.A.; van der Vorst, E.P.C. Interaction between high-density lipoproteins and inflammation: Function matters more than concentration! Adv. Drug Deliv. Rev. 2020, 159, 94–119. [Google Scholar] [CrossRef]
- Annema, W.; DIkkers, A.; de Boer, J.F.; Dullaart, R.P.F.; Sanders, J.S.F.; Bakker, S.J.L.; Tietge, U.J.F. HDL cholesterol efflux predicts graft failure in renal transplant recipients. J. Am. Soc. Nephrol. 2016, 27, 595–603. [Google Scholar] [CrossRef]
- Shea, S.; Stein, J.H.; Jorgensen, N.W.; McClelland, R.L.; Tascau, L.; Shrager, S.; Heinecke, J.W.; Yvan-Charvet, L.; Tall, A.R. Cholesterol Mass Efflux Capacity, Incident Cardiovascular Disease, and Progression of Carotid Plaque. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 89–96. [Google Scholar] [CrossRef]
- Garg, P.K.; Jorgensen, N.W.; McClelland, R.L.; Allison, M.; Stein, J.H.; Yvan-Chavret, L.; Tall, A.R.; Shea, S. Cholesterol mass efflux capacity and risk of peripheral artery disease: The Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 2020, 297, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Hobbs, H.H. Disorders of Lipoprotein Metabolism. Harrison’s Princ. Intern. Med. 2008, 2416–2429. [Google Scholar]
- Oldoni, F.; Sinke, R.J.; Kuivenhoven, J.A. Mendelian disorders of high-density lipoprotein metabolism. Circ. Res. 2014, 114, 124–142. [Google Scholar] [CrossRef]
- Franceschini, G.; Sirtori, C.; Capurso, A.; Weisgraber, K.H.; Mahley, R.W. A-IMilano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. J. Clin. Investig. 1980, 66, 892–900. [Google Scholar] [CrossRef]
- Sirtori, C.R.; Calabresi, L.; Franceschini, G.; Baldassarre, D.; Amato, M.; Johansson, J.; Salvetti, M.; Monteduro, C.; Zulli, R.; Muiesan, M.L.; et al. Cardiovascular status of carriers of the apolipoprotein A-IMilano mutant: The limone sul garda study. Circulation 2001, 103, 1949–1954. [Google Scholar] [CrossRef]
- Nilsson, O.; Lindvall, M.; Obici, L.; Ekström, S.; Lagerstedt, J.O.; del Giudice, R. Structure dynamics of ApoA-I amyloidogenic variants in small HDL increase their ability to mediate cholesterol efflux. J. Lipid Res. 2020, 1–36. [Google Scholar] [CrossRef]
- Gkolfinopoulou, C.; Bourtsala, A.; Chroni, A. Structural and functional basis for increased HDL-cholesterol levels due to the naturally occurring V19L mutation in human apolipoprotein A-I. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2020, 1865. [Google Scholar] [CrossRef] [PubMed]
- Pisciotta, L.; Vitali, C.; Favari, E.; Fossa, P.; Adorni, M.P.; Leone, D.; Artom, N.; Fresa, R.; Calabresi, L.; Calandra, S.; et al. A complex phenotype in a child with familial HDL deficiency due to a novel frameshift mutation in APOA1 gene (apoA-IGuastalla). J. Clin. Lipidol. 2015, 9, 837–846. [Google Scholar] [CrossRef]
- Kono, M.; Tanaka, T.; Tanaka, M.; Vedhachalam, C.; Chetty, P.S.; Nguyen, D.; Dhanasekaran, P.; Lund-Katz, S.; Phillips, M.C.; Saito, H. Disruption of the C-terminal helix by single amino acid deletion is directly responsible for impaired cholesterol effl ux ability of apolipoprotein A-I Nichinan. J. Lipid Res. 2010, 51, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Anthanont, P.; Asztalos, B.F.; Polisecki, E.; Zachariah, B.; Schaefer, E.J. Case report: A novel apolipoprotein A-I missense mutation apoA-I (Arg149Ser)Boston associated with decreased lecithin-cholesterol acyltransferase activation and cellular cholesterol efflux. J. Clin. Lipidol. 2015, 9, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Miccoli, R.; Zhu, Y.; Daum, U.; Wessling, J.; Huang, Y.; Navalesi, R.; Assmann, G.; von Eckardstein, A. A natural apolipoprotein A-I variant, apoA-I(L141R)(Pisa), interferes with the formation of α-high density lipoproteins (HDL) but not with the formation of prβ1-HDL and influences efflux of cholesterol into plasma. J. Lipid Res. 1997, 38, 1242–1253. [Google Scholar] [CrossRef]
- Leren, T.P.; Bakken, K.S.; Daum, U.; Ose, L.; Berg, K.; Assmann, G.; von Eckardstein, A. Heterozygosity for apolipoprotein A-I(R160L)(Oslo) is associated with low levels of high density lipoprotein cholesterol and HDL-subclass LpA- I/A-II but normal levels of HDL-subclass LpA-I. J. Lipid Res. 1997, 38, 121–131. [Google Scholar] [CrossRef]
- Von Eckardstein, A.; Funke, H.; Henke, A.; Altland, K.; Benninghoven, A.; Assmann, G. Apolipoprotein A-I variants. Naturally occurring substitutions of proline residues affect plasma concentration of apolipoprotein A-I. J. Clin. Investig. 1989, 84, 1722–1730. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Calabresi, L.; Simonelli, S.; Gomaraschi, M.; Franceschini, G. Genetic lecithin: Cholesterol acyltransferase deficiency and cardiovascular disease. Atherosclerosis 2012, 222, 299–306. [Google Scholar] [CrossRef]
- Asztalos, B.F.; Schaefer, E.J.; Horvath, K.V.; Yamashita, S.; Miller, M.; Franceschini, G.; Calabresi, L. Role of LCAT in HDL remodeling: Investigation of LCAT deficiency states. J. Lipid Res. 2007, 48, 592–599. [Google Scholar] [CrossRef]
- Bérard, A.M.; Clerc, M.; Brewer, B.; Santamarina-Fojo, S. A normal rate of cellular cholesterol removal can be mediated by plasma from a patient with familial lecithin-cholesterol acyltransferase (LCAT) deficiency. Clin. Chim. Acta 2001, 314, 131–139. [Google Scholar] [CrossRef]
- Inazu, A.; Brown, M.; Helsler, C.; Agellon, L.; Koizumi, J.; Takata, K.; Maruhama, Y.; Mabuchi, H.; Tall, A.R. Increased High-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. New English J. Med. 1990, 323, 1234–1238. [Google Scholar] [CrossRef]
- Calabresi, L.; Gomaraschi, M.; Simonelli, S.; Bernini, F.; Franceschini, G. HDL and atherosclerosis: Insights from inherited HDL disorders. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2015, 1851, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, L.; Nilsson, P.; Pinotti, E.; Gomaraschi, M.; Favari, E.; Adorni, M.P.; Bernini, F.; Sirtori, C.R.; Calandra, S.; Franceschini, G.; et al. A novel homozygous mutation in CETP gene as a cause of CETP deficiency in a caucasian kindred. Atherosclerosis 2009, 205, 506–511. [Google Scholar] [CrossRef]
- Miwa, K.; Inazu, A.; Kawashiri, M.; Nohara, A.; Higashikata, T.; Kobayashi, J.; Koizumi, J.; Nakajima, K.; Nakano, T.; Niimi, M.; et al. Cholesterol efflux from J774 macrophages and Fu5AH hepatoma cells to serum is preserved in CETP-deficient patients. Clin. Chim. Acta 2009, 402, 19–24. [Google Scholar] [CrossRef]
- Plengpanich, W.; le Goff, W.; Poolsuk, S.; Julia, Z.; Guerin, M.; Khovidhunkit, W. CETP deficiency due to a novel mutation in the CETP gene promoter and its effect on cholesterol efflux and selective uptake into hepatocytes. Atherosclerosis 2011, 216, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Vergeer, M.; Korporaal, S.J.A.; Franssen, R.; Meurs, I.; Out, R.; Hovingh, G.K.; Hoekstra, M.; Sierts, J.A.; Dallinga-Thie, G.M.; Motazacker, M.M.; et al. Genetic Variant of the Scavenger Receptor BI in Humans. N. Engl. J. Med. 2011, 364, 136–145. [Google Scholar] [CrossRef]
- Chadwick, A.C.; Sahoo, D. Functional Characterization of Newly-Discovered Mutations in Human SR-BI. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Chapman, M.J. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol. Rev. 2006, 58, 342–374. [Google Scholar] [CrossRef]
- Ottestad, I.O.; Halvorsen, B.; Balstad, T.R.; Otterdal, K.; Borge, G.I.; Brosstad, F.; Myhre, A.M.; Ose, L.; Nenseter, M.S.; Holven, K.B. Triglyceride-rich HDL3 from patients with familial hypercholesterolemia are less able to inhibit cytokine release or to promote cholesterol efflux. J. Nutr. 2006, 136, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, N.; Orsoni, A.; Julia, Z.; Fournier, N.; Frisdal, E.; Duchene, E.; Bruckert, E.; Carrie, A.; Bonnefont-Rousselot, D.; Pirault, J.; et al. Atheroprotective reverse cholesterol transport pathway is defective in familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1675–1681. [Google Scholar] [CrossRef]
- Versmissen, J.; Vongpromek, R.; Yahya, R.; van der Net, J.B.; van Vark-van der Zee, L.; Blommesteijn-Touw, J.; Wattimena, D.; Rietveld, T.; Pullinger, C.R.; Christoffersen, C.; et al. Familial hypercholesterolaemia: Cholesterol efflux and coronary disease. Eur. J. Clin. Investig. 2016, 46, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Dana, S.E.; Faust, J.R.; Beaudet, A.L.; Brown, M.S. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J. Biol. Chem. 1975, 250, 8487–8495. [Google Scholar] [CrossRef]
- Guardamagna, O.; Nair, D.; Soran, H.; Hovingh, K.; Calandra, S.; Hamilton, J.; Bertolini, S.; Jones, S.; Cori, M.; Eagleton, T.; et al. Lysosomal acid lipase de fi ciency e An under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014, 235, 21–30. [Google Scholar] [CrossRef]
- Zimetti, F.; Favari, E.; Cagliero, P.; Pia, M.; Ronda, N.; Bonardi, R.; Gomaraschi, M.; Calabresi, L. Cholesterol traf fi cking-related serum lipoprotein functions in children with cholesteryl ester storage disease. Atherosclerosis 2015, 242, 443–449. [Google Scholar] [CrossRef]
- Shimamura, M.; Matsuda, M.; Yasumo, H.; Okazaki, M.; Fujimoto, K.; Kono, K.; Shimizugawa, T.; Ando, Y.; Koishi, R.; Kohama, T.; et al. Angiopoietin-Like Protein3 Regulates Plasma HDL Cholesterol Through Suppression of Endothelial Lipase Mitsuru. Arter. Thromb. Vasc. Biol. 2007, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Pirruccello, J.P.; Do, R.; Peloso, G.M.; Guiducci, C.; Sougnez, C.; Garimella, K.V.; Fisher, S.; Abreu, J.; Barry, A.J.; et al. Exome Sequencing, ANGPTL3 Mutations, and Familial Combined Hypolipidemia. N. Engl. J. Med. 2010, 363, 2220–2227. [Google Scholar] [CrossRef] [PubMed]
- Pisciotta, L.; Favari, E.; Magnolo, L.; Simonelli, S.; Adorni, M.P.; Sallo, R.; Fancello, T.; Zavaroni, I.; Ardigo, D.; Bernini, F.; et al. Characterization of three kindreds with familial combined hypolipidemia caused by loss-of-function mutations of ANGPTL3. Circ. Cardiovasc. Genet. 2012, 5, 42–50. [Google Scholar] [CrossRef]
- Minicocci, I.; Cantisani, V.; Poggiogalle, E.; Favari, E.; Zimetti, F.; Montali, A.; Labbadia, G.; Pigna, G.; Pannozzo, F.; Zannella, A.; et al. Functional and morphological vascular changes in subjects with familial combined hypolipidemia: An exploratory analysis. Int. J. Cardiol. 2013, 168, 4375–4378. [Google Scholar] [CrossRef] [PubMed]
- Podkowińska, A.; Formanowicz, D. Chronic kidney disease as oxidative stress-and inflammatory-mediated cardiovascular disease. Antioxidants 2020, 9, 752. [Google Scholar] [CrossRef]
- Rysz, J.; Gluba-Brzózka, A.; Rysz-Górzyńska, M.; Franczyk, B. The role and function of HDL in patients with chronic kidney disease and the risk of cardiovascular disease. Int. J. Mol. Sci. 2020, 21, 601. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Birner-Gruenberger, R.; Stojakovic, T.; El-Gamal, D.; Binder, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Uremia alters HDL composition and function. J. Am. Soc. Nephrol. 2011, 22, 1631–1641. [Google Scholar] [CrossRef]
- Holzer, M.; Schilcher, G.; Curcic, S.; Trieb, M.; Ljubojevic, S.; Stojakovic, T.; Scharnagl, H.; Kopecky, C.M.; Rosenkranz, A.R.; Heinemann, A.; et al. Dialysis Modalities and HDL Composition and Function. J. Am. Soc. Nephrol. 2015, 26, 2267–2276. [Google Scholar] [CrossRef]
- Yamamoto, S.; Yancey, P.G.; Ikizler, T.A.; Jerome, W.G.; Kaseda, R.; Cox, B.; Bian, A.; Shintani, A.; Fogo, A.B.; Linton, M.F.; et al. Dysfunctional high-density lipoprotein in patients on chronic hemodialysis. J. Am. Coll. Cardiol. 2012, 60, 2372–2379. [Google Scholar] [CrossRef]
- Meier, S.M.; Wultsch, A.; Hollaus, M.; Ammann, M.; Pemberger, E.; Liebscher, F.; Lambers, B.; Fruhwürth, S.; Stojakovic, T.; Scharnagl, H.; et al. Effect of chronic kidney disease on macrophage cholesterol efflux. Life Sci. 2015, 136, 1–6. [Google Scholar] [CrossRef]
- Wang, K.; Zelnick, L.R.; Hoofnagle, A.N.; Vaisar, T.; Henderson, C.M.; Imrey, P.B.; Robinson-Cohen, C.; de Boer, I.H.; Shiu, Y.-T.; Himmelfarb, J.; et al. Alteration of HDL Protein Composition with Hemodialysis Initiation. Clin. J. Am. Soc. Nephrol. 2018, 13, 1225–1233. [Google Scholar] [CrossRef]
- Shao, B.; Tang, C.; Heinecke, J.W.; Oram, J.F. Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J. Lipid Res. 2010, 51, 1849–1858. [Google Scholar] [CrossRef]
- Gipson, G.T.; Carbone, S.; Wang, J.; Dixon, D.L.; Jovin, I.S.; Carl, D.E.; Gehr, T.W.; Ghosh, S. Impaired Delivery of Cholesterol Effluxed From Macrophages to Hepatocytes by Serum From CKD Patients May Underlie Increased Cardiovascular Disease Risk. Kidney Int. Reports 2020, 5, 199–210. [Google Scholar] [CrossRef]
- Stadler, J.T.; Marsche, G. Obesity-related changes in high-density lipoprotein metabolism and function. Int. J. Mol. Sci. 2020, 21, 8985. [Google Scholar] [CrossRef] [PubMed]
- Talbot, C.P.J.; Plat, J.; Joris, P.J.; Konings, M.; Kusters, Y.H.A.M.; Schalkwijk, C.G.; Ritsch, A.; Mensink, R.P. HDL cholesterol efflux capacity and cholesteryl ester transfer are associated with body mass, but are not changed by diet-induced weight loss: A randomized trial in abdominally obese men. Atherosclerosis 2018, 274, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Marsche, G.; Zelzer, S.; Meinitzer, A.; Kern, S.; Meissl, S.; Pregartner, G.; Weghuber, D.; Almer, G.; Mangge, H. Adiponectin predicts high-density lipoprotein cholesterol efflux capacity in adults irrespective of body mass index and fat distribution. J. Clin. Endocrinol. Metab. 2017, 102, 4117–4123. [Google Scholar] [CrossRef]
- Vazquez, E.; Sethi, A.A.; Freeman, L.; Zalos, G.; Chaudhry, H.; Haser, E.; Aicher, B.O.; Aponte, A.; Gucek, M.; Kato, G.J.; et al. High-density lipoprotein cholesterol efflux, nitration of apolipoprotein A-I, and endothelial function in obese women. Am. J. Cardiol. 2012, 109, 527–532. [Google Scholar] [CrossRef][Green Version]
- Adams, T.; Gress, R.; Smith, S.; Halverson, R.; Simper, S.; Rosamond, W.; LaMonte, M.; Stroup, A.; Hunt, S. Long-Term Mortality After Gastric Bypass Surgery. N. Engl. J. Med. 2007, 357, 753–761. [Google Scholar] [CrossRef]
- Kjellmo, C.A.; Karlsson, H.; Nestvold, T.K.; Ljunggren, S.; Cederbrant, K.; Marcusson-Ståhl, M.; Mathisen, M.; Lappegård, K.T.; Hovland, A. Bariatric surgery improves lipoprotein profile in morbidly obese patients by reducing LDL cholesterol, apoB, and SAA/PON1 ratio, increasing HDL cholesterol, but has no effect on cholesterol efflux capacity. J. Clin. Lipidol. 2018, 12, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Julia, Z.; Poitou, C.; Bouillot, J.L.; Basdevant, A.; Chapman, M.J.; Clement, K.; Guerin, M. Effect of bariatric surgery-induced weight loss on SR-BI-, ABCG1-, and ABCA1-mediated cellular cholesterol efflux in obese women. J. Clin. Endocrinol. Metab. 2011, 96, 1151–1159. [Google Scholar] [CrossRef]
- Lorkowski, S.W.; Brubaker, G.; Rotroff, D.M.; Kashyap, S.R.; Bhatt, D.L.; Nissen, S.E.; Schauer, P.R.; Aminian, A.; Smith, J.D. Bariatric surgery improves hdl function examined by apoa1 exchange rate and cholesterol efflux capacity in patients with obesity and type 2 diabetes. Biomolecules 2020, 10, 551. [Google Scholar] [CrossRef] [PubMed]
- Heffron, S.P.; Lin, B.; Parikh, M.; Adelman, S.J.; Collins, H.L.; Berger, J.S.; Fisher, E.A.; Nutrition, E.; Paulo, S.; Meeting, P. Changes in HDL Cholesterol Efflux Capacity following Bariatric Surgery are Procedure Dependent. Arter. Thromb. Vasc. Biol. 2018, 38, 245–254. [Google Scholar] [CrossRef]
- Cardner, M.; Yalcinkaya, M.; Goetze, S.; Luca, E.; Balaz, M.; Hunjadi, M.; Hartung, J.; Shemet, A.; Kränkel, N.; Radosavljevic, S.; et al. Structure-function relationships of HDL in diabetes and coronary heart disease. JCI Insight 2020, 5, 1–18. [Google Scholar] [CrossRef]
- Wong, N.K.P.; Nicholls, S.J.; Tan, J.T.M.; Bursill, C.A. The role of high-density lipoproteins in diabetes and its vascular complications. Int. J. Mol. Sci. 2018, 19, 1680. [Google Scholar] [CrossRef] [PubMed]
- Apro, J.; Tietge, U.J.F.; Dikkers, A.; Parini, P.; Angelin, B.; Rudling, M. Impaired cholesterol efflux capacity of high-density lipoprotein isolated from interstitial fluid in type 2 diabetes mellitus—brief report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 787–791. [Google Scholar] [CrossRef]
- Shiu, S.W.; Wong, Y.; Tan, K.C. Pre-β1 HDL in type 2 diabetes mellitus. Atherosclerosis 2017, 263, 24–28. [Google Scholar] [CrossRef]
- He, Y.; Ronsein, G.E.; Tang, C.; Jarvik, G.P.; Davidson, W.S.; Kothari, V.; Song, H.D.; Segrest, J.P.; Bornfeldt, K.E.; Heinecke, J.W. Diabetes impairs cellular cholesterol efflux from ABCA1 to small HDL particles. Circ. Res. 2020, 1198–1210. [Google Scholar] [CrossRef]
- Blanco-Rojo, R.; Perez-Martinez, P.; Lopez-Moreno, J.; Martinez-Botas, J.; Delgado-Lista, J.; Van-Ommen, B.; Yubero-Serrano, E.; Camargo, A.; Ordovas, J.M.; Perez-Jimenez, F.; et al. HDL cholesterol efflux normalised to apoA-I is associated with future development of type 2 diabetes: From the CORDIOPREV trial. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Machado-Lima, A.; Iborra, R.T.; Pinto, R.S.; Castilho, G.; Sartori, C.H.; Oliveira, E.R.; Okuda, L.S.; Nakandakare, E.R.; Giannella-Neto, D.; Machado, U.F.; et al. In Type 2 Diabetes Mellitus Glycated Albumin Alters Macrophage Gene Expression Impairing ABCA1-Mediated Cholesterol Efflux. J. Cell. Physiol. 2015, 230, 1250–1257. [Google Scholar] [CrossRef]
- Dullaart, R.P.F.; Pagano, S.; Perton, F.G.; Vuilleumier, N. Antibodies against the C-terminus of ApoA-1 are inversely associated with cholesterol efflux capacity and HDL metabolism in subjects with and without type 2 diabetes mellitus. Int. J. Mol. Sci. 2019, 20, 732. [Google Scholar] [CrossRef]
- Dullaart, R.P.F.; Annema, W.; de Boer, J.F.; Tietge, U.J.F. Pancreatic β-cell function relates positively to HDL functionality in well-controlled Type 2 diabetes mellitus. Atherosclerosis 2012, 222, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.C.; Tai, E.S.; Sviridov, D.; Nestel, P.J.; Ng, C.; Chan, E.; Teo, Y.; Wai, D.C.H. Relationships between cholesterol efflux and high-density lipoprotein particles in patients with type 2 diabetes mellitus. J. Clin. Lipidol. 2011, 5, 467–473. [Google Scholar] [CrossRef] [PubMed]
- de Vries, R.; Groen, A.K.; Perton, F.G.; Dallinga-Thie, G.M.; van Wijland, M.J.A.; Dikkeschei, L.D.; Wolffenbuttel, B.H.R.; van Tol, A.; Dullaart, R.P.F. Increased cholesterol efflux from cultured fibroblasts to plasma from hypertriglyceridemic type 2 diabetic patients: Roles of pre β-HDL, phospholipid transfer protein and cholesterol esterification. Atherosclerosis 2008, 196, 733–741. [Google Scholar] [CrossRef]
- Machado-Lima, A.; Iborra, R.T.; Pinto, R.; Sartori, C.; Oliveira, E.R.; Nakandakare, E.R.; Stefano, J.; Giannella-Neta, D.; Corrêa-Giannella, M.L.C.; Passarelli, M. Advanced glycated albumin isolated from poorly controlled type 1 diabetes mellitus patients alters macrophage gene expression impairing ABCA-1- mediated reverse cholesterol transport. Diabetes. Metab. Res. Rev. 2013, 29, 66–76. [Google Scholar] [CrossRef]
- Guy, J.; Ogden, L.; Wadwa, R.P.; Hamman, R.F.; Mayer-Davis, E.J.; LIese, A.D.; D’Agostino, R.; Marcovina, S.; Dabelea, D. Lipid and lipoprotein profiles in youth with and without type 1 diabetes: The SEARCH for diabetes in youth case-control study. Diabetes Care 2009, 32, 416–420. [Google Scholar] [CrossRef]
- Gourgari, E.; Playford, M.P.; Campia, U.; Dey, A.K.; Cogen, F.; Gubb-Weiser, S.; Mete, M.; Desale, S.; Sampson, M.; Taylor, A.; et al. Low cholesterol efflux capacity and abnormal lipoprotein particles in youth with type 1 diabetes: A case control study. Cardiovasc. Diabetol. 2018, 17, 1–10. [Google Scholar] [CrossRef]
- Ahmed, M.O.; Byrne, R.E.; Pazderska, A.; Segurado, R.; Guo, W.; Gunness, A.; Frizelle, I.; Sherlock, M.; Ahmed, K.S.; McGowan, A.; et al. HDL particle size is increased and HDL-cholesterol efflux is enhanced in type 1 diabetes: A cross-sectional study. Diabetologia 2020. [Google Scholar] [CrossRef]
- Roever, L.; Resende, E.S.; Diniz, A.L.D.; Penha-Silva, N.; O’Connell, J.L.; Gomes, P.F.S.; Zanetti, H.R.; Roerver-Borges, A.S.; Veloso, F.C.; de Souza, F.R.; et al. High-density lipoprotein-cholesterol functionality and metabolic syndrome: Protocol for review and meta-analysis. Medicine 2018, 97, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Lee, D.K.; Liu, M.; Portincasa, P.; Wang, D.Q.H. Novel insights into the pathogenesis and management of the metabolic syndrome. Pediatr. Gastroenterol. Hepatol. Nutr. 2020, 23, 189–230. [Google Scholar] [CrossRef]
- Kontush, A. HDL-mediated mechanisms of protection in cardiovascular disease. Cardiovasc. Res. 2014, 103, 341–349. [Google Scholar] [CrossRef]
- Lucero, D.; Svidirov, D.; Freeman, L.; López, G.I.; Fassio, E.; Remaley, A.T.; Schreier, L. Increased cholesterol efflux capacity in metabolic syndrome: Relation with qualitative alterations in HDL and LCAT. Atherosclerosis 2015, 242, 236–242. [Google Scholar] [CrossRef]
- Gall, J.; Frisdal, E.; Bittar, R.; le Goff, W.; Bruckert, E.; Lesnik, P.; Guerin, M.; Giral, P. Association of cholesterol efflux capacity with clinical features of metabolic syndrome: Relevance to atherosclerosis. J. Am. Heart Assoc. 2016, 5, 1–13. [Google Scholar] [CrossRef]
- Annema, W.; Dikkers, A.; de Boer, J.F.; van Greevenbroek, M.M.J.; van der Kallen, C.J.H.; Schalkwijk, C.G.; Stehouwer, C.D.A.; Dullaart, R.P.F.; Tietge, U.J.F. Impaired HDL cholesterol efflux in metabolic syndrome is unrelated to glucose tolerance status: The CODAM study. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, E.H.; Gruppen, E.G.; Ebtehaj, S.; Bakker, S.J.L.; Tietge, U.J.F.; Dullaart, R.P.F. Cholesterol efflux capacity is impaired in subjects with an elevated Fatty Liver Index, a proxy of non-alcoholic fatty liver disease. Atherosclerosis 2018, 277, 21–27. [Google Scholar] [CrossRef]
- Di Costanzo, A.; Ronca, A.; D’erasmo, L.; Manfredini, M.; Baratta, F.; Pastori, D.; di Martino, M.; Ceci, F.; Angelico, F.; del Ben, M.; et al. Hdl-mediated cholesterol efflux and plasma loading capacities are altered in subjects with metabolically-but not genetically driven non-alcoholic fatty liver disease (Nafld). Biomedicines 2020, 8, 625. [Google Scholar] [CrossRef]
- Khan, A.A.; Mundra, P.A.; Straznicky, N.E.; Nestel, P.J.; Wong, G.; Tan, R.; Huynh, K.; Ng, T.W.; Mellett, N.A.; Weir, J.M.; et al. Weight Loss and Exercise Alter the High-Density Lipoprotein Lipidome and Improve High-Density Lipoprotein Functionality in Metabolic Syndrome. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Zhang, Z.; Li, H.; Bai, P.; Cao, X.; Dobs, A.S. Analysis of cardiovascular risk factors associated with serum testosterone levels according to the US 2011–2012 National Health and Nutrition Examination Survey. Aging Male 2019, 22, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Adorni, M.P.; Zimetti, F.; Cangiano, B.; Vezzoli, V.; Bernini, F.; Caruso, D.; Corsini, A.; Sirtori, C.R.; Cariboni, A.; Bonomi, M.; et al. High-Density Lipoprotein Function Is Reduced in Patients Affected by Genetic or Idiopathic Hypogonadism. J. Clin. Endocrinol. Metab. 2019, 104, 3097–3107. [Google Scholar] [CrossRef] [PubMed]
- Sankaramarayanan, S.; Oram, J.F.; Asztalos, B.F.; Vaughan, A.M.; Lund-Katz, S.; Adorni, M.P.; Phillips, M.C.; Rothblat, G.H. Effects of acceptor composition and mechanism of ABCG1-mediated cellular free cholesterol efflux. J. Lipid Res. 2009, 50, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Asztalos, B.F.; Horvath, K.V.; Mehan, M.; Yokota, Y.; Schaeer, E.J. Influence of HDL particles on cell-cholesterol efflux under various pathological conditions. J. Lipid Res. 2017, 58, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Rubinow, K.B.; Vaisar, T.; Chao, J.H.; Heinecke, J.W.; Page, S.T. Sex Steroids Mediate Discrete Effects on HDL Cholesterol Efflux Capacity and Particle Concentration in Healthy Men. J. Clin. Lipidol. 2018, 12, 1072–1082. [Google Scholar] [CrossRef]
- Rubinow, K.B.; Tang, C.; Hoofnagle, A.N.; Snyder, C.N.; Amory, J.K.; Heinecke, J.W.; Page, S.T. Acute Sex Steroid Withdrawal Increases Cholesterol Efflux Capacity and HDL-Associated Clusterin in Men Katya. Steroids 2012, 77, 454–460. [Google Scholar] [CrossRef][Green Version]
- Rubinow, K.B.; Vaisar, T.; Tang, C.; Matsumoto, A.M.; Heinecke, J.W.; Page, S.T. Testosterone replacement in hypogonadal men alters the HDL proteome but not HDL cholesterol efflux capacity. J. Lipid Res. 2012, 53, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Biondi, B.; Klein, I. Hypothyroidism as a risk factor for cardiovascular disease. Endocrine 2004, 24, 1–13. [Google Scholar] [CrossRef]
- van der Boom, T.; Jia, C.; Lefrandt, J.D.; Connelly, M.A.; Links, T.P.; Tietge, U.J.F.; Dullaart, R.P.F. HDL cholesterol efflux capacity is impaired in severe short-term hypothyroidism despite increased HDL cholesterol. J. Clin. Endocrinol. Metab. 2020, 105, E3355–E3362. [Google Scholar] [CrossRef] [PubMed]
- Roe, A.; Hillman, J.; Butts, S.; Smith, M.; Rader, D.; Playford, M.; Mehta, N.N.; Dokras, A. Decreased cholesterol efflux capacity and atherogenic lipid profile in young women with PCOS. J. Clin. Endocrinol. Metab. 2014, 99, 841–847. [Google Scholar] [CrossRef]
- Tedesco, S.; Adorni, M.P.; Ronda, N.; Cappellari, R.; Mioni, R.; Barbot, M.; Pinelli, S.; Plebani, M.; Bolego, C.; Scaroni, C.; et al. Activation profiles of monocyte-macrophages and HDL function in healthy women in relation to menstrual cycle and in polycystic ovary syndrome patients. Endocrine 2019, 66, 360–369. [Google Scholar] [CrossRef]
- Gidwani, S.; Phelan, N.; McGill, J.; McGowan, A.; O’Connor, A.; Young, I.S.; Gibney, J.; McEneny, J. Polycystic ovary syndrome influences the level of serum amyloid A and activity of phospholipid transfer protein in HDL2 and HDL3. Hum. Reprod. 2014, 29, 1518–1525. [Google Scholar] [CrossRef][Green Version]
- Miilunpohja, M.; Uphoff, A.; Somerharju, P.; Tiitinen, A.; Wähälä, K.; Tikkanen, M.J. Fatty acid esterification of lipoprotein-associated estrone in human plasma and follicular fluid. J. Steroid Biochem. Mol. Biol. 2006, 100, 59–66. [Google Scholar] [CrossRef]
- Dokras, A.; Playford, M.; Kris-Etherton, P.M.; Kunselman, A.R.; Stetter, C.M.; Williams, N.I.; Gnatuk, C.L.; Estes, S.J.; Sarwer, D.B.; Allison, K.C.; et al. Impact of hormonal contraception and weight loss on high-density lipoprotein cholesterol efflux and lipoprotein particles in women with polycystic ovary syndrome. Clin. Endocrinol. 2017, 86, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Lightbody, R.J.; Taylor, J.M.W.; Dempsie, Y.; Graham, A. MicroRNA sequences modulating inflammation and lipid accumulation in macrophage “foam” cells: Implications for atherosclerosis. World J. Cardiol. 2020, 12, 303–333. [Google Scholar] [CrossRef]
- Transporter, G.A.; Panousis, C.G.; Zuckerman, S.H. Interferon-r Induces Downregulation of Tangier Disease. 2011, 1565–1571. Arterioscler. Thromb. Vasc. Biol. 2011, 1565–1571. [Google Scholar]
- Catapano, A.L.; Pirillo, A.; Bonacina, F.; Norata, G.D. HDL in innate and adaptive immunity. Cardiovasc. Res. 2014, 103, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Gautier, E.L.; Ganda, A.; Molusky, M.M.; Wang, W.; Fotakis, P.; Randolph, G.J.; D’Agati, V.D.; Yvan-charvet, L.; Laurent-Tall, A.R. Cholesterol Accumulation in Dendritic Cells Links the Inflammasome to Acquired Immunity. Cell Metab 2017, 25, 1294–1304. [Google Scholar] [CrossRef]
- Tanaka, S.; Couret, D.; Tran-Dinh, A.; Duranteau, J.; Montravers, P.; Schwendeman, A.; Meilhac, O. High-density lipoproteins during sepsis: From bench to bedside. Crit. Care 2020, 24, 1–11. [Google Scholar] [CrossRef]
- Vaisar, T.; Tang, C.; Babenko, I.; Hutchins, P.; Wimberger, J.; Suffredini, A.F.; Heinecke, J.W. Inflammatory remodeling of the HDL proteome impairs cholesterol efflux capacity. J. Lipid Res. 2015, 56, 1519–1530. [Google Scholar] [CrossRef]
- Sharma, N.K.; Tashima, A.K.; Brunialti, M.K.C.; Ferreira, E.R.; Torquato, R.J.S.; Mortara, R.A.; MacHado, F.R.; Assuncao, M.; Rigato, O.; Salomao, R. Proteomic study revealed cellular assembly and lipid metabolism dysregulation in sepsis secondary to community-acquired pneumonia. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- de la Llera Moya, M.; McGillicuddy, F.C.; Hinkle, C.C.; Byrne, M.; Joshi, M.R.; Nguyen, V.; Tabita-Martinez, J.; Wolfe, M.L.; Badellino, K.; Pruscino, L.; et al. Inflammation modulates human HDL composition and function in vivo. Atherosclerosis 2012, 222, 390–394. [Google Scholar] [CrossRef]
- Zimetti, F.; de Vuono, S.; Gomaraschi, M.; Adorni, M.P.; Favari, E.; Ronda, N.; Ricci, M.A.; Veglia, F.; Calabresi, L.; Lupattelli, G. Plasma cholesterol homeostasis, HDL remodeling and function during the acute phase reaction. J. Lipid Res. 2017, 58, 2051–2060. [Google Scholar] [CrossRef]
- Jahangiri, A.; de Beer, M.C.; Noffsinger, V.; Tannock, L.R.; Ramaiah, C.; Webb, N.R.; van der Westhuyzen, D.R.; de Beer, F.C. HDL remodeling during the acute phase response. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 261–267. [Google Scholar] [CrossRef]
- Guirgis, F.W.; Leeuwenburgh, C.; Grijalva, V.; Bowman, J.; Kalynych, C.; Moldawer, L.; Moore, F.A.; Reddy, S.T. HDL Cholesterol Efflux is Impaired in Older Patients with Early Sepsis: A Subanalysis of a Prospective Pilot Study. Shock 2018, 50, 66–70. [Google Scholar] [CrossRef]
- Pussinen, P.J.; Jauhiainen, M.; Vilkuna-Rautiainen, T.; Sundvall, J.; Vesanen, M.; Mattila, K.; Palosuo, T.; Alfthan, G.; Asikainen, S. Periodontitis decreases the antiatherogenic potency of high density lipoprotein. J. Lipid Res. 2004, 45, 139–147. [Google Scholar] [CrossRef]
- Hollan, I.; Meroni, P.L.; Ahearn, J.M.; Cohen-Tervaert, J.W.; Curran, S.; Goodyear, C.S.; Hestad, K.A.; Kahaleh, B.; Riggio, M.; Shields, K.; et al. Cardiovascular disease in autoimmune rheumatic diseases. Autoimmun. Rev. 2013, 12, 1004–1015. [Google Scholar] [CrossRef]
- Myasoedova, E.; Crowson, C.S.; Kremers, H.M.; Roger, V.L.; Fitz-Gibbon, P.; Therneau, T.M.; Gabriel, S.E. Lipid paradox in rheumatoid arthritis: The impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Ann Rheum Dis 2011, 70, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Säemann, M.D.; Poglitsch, M.; Kopecky, C.; Haidinger, M.; Hörl, W.H.; Weichhart, T. The versatility of HDL: A crucial anti-inflammatory regulator. Eur. J. Clin. Investig. 2010, 40, 1131–1143. [Google Scholar] [CrossRef]
- Montecucco, F.; Favari, E.; Norata, G.D.; Ronda, N.; Nofer, J.; Vuilleumier, N. Impact of systemic inflammation and autoimmune diseases on apoA-I and HDL plasma levels and functions. Handb. Exp. Pharmacol. 2015, 224, 455–482. [Google Scholar] [PubMed]
- Charles-Schoeman, C.; Lee, Y.Y.; Grijalva, V.; Amjadi, S.; FitzGerald, J.; Ranganath, V.K.; Taylor, M.; McMahon, M.; Paulus, H.E.; Reddy, S.T. Cholesterol efflux by high density lipoproteins is impaired in patients with active rheumatoid arthritis. Ann. Rheum. Dis. 2012, 71, 1157–1162. [Google Scholar] [CrossRef]
- Ronda, N.; Favari, E.; Borghi, M.O.; Ingegnoli, F.; Gerosa, M.; Chighizola, C.; Zimetti, F.; Adorni, M.P.; Bernini, F.; Meroni, P.L. Impaired serum cholesterol efflux capacity in rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 2014, 73, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Bozzolo, E.P.; Contri, R.; Baragetti, A.; Palini, A.G.; Cianflone, D.; Banfi, M.; Uboldi, P.; Bottoni, G.; Scotti, I.; et al. Cardiometabolic and immune factors associated with increased common carotid artery intima-media thickness and cardiovascular disease in patients with systemic lupus erythematosus. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 751–759. [Google Scholar] [CrossRef]
- Quevedo-Abeledo, J.C.; Sánchez-Pérez, H.; Tejera-Segura, B.; de Armas-Rillo, L.; Armas-González, E.; Machado, J.D.; González-Gay, M.A.; Díaz-González, F.; Ferraz-Amaro, I. Differences in HDL-Cholesterol Efflux Capacity Between Patients with Systemic Lupus Erythematosus and Rheumatoid Arthritis. Arthritis Care Res. 2020, 3. [Google Scholar] [CrossRef]
- Ferraz-Amaro, I.; Hernández-Hernández, M.V.; Armas-González, E.; Sánchez-Pérez, H.; Machado, J.D.; Díaz-González, F. HDL cholesterol efflux capacity is related to disease activity in psoriatic arthritis patients. Clin. Rheumatol. 2020, 39, 1871–1880. [Google Scholar] [CrossRef]
- Sánchez-Pérez, H.; Quevedo-Abeledo, J.C.; de Armas-Rillo, L.; Rua-Figueroa, Í.; Tejera-Segura, B.; Armas-González, E.; Machado, J.D.; García-Dopico, J.A.; Jimenez-Sosa, A.; Rodríguez-Lozano, C.; et al. Impaired HDL cholesterol efflux capacity in systemic lupus erythematosus patients is related to subclinical carotid atherosclerosis. Rheumatology 2020, 59, 2847–2856. [Google Scholar] [CrossRef]
- Feingold, K.R.; Grunfeld, C. Effect of inflammation on HDL structure and function. Curr. Opin. Lipidol. 2016, 27, 521–530. [Google Scholar] [CrossRef]
- Charles-Schoeman, C.; Meriwether, D.; Lee, Y.Y.; Shahbazian, A.; Reddi, S.T. High levels of oxidized fatty acids in HDL are associated with impaired HDL function in patients with active rheumatoid arthritis. Clin Rheumatol 2018, 37, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Wolf, P.; Curcic, S.; Birner-Gruenberger, R.; Weger, W.; Inzinger, M.; El-Gamal, D.; Wadsack, C.; Heinemann, A.; Marsche, G. Psoriasis alters HDL composition and cholesterol efflux capacity. J. Lipid Res. 2012, 53, 1618–1624. [Google Scholar] [CrossRef]
- Gkolfinopoulou, C.; Stratikos, E.; Theofilatos, D.; Kardassis, D.; Voulgari, P.V.; Drosos, A.A.; Chroni, A. Impaired antiatherogenic functions of high-density lipoprotein in patients with ankylosing spondylitis. J. Rheumatol. 2015, 42, 1652–1660. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Sehgal, A.; Zengin, G.; Brisc, C.; Brisc, M.C.; Munteanu, M.A.; Nistor-Cseppento, D.C.; Bungau, S. The lipid paradox as a metabolic checkpoint and its therapeutic significance in ameliorating the associated cardiovascular risks in rheumatoid arthritis patients. Int. J. Mol. Sci. 2020, 21, 9505. [Google Scholar] [CrossRef]
- Naerr, G.W.; Rein, P.; Saely, C.H.; Drexel, H. Effects of synthetic and biological disease modifying antirheumatic drugs on lipid and lipoprotein parameters in patients with rheumatoid arthritis. Vascul. Pharmacol. 2016, 81, 22–30. [Google Scholar] [CrossRef]
- Charles-Schoeman, C.; Gonzalez-Gay, M.A.; Kaplan, I.; Boy, M.; Geier, J.; Luo, Z.; Zuckerman, A.; Riese, R. Effects of tofacitinib and other DMARDs on lipid profiles in rheumatoid arthritis: Implications for the rheumatologist. Semin. Arthritis Rheum. 2016, 46, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Karpouzas, G.A.; Ormseth, S.R.; Hernandez, E.; Budoff, M.J. Biologics May Prevent Cardiovascular Events in Rheumatoid Arthritis by Inhibiting Coronary Plaque Formation and Stabilizing High-Risk Lesions. Arthritis Rheumatol. 2020, 72, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Thompson, L.; Giles, J.T.; Bathon, J.M.; Salmon, J.E.; Beaulieu, A.D.; Codding, C.E.; Carlson, T.H.; Delles, C.; Lee, J.S.; et al. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann. Rheum. Dis. 2015, 74, 694–702. [Google Scholar] [CrossRef]
- Raterman, H.G.; Levels, H.; Voskuyl, A.E.; Lems, W.F.; Dijkmans, B.A.; Nurmohamed, M.T. HDL protein composition alters from proatherogenic into less atherogenic and proinflammatory in rheumatoid arthritis patients responding to rituximab. Ann. Rheum. Dis. 2013, 72, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Charles-Schoeman, C.; Lim, L.L.; Shahbazian, A.; Wang, X.; Elashoff, D.; Curtis, J.R.; Navarro-Millàn, I.; Yang, L.; Cofield, S.S.; Moreland, L.W.; et al. Improvement in HDL Function in Early Rheumatoid Arthritis Patients Treated with Methotrexate Monotherapy or Combination Therapy in the TEAR Trial. Arthritis Rheumatol. 2017, 69, 46–57. [Google Scholar] [CrossRef]
- Charles-Schoeman, C.; Gugiu, G.; Ge, H.; Shahbazian, A.; Lee, Y.; Wang, X.; Furst, D.; Ranganath, V.; Maldonado, M.; Lee, T.; et al. Remodiling of the HDL Proteome with Treatment Response to Abatacept or Adalimumab in the AMPLE Trial of Patients with Rheumatoid Arthritis. Atherosclerosis 2018, 275, 107–114. [Google Scholar] [CrossRef]
- Ferraz-Amaro, I.; Hernández-Hernández, M.V.; Tejera-Segura, B.; Delgado-Frías, E.; Macía-Díaz, M.; Machado, J.D.; Diaz-González, F. Effect of IL-6 Receptor Blockade on Proprotein Convertase Subtilisin/Kexin Type-9 and Cholesterol Efflux Capacity in Rheumatoid Arthritis Patients. Horm. Metab. Res. 2019, 51, 200–209. [Google Scholar] [CrossRef]
- Cacciapaglia, F.; Perniola, S.; Venerito, V.; Anelli, M.G.; Härdfeldt, J.; Fornaro, M.; Moschetta, A.; Iannone, F. The Impact of Biologic Drugs on High-Density Lipoprotein Cholesterol Efflux Capacity in Rheumatoid Arthritis Patients. J. Clin. Rheumatol. 2020. [Google Scholar] [CrossRef]
- Holzer, M.; Wolf, P.; Inzinger, M.; Trieb, M.; Curcic, S.; Pasterk, L.; Weger, W.; Heinemann, A.; Marsche, G. Anti-psoriatic therapy recovers high-density lipoprotein composition and function. J. Investig. Dermatol. 2014, 134, 635–642. [Google Scholar] [CrossRef]
- Mani, P.; Uno, K.; Duong, M.; Wolski, K.; Spalding, S.; Elaine Husni, M.; Nicholls, S.J. HDL function and subclinical atherosclerosis in juvenile idiopathic arthritis. Cardiovasc. Diagn. Ther. 2016, 6, 34–43. [Google Scholar] [CrossRef]
- Ronda, N.; Greco, D.; Adorni, M.P.; Zimetti, F.; Favari, E.; Hjeltnes, G.; Mikkelsen, K.; Borghi, M.O.; Favalli, E.G.; Gatti, R.; et al. Newly identified antiatherosclerotic activity of methotrexate and adalimumab: Complementary effects on lipoprotein function and macrophage cholesterol metabolism. Arthritis Rheumatol. 2015, 67, 1155–1164. [Google Scholar] [CrossRef]
- Greco, D.; Gualtierotti, R.; Agosti, P.; Adorni, M.P.; Ingegnoli, F.; Rota, M.; Bernini, F.; Meroni, P.L.; Ronda, N. Anti-atherogenic Modification of Serum Lipoprotein Function in Patients with Rheumatoid Arthritis after Tocilizumab Treatment, a Pilot Study. J. Clin. Med. 2020, 9, 2157. [Google Scholar] [CrossRef]
- O’Neill, F.; Charakida, M.; Topham, E.; McLoughlin, E.; Patel, N.; Sutill, E.; Kay, C.W.M.; D’Aiuto, F.; Landmesser, U.; Taylor, P.C.; et al. Anti-inflammatory treatment improves high-density lipoprotein function in rheumatoid arthritis. Heart 2017, 103, 766–773. [Google Scholar] [CrossRef]
- Hunjadi, M.; Lamina, C.; Kahler, P.; Bernscherer, T.; Viikari, J.; Lehtimäki, T.; Kähönen, M.; Hurme, M.; Juonala, M.; Taittonen, L.; et al. HDL cholesterol efflux capacity is inversely associated with subclinical cardiovascular risk markers in young adults: The cardiovascular risk in Young Finns study. Sci. Rep. 2020, 10, 19223. [Google Scholar] [CrossRef]
- Mutharasan, R.K.; Thaxton, C.S.; Berry, J.; Daviglus, M.L.; Yuan, C.; Sun, J.; Ayers, C.; Lloyd-Jones, D.M.; Wilkins, J.T. HDL efflux capacity, HDL particle size, and high-risk carotid atherosclerosis in a cohort of asymptomatic older adults: The Chicago Healthy Aging Study. J. Lipid Res. 2017, 58, 600–606. [Google Scholar] [CrossRef]
- Berrougui, H.; Isabelle, M.; Cloutier, M.; Grenier, G.; Khalil, A. Age-related impairment of HDL-mediated cholesterol efflux. J. Lipid Res. 2007, 48, 328–336. [Google Scholar] [CrossRef]
- Zimetti, F.; Freitas, W.M.; Campos, A.M.; Daher, M.; Adorni, M.P.; Bernini, F.; Sposito, A.C.; Zanotti, I. Cholesterol efflux capacity does not associate with coronary calcium, plaque vulnerability, and telomere length in healthy octogenarians. J. Lipid Res. 2018, 59, 714–721. [Google Scholar] [CrossRef]
- Ritsch, A.; Duerr, A.; Kahler, P.; Hunjadi, M.; Stojakovic, T.; Silbernagel, G.; Scharnagl, H.; Kleber, M.E.; März, W. Cholesterol Efflux Capacity and Cardiovascular Disease: The Ludwigshafen Risk and Cardiovascular Health (LURIC) Study. Biomedicines 2020, 8, 524. [Google Scholar] [CrossRef]
- Guerin, M.; Silvain, J.; Gall, J.; Darabi, M.; Berthet, M.; Frisdal, E.; Hauguel-Moreau, M.; Zeitouni, M.; Kerneis, M.; Lattuca, B.; et al. Association of Serum Cholesterol Efflux Capacity With Mortality in Patients With ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 72, 3259–3269. [Google Scholar] [CrossRef]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef]
- Hernáez, Á.; Castañer, O.; Elosua, R.; Pintó, X.; Estruch, R.; Salas-Salvadó, J.; Corella, D.; Arós, F.; Serra-Majem, L.; Fiol, M.; et al. Mediterranean Diet Improves High-Density Lipoprotein Function in High-Cardiovascular-Risk Individuals: A Randomized Controlled Trial. Circulation 2017, 135, 633–643. [Google Scholar] [CrossRef]
- Liu, X.; Garban, J.; Jones, P.J.; Vanden-Heuvel, J.; Lamarche, B.; Jenkins, D.J.; Connelly, P.W.; Couture, P.; Pu, S.; Fleming, J.A.; et al. Diets Low in Saturated Fat with Different Unsaturated Fatty Acid Profiles Similarly Increase Serum-Mediated Cholesterol Efflux from THP-1 Macrophages in a Population with or at Risk for Metabolic Syndrome: The Canola Oil Multicenter Intervention Trial. J. Nutr. 2018, 148, 721–728. [Google Scholar] [CrossRef]
- Berryman, C.E.; Grieger, J.A.; West, S.G.; Chen, C.-Y.O.; Blumberg, J.B.; Rothblat, G.H.; Sankaranarayanan, S.; Kris-Etherton, P.M. Acute consumption of walnuts and walnut components differentially affect postprandial lipemia, endothelial function, oxidative stress, and cholesterol efflux in humans with mild hypercholesterolemia. J. Nutr. 2013, 143, 788–794. [Google Scholar] [CrossRef]
- Tanaka, N.; Ishida, T.; Nagao, M.; Mori, T.; Monguchi, T.; Sasaki, M.; Mori, K.; Kondo, K.; Nakajima, H.; Honjo, T.; et al. Administration of high dose eicosapentaenoic acid enhances anti-inflammatory properties of high-density lipoprotein in Japanese patients with dyslipidemia. Atherosclerosis 2014, 237, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Brassard, D.; Arsenault, B.J.; Boyer, M.; Bernic, D.; Tessier-Grenier, M.; Talbot, D.; Tremblay, A.; Levy, E.; Asztalos, B.; Jones, P.J.H.; et al. Saturated Fats from Butter but Not from Cheese Increase HDL-Mediated Cholesterol Efflux Capacity from J774 Macrophages in Men and Women with Abdominal Obesity. J. Nutr. 2018, 148, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Tindall, A.M.; Kris-Etherton, P.M.; Petersen, K.S. Replacing Saturated Fats with Unsaturated Fats from Walnuts or Vegetable Oils Lowers Atherogenic Lipoprotein Classes Without Increasing Lipoprotein(a). J. Nutr. 2020, 150, 818–825. [Google Scholar] [CrossRef]
- Manninen, S.; Lankinen, M.; Erkkilä, A.; Nguyen, S.D.; Ruuth, M.; de Mello, V.; Öörni, K.; Schwab, U. The effect of intakes of fish and Camelina sativa oil on atherogenic and anti-atherogenic functions of LDL and HDL particles: A randomized controlled trial. Atherosclerosis 2019, 281, 56–61. [Google Scholar] [CrossRef] [PubMed]
- van der Gaag, M.S.; van Tol, A.; Vermunt, S.H.; Scheek, L.M.; Schaafsma, G.; Hendriks, H.F. Alcohol consumption stimulates early steps in reverse cholesterol transport. J. Lipid Res. 2001, 42, 2077–2083. [Google Scholar] [CrossRef]
- Sierksma, A.; Vermunt, S.H.F.; Lankhuizen, I.M.; van der Gaag, M.S.; Scheek, L.M.; Grobbee, D.E.; van Tol, A.; Hendriks, H.F.J. Effect of moderate alcohol consumption on parameters of reverse cholesterol transport in postmenopausal women. Alcohol. Clin. Exp. Res. 2004, 28, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.J.; Sierksma, A.; van Tol, A.; Fournier, N.; van Gent, T.; Paul, J.-L.; Hendriks, H.F.J. Moderate alcohol consumption increases cholesterol efflux mediated by ABCA1. J. Lipid Res. 2004, 45, 1716–1723. [Google Scholar] [CrossRef]
- Serdyuk, A.P.; Metelskaya, V.A.; Ozerova, I.N.; Kovaltchouk, N.V.; Olferiev, A.M.; Bubnova, M.G.; Perova, N.V.; Jauhiainen, M.; Lasselin, C.; Castro, G. Effects of alcohol on the major steps of reverse cholesterol transport. Biochemistry 2000, 65, 1310–1315. [Google Scholar] [PubMed]
- Marmillot, P.; Munoz, J.; Patel, S.; Garige, M.; Rosse, R.B.; Lakshman, M.R. Long-term ethanol consumption impairs reverse cholesterol transport function of high-density lipoproteins by depleting high-density lipoprotein sphingomyelin both in rats and in humans. Metabolism 2007, 56, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.N.; Liu, Q.H.; Marmillot, P.; Seeff, L.B.; Strader, D.B.; Lakshman, M.R. High-density lipoproteins from human alcoholics exhibit impaired reverse cholesterol transport function. Metabolism 2000, 49, 1406–1410. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; de Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ramie, J.J.; Barber, J.L.; Sarzynski, M.A. Effects of exercise on HDL functionality. Curr. Opin. Lipidol. 2019, 30, 16–23. [Google Scholar] [CrossRef]
- Hernáez, Á.; Soria-Florido, M.T.; Castañer, O.; Pintó, X.; Estruch, R.; Salas-Salvadó, J.; Corella, D.; Alonso-Gómez, Á.; Martínez-González, M.Á.; Schröder, H.; et al. Leisure time physical activity is associated with improved HDL functionality in high cardiovascular risk individuals: A cohort study. Eur. J. Prev. Cardiol. 2020, 2047487320925625. [Google Scholar] [CrossRef]
- Trakaki, A.; Scharnagl, H.; Trieb, M.; Holzer, M.; Hinghofer-Szalkay, H.; Goswami, N.; Marsche, G. Prolonged bedrest reduces plasma high-density lipoprotein levels linked to markedly suppressed cholesterol efflux capacity. Sci. Rep. 2020, 10, 15001. [Google Scholar] [CrossRef]
- Prochaska, J.J.; Benowitz, N.L. Smoking cessation and the cardiovascular patient. Curr. Opin. Cardiol. 2015, 30, 506–511. [Google Scholar] [CrossRef]
- Millar, C.L.; Duclos, Q.; Blesso, C.N. Effects of dietary flavonoids on reverse cholesterol transport, HDL metabolism, and HDL function. Adv. Nutr. 2017, 8, 226–239. [Google Scholar] [CrossRef]
- Zhu, Y.; Huang, X.; Zhang, Y.; Wang, Y.; Liu, Y.; Sun, R.; Xia, M. Anthocyanin supplementation improves HDL-Associated paraoxonase 1 activity and enhances cholesterol efflux capacity in subjects with hypercholesterolemia. J. Clin. Endocrinol. Metab. 2014, 99, 561–569. [Google Scholar] [CrossRef]
- Favari, E.; Angelino, D.; Cipollari, E.; Adorni, M.P.; Zimetti, F.; Bernini, F.; Ronda, N.; Pellegrini, N. Functional pasta consumption in healthy volunteers modulates ABCG1-mediated cholesterol efflux capacity of HDL. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Greco, D.; Kocyigit, D.; Adorni, M.P.; Marchi, C.; Ronda, N.; Bernini, F.; Gurses, K.M.; Canpinar, H.; Guc, D.; Oguz, S.H.; et al. Vitamin D replacement ameliorates serum lipoprotein functions, adipokine profile and subclinical atherosclerosis in pre-menopausal women. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Adorni, M.P.; Ferri, N.; Marchianò, S.; Trimarco, V.; Rozza, F.; Izzo, R.; Bernini, F.; Zimetti, F. Effect of a novel nutraceutical combination on serum lipoprotein functional profile and circulating PCSK9. Ther. Clin. Risk Manag. 2017, 13, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- van Capelleveen, J.C.; Kastelein, J.J.P.; Zwinderman, A.H.; van Deventer, S.J.H.; Collins, H.L.; Adelman, S.J.; Round, P.; Ford, J.; Rader, D.J.; Hovingh, G.K. Effects of the cholesteryl ester transfer protein inhibitor, TA-8995, on cholesterol efflux capacity and high-density lipoprotein particle subclasses. J. Clin. Lipidol. 2016, 10, 1137–1144. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ruotolo, G.; Brewer, H.B.; Kane, J.P.; Wang, M.-D.; Krueger, K.A.; Adelman, S.J.; Nissen, S.E.; Rader, D.J. Cholesterol Efflux Capacity and Pre-Beta-1 HDL Concentrations Are Increased in Dyslipidemic Patients Treated With Evacetrapib. J. Am. Coll. Cardiol. 2015, 66, 2201–2210. [Google Scholar] [CrossRef]
- Gomaraschi, M.; Adorni, M.P.; Banach, M.; Bernini, F.; Franceschini, G.; Calabresi, L. Effects of established hypolipidemic drugs on HDL concentration, subclass distribution, and function. Handb. Exp. Pharmacol. 2015, 224, 593–615. [Google Scholar]
- Gordon, S.M.; Amar, M.J.; Jeiran, K.; Stagliano, M.; Staller, E.; Playford, M.P.; Mehta, N.N.; Vaisar, T.; Remaley, A.T. Effect of niacin monotherapy on high density lipoprotein composition and function. Lipids Health Dis. 2020, 19, 190. [Google Scholar] [CrossRef]
- Adorni, M.P.; Zimetti, F.; Puntoni, M.; Bigazzi, F.; Sbrana, F.; Minichilli, F.; Bernini, F.; Ronda, N.; Favari, E.; Sampietro, T. Cellular cholesterol efflux and cholesterol loading capacity of serum: Effects of LDL-apheresis. J. Lipid Res. 2012, 53, 984–989. [Google Scholar] [CrossRef]
- Nenseter, M.S.; Narverud, I.; Græsdal, A.; Bogsrud, M.P.; Aukrust, P.; Retterstøl, K.; Ose, L.; Halvorsen, B.; Holven, K.B. Cholesterol efflux mediators in homozygous familial hypercholesterolemia patients on low-density lipoprotein apheresis. J. Clin. Lipidol. 2013, 7, 109–116. [Google Scholar] [CrossRef]
- Orsoni, A.; Villard, E.F.; Bruckert, E.; Robillard, P.; Carrie, A.; Bonnefont-Rousselot, D.; Chapman, M.J.; Dallinga-Thie, G.M.; le Goff, W.; Guerin, M. Impact of LDL apheresis on atheroprotective reverse cholesterol transport pathway in familial hypercholesterolemia. J. Lipid Res. 2012, 53, 767–775. [Google Scholar] [CrossRef]
- Gou, S.; Wang, L.; Zhong, C.; Chen, X.; Ouyang, X.; Li, B.; Bao, G.; Liu, H.; Zhang, Y.; Ni, J. A novel apoA-I mimetic peptide suppresses atherosclerosis by promoting physiological HDL function in apoE−/− mice. Br. J. Pharmacol. 2020, 177, 4627–4644. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Puri, R.; Ballantyne, C.M.; Jukema, J.W.; Kastelein, J.J.P.; Koenig, W.; Wright, R.S.; Kallend, D.; Wijngaard, P.; Borgman, M.; et al. Effect of infusion of high-density lipoprotein mimetic containing recombinant apolipoprotein A-I Milano on coronary disease in patients with an acute coronary syndrome in the MILANO-PILOT trial: A randomized clinical trial. JAMA Cardiol. 2018, 3, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.H.; Kaiser, Y.; van Olden, C.C.; Santos, R.D.; Dasseux, J.L.; Genest, J.; Gaudet, D.; Westerink, J.; Keyserling, C.; Verberne, H.J.; et al. No benefit of HDL mimetic CER-001 on carotid atherosclerosis in patients with genetically determined very low HDL levels. Atherosclerosis 2020, 311, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Gille, A.; Easton, R.; D’Andrea, D.; Wright, S.D.; Shear, C.L. CSL112 enhances biomarkers of reverse cholesterol transport after single and multiple infusions in healthy subjects. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2106–2114. [Google Scholar] [CrossRef]
- Tricoci, P.; D’Andrea, D.M.; Gurbel, P.A.; Yao, Z.; Cuchel, M.; Winston, B.; Schott, R.; Weiss, R.; Blazing, M.A.; Cannon, L.; et al. Infusion of Reconstituted High-Density Lipoprotein, CSL112, in Patients With Atherosclerosis: Safety and Pharmacokinetic Results From a Phase 2a Randomized Clinical Trial. J. Am. Heart Assoc. 2015, 4, e002171. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.M.; Korjian, S.; Tricoci, P.; Daaboul, Y.; Yee, M.; Jain, P.; Alexander, J.H.; Steg, P.G.; Lincoff, A.M.; Kastelein, J.J.P.; et al. Safety and Tolerability of CSL112, a Reconstituted, Infusible, Plasma-Derived Apolipoprotein A-I, after Acute Myocardial Infarction: The AEGIS-I Trial (ApoA-I Event Reducing in Ischemic Syndromes I). Circulation 2016, 134, 1918–1930. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Cross-Sectional Studies | |||
|---|---|---|---|
| Study | Study Population | Main Findings | OD/HR/r and p Value |
| Khera A.V. et al., 2011 [37] | 203 healthy volunteers; 442 patients with and 351 without angiographically confirmed CAD | HDL CEC was a strong inverse predictor of CAD also after adjustment for HDL-C 1or apoA-I levels 2 | 1 OR: 0.75; p = 0.002 2 OR: 0.75; p = 0.002 |
| Ishikawa T. et al., 2015 [60] | 182 patients with and 72 without CAD; | HDL CEC, but not HDL-C or apoA-I levels, was a significant predictor of CAD. | OR: 0.23; p = 0.037 |
| Ogura M. et al., 2016 [61] | 227 HeFH patients of which 76 had ASCVD | Increased HDL CEC was associated with decreased risk of ASCVD even after the addition of HDL-C level as a covariate and after adjustment for age, sex and traditional CV risk factors | OR: 0.95; p < 0.05 |
| Thakkar H. et al., 2020 [64] | 150 ACS patients; 110 controls | HDL CEC was associated with a higher OR of ACS even after adjustment for confounding factors. | OR: 0.49; p = 0.006 |
| Favari E. et al., 2013 [65] | 167 healthy subjects | ABCA1-dependent CEC was inversely correlated with PWV | r = −0.183; p = 0.018 |
| Vigna G. B. et al., 2014 [66] | 20 subjects with HAL; 20 controls | ABCG1-CEC was directly correlated with the FMD | r = 0.377; p < 0.05 |
| Josefs T. et al., 2020 [71] | 574 subjects from CODAM cohort with T2DM and CVD | HDL CEC was not associated with either markers of atherosclerosis cIMT and EnD, nor with CVD or CVE | p = 0.332 |
| Li X.M. et al., 2013 [70] | Cohort A: Stable Angiographic Case—Control Cohort (n = 1150); Cohort B: Outpatient Case—Control Cohort (n = 577) | Higher CEC was paradoxically associated with increased risk of myocardial infarction/stroke1 and major adverse CVE2 | 1 HR: 2.19 2 HR: 1.85 |
| Longitudinal Studies | |||
|---|---|---|---|
| Study | Study Population | Main Findings | OD/HR/r and p Value |
| Rohatgi A. et al., 2014 [58] | 2924 subjects free from CVD | HDL CEC was inversely associated with the incidence of CV events after adjustment for traditional risk factors. | OR: 0.33 |
| Saleheen D. et al., 2015 [59] | 1745 initially healthy subjects who later developed fatal or non-fatal CHD; 1749 controls | HDL CEC was inversely associated with incidence of CHD events after the adjustment for CV risk factors and HDL-C. | OR: 0.64 |
| Patel P.J. et al., 2013 [62] | 23 subjects with CAD and EF < 50%; 46 control subjects without CAD and EF > 55% | Low HDL CEC was a significant risk factors for HF | OR: 2.1 p = 0.03 |
| Khera A.V. et al., 2017 [72] | 314 subjects with CVD; 314 controls | On-statin HDL CEC was inversely associated with the incidence of CVD 1, although HDL particle number emerged as the strongest predictor 2. | 1 OR: 0.62 p = 0.02 2 OR:0.51 p < 0.001 |
| Ritsch A. et al., 2015 [73] | 2450 healthy subjects undergoing coronary angiography | Inverse correlation between CEC and CV mortality in a fully adjusted model that included traditional CV risk factor. | HR for Q4 vs. Q1: 0.64 |
| Chindhy et al., 2018 [74] | 2895 subjects without baseline CVD; 210 of them with baseline CKD | No significant interaction between CEC and CKD on associations with ASCVD 1 and total CVD 2. | 1 HR: 1.30 p = 0.01 2 HR: 1.15 p = 0.05 |
| Javaheri et al., 2016 [75] | 35 patients with CAV 1 year after heart transplantation | Reduced CEC was independently associated with disease progression and mortality in CAV patients. | OR: 0.35 p = 0.048 |
| Kopecki et al., 2017 [76] | 1147 patients with T2DM and undergoing hemodialysis | No association between CEC and CVD mortality 1, cardiac events 2, and all-cause mortality 3. | 1 HR: 0.96 p = 0.42 2 HR: 0.92 p = 0.11 3 HR: 0.96 p = 0.39 |
| Liu et al., 2016 [77] | 1737 patients with CAD | CEC was an independent factor to predict all-cause 1 and CV mortality 2 in patients with CAD | Q4 vs. Q1 1 HR: 0.24 p = 0.001 2 HR: 0.17 p = 0.001 |
| Mody et al., 2016 [78] | 1972 patients with/without CAD | CEC was inversely associated with ASCVD among subjects with CAC 1, FH 2 and elevated hsCRP 3. | 1 HR: 0.40 2 HR: 0.31 3 HR: 0.37 |
| Soria-Florido et al., 2020 [79] | 167 patients with ACS; 334 controls | CEC was inversely associated with ACS incidence 1 and MI 2. | 1 OR: 0.58 2 OR: 0.33 |
| Ebtehaj S. et al., 2019 [71] | 351 subjects with CVD developed during follow-up; 354 controls | CEC was significantly associated with the future development of CVD events independently of HDL-C and ApoA-I plasma levels | OR: 0.73 p < 0.001 |
| Riggs K.A. et al., 2019 [80] | 2643 health subjects with < 65 years | GlycA was directly associated with HDL-C and ApoA-I, while it was inversely correlated with CEC | HR for Q4 vs. Q1: 3.00 |
| Annema et al., 2016 [82] | 495 patients that underwent renal transplantation | CEC was not associated with future CV mortality 1 or all-cause mortality 2, while it was found to predict graft failure 3. | 1 HR: 1.014 p = 0.92 2 HR: 0.908 p = 0.35 3 HR: 0.428 p = 0.001 |
| Shea S. et al., 2019 [83] | 465 cases with incident CV events; 465 controls | CEC was significantly associated with lower odds of CVD 1, higher CEC was associated with lower risk of incident CHD 2. | 1 OR = 0.82 p = 0.031 2 OR = 0.72 p = 0.007 |
| Garg p.K. et al., 2020 [84] | 1458 patients that developed incident clinical or subclinical PAD during 6 years of follow-up | High CEC was not significantly associated with incident clinical PAD 1, or subclinical PAD 2 | 1 HR: 1.25 2 HR: 1.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adorni, M.P.; Ronda, N.; Bernini, F.; Zimetti, F. High Density Lipoprotein Cholesterol Efflux Capacity and Atherosclerosis in Cardiovascular Disease: Pathophysiological Aspects and Pharmacological Perspectives. Cells 2021, 10, 574. https://doi.org/10.3390/cells10030574
Adorni MP, Ronda N, Bernini F, Zimetti F. High Density Lipoprotein Cholesterol Efflux Capacity and Atherosclerosis in Cardiovascular Disease: Pathophysiological Aspects and Pharmacological Perspectives. Cells. 2021; 10(3):574. https://doi.org/10.3390/cells10030574
Chicago/Turabian StyleAdorni, Maria Pia, Nicoletta Ronda, Franco Bernini, and Francesca Zimetti. 2021. "High Density Lipoprotein Cholesterol Efflux Capacity and Atherosclerosis in Cardiovascular Disease: Pathophysiological Aspects and Pharmacological Perspectives" Cells 10, no. 3: 574. https://doi.org/10.3390/cells10030574
APA StyleAdorni, M. P., Ronda, N., Bernini, F., & Zimetti, F. (2021). High Density Lipoprotein Cholesterol Efflux Capacity and Atherosclerosis in Cardiovascular Disease: Pathophysiological Aspects and Pharmacological Perspectives. Cells, 10(3), 574. https://doi.org/10.3390/cells10030574