
Synthesis and Biological Evaluation of 1-(Diarylmethyl)-1H-1,2,4-triazoles and 1-(Diarylmethyl)-1H-imidazoles as a Novel Class of Anti-Mitotic Agent for Activity in Breast Cancer

 ,
,  and
and 
Abstract
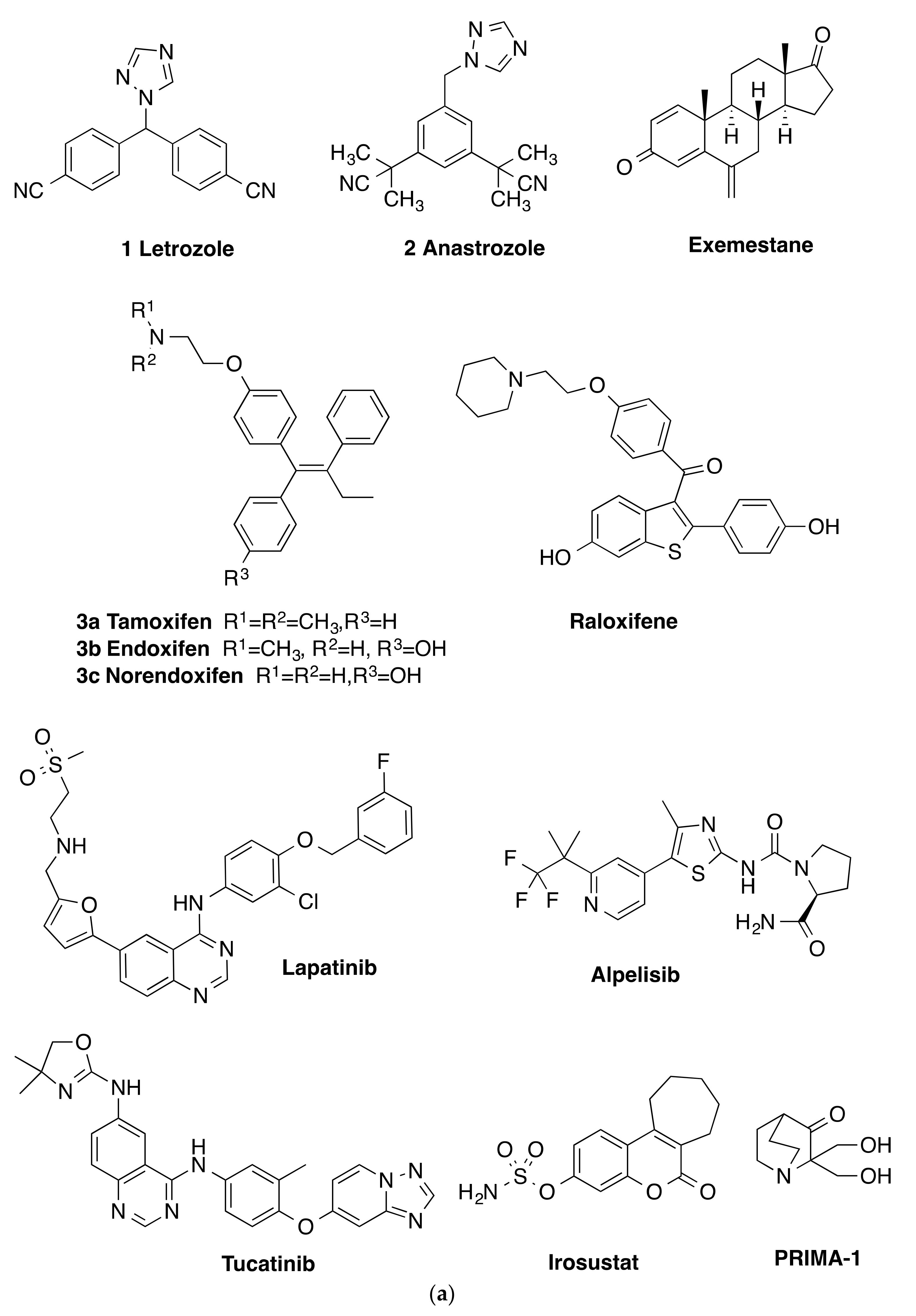
:1. Introduction
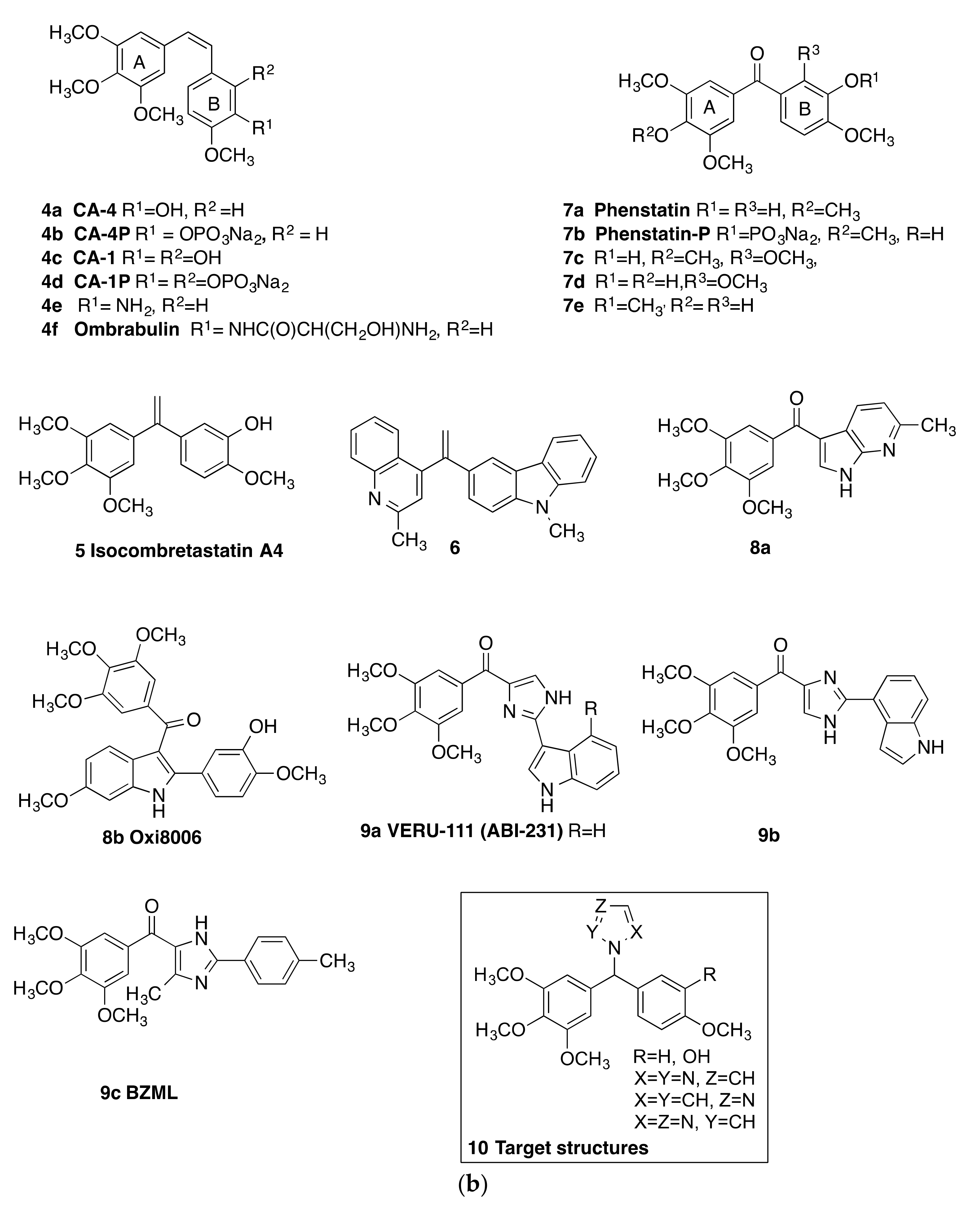
2. Results and Discussion
2.1. Chemistry
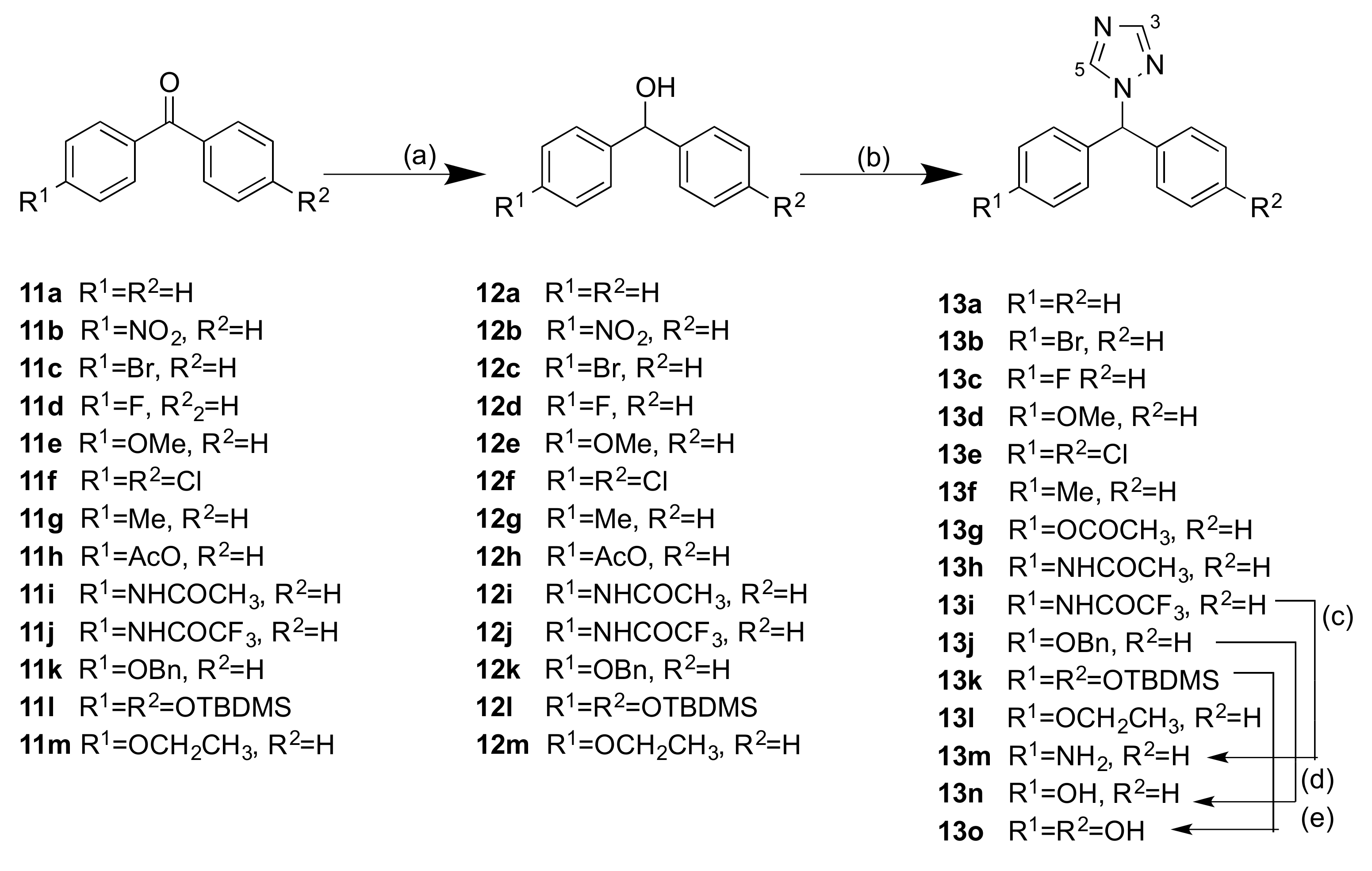
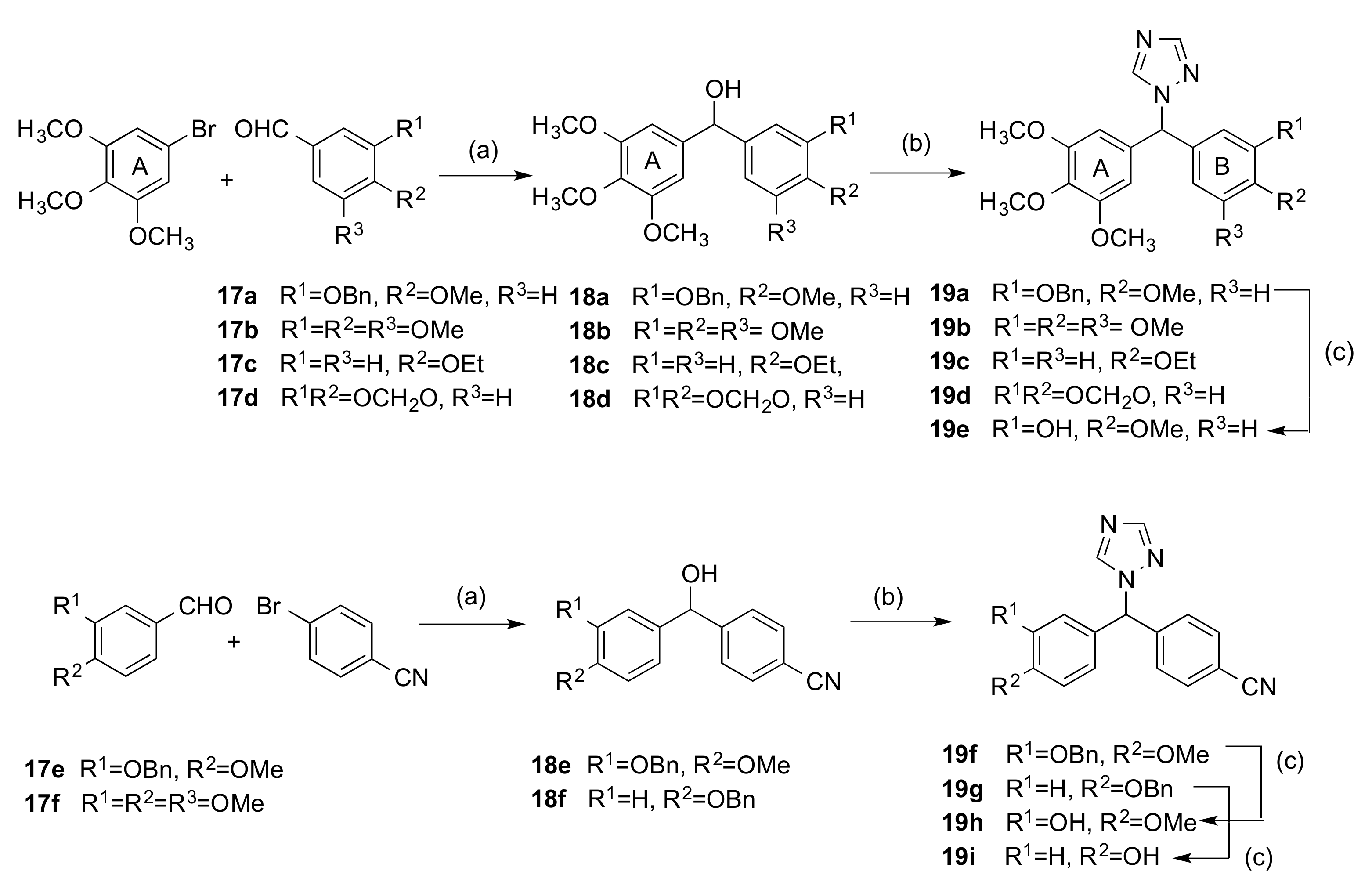
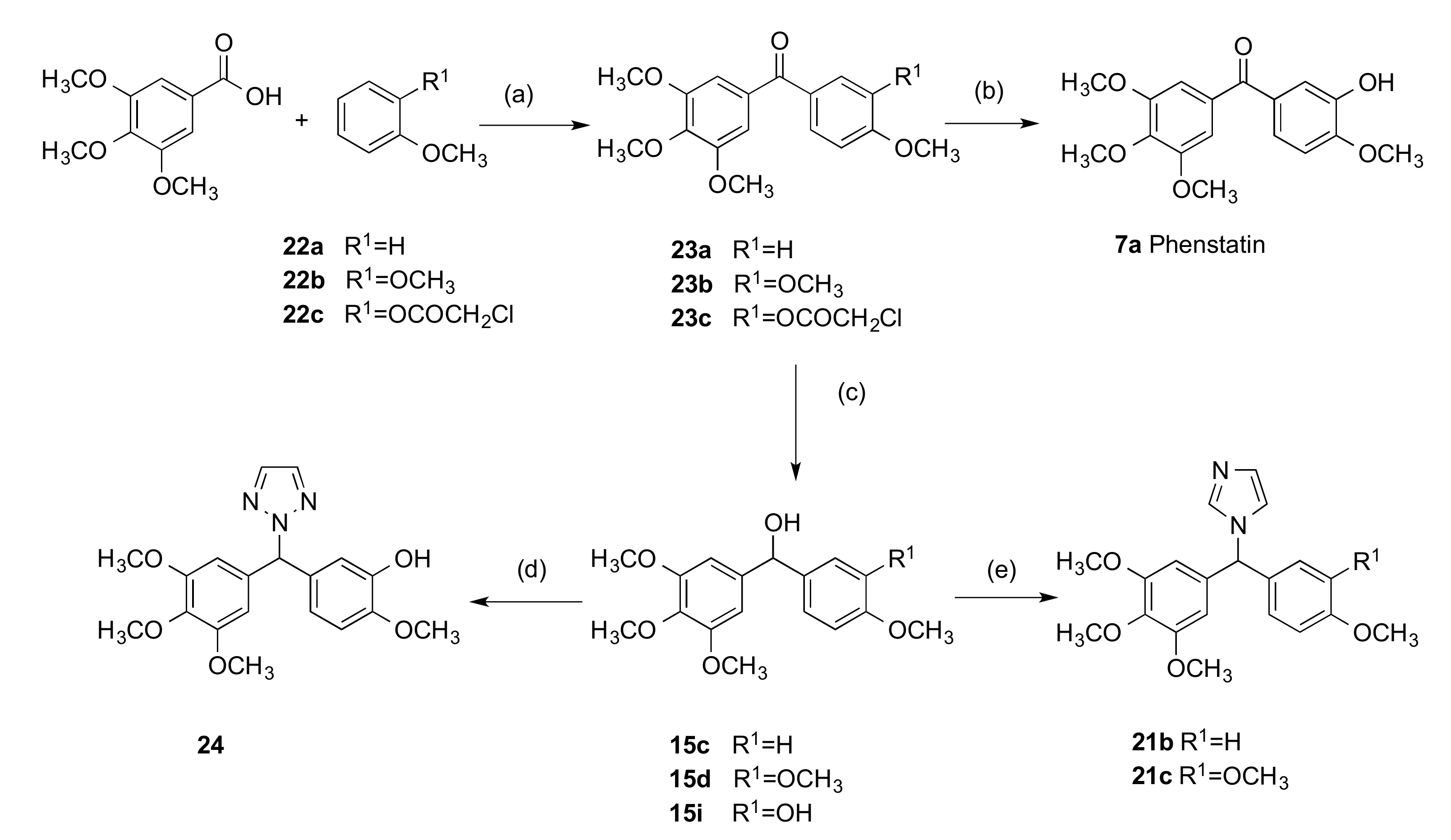
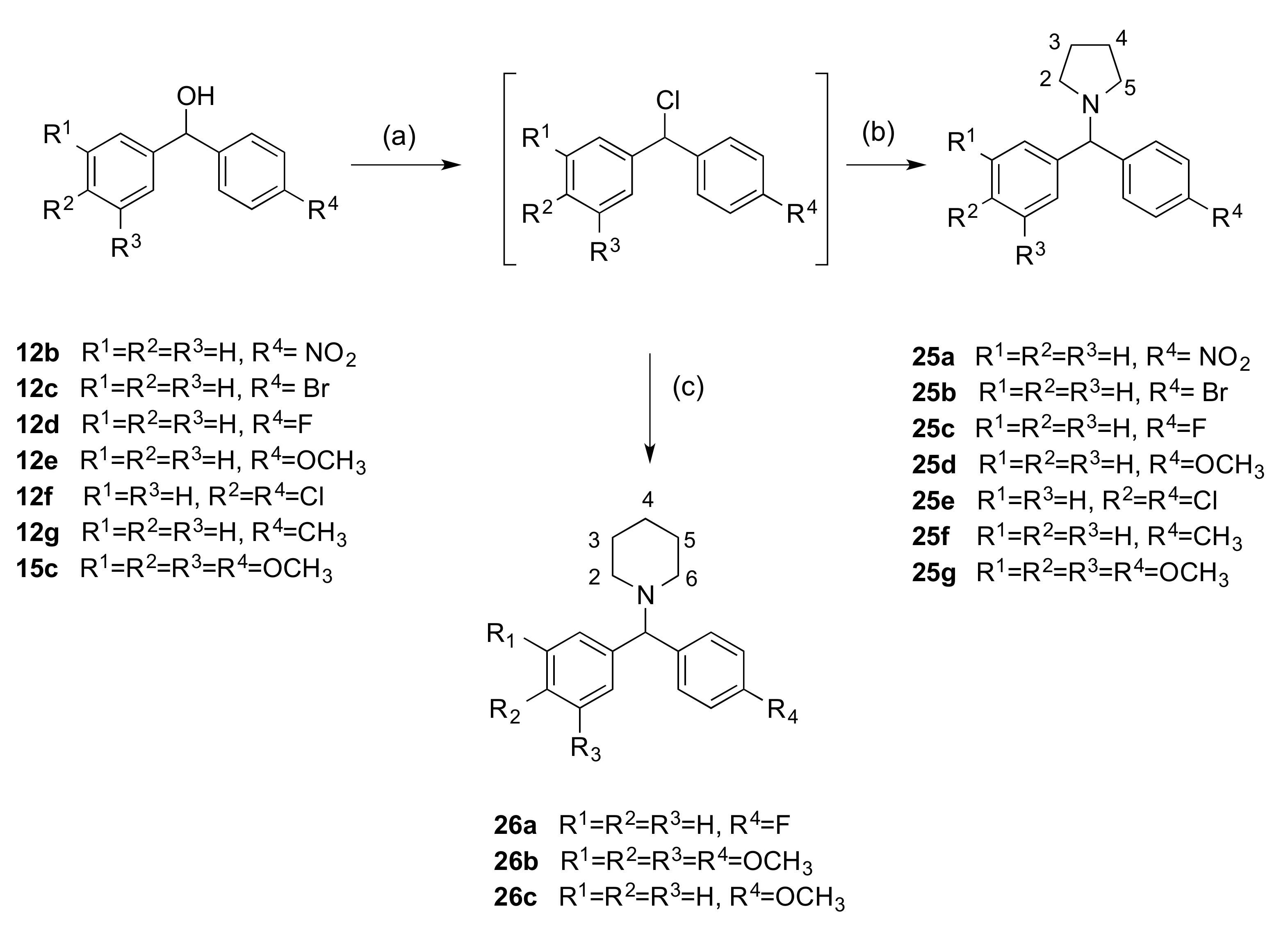
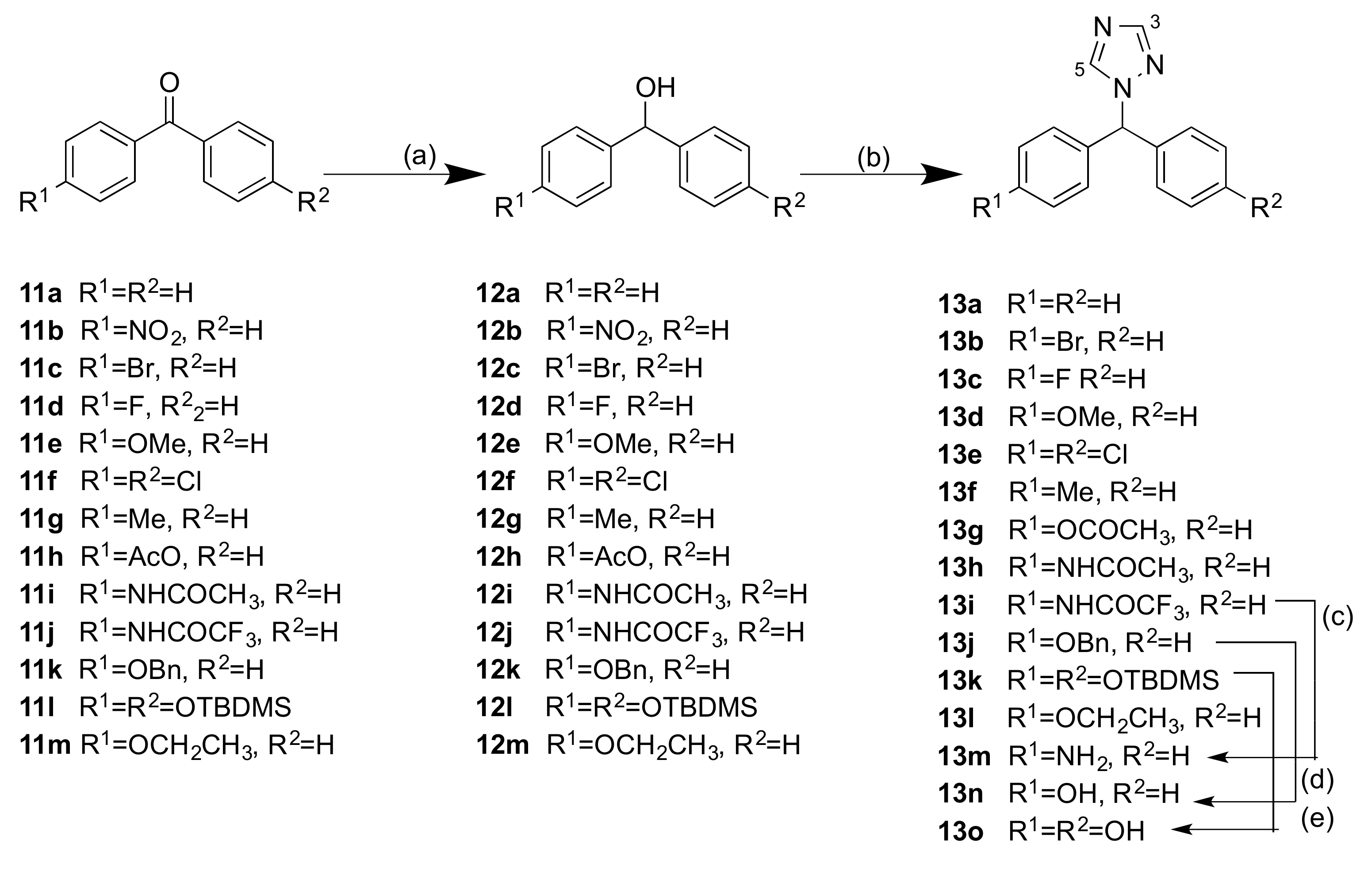
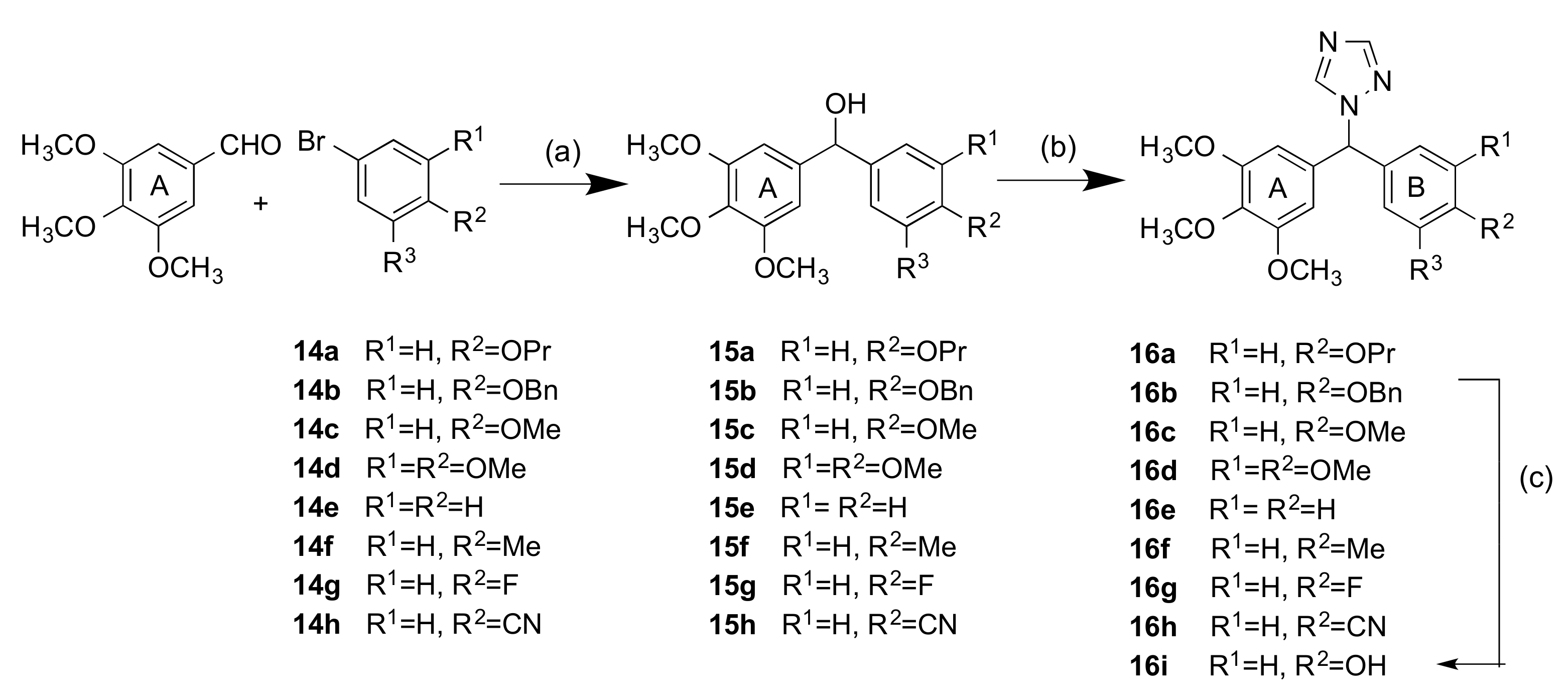
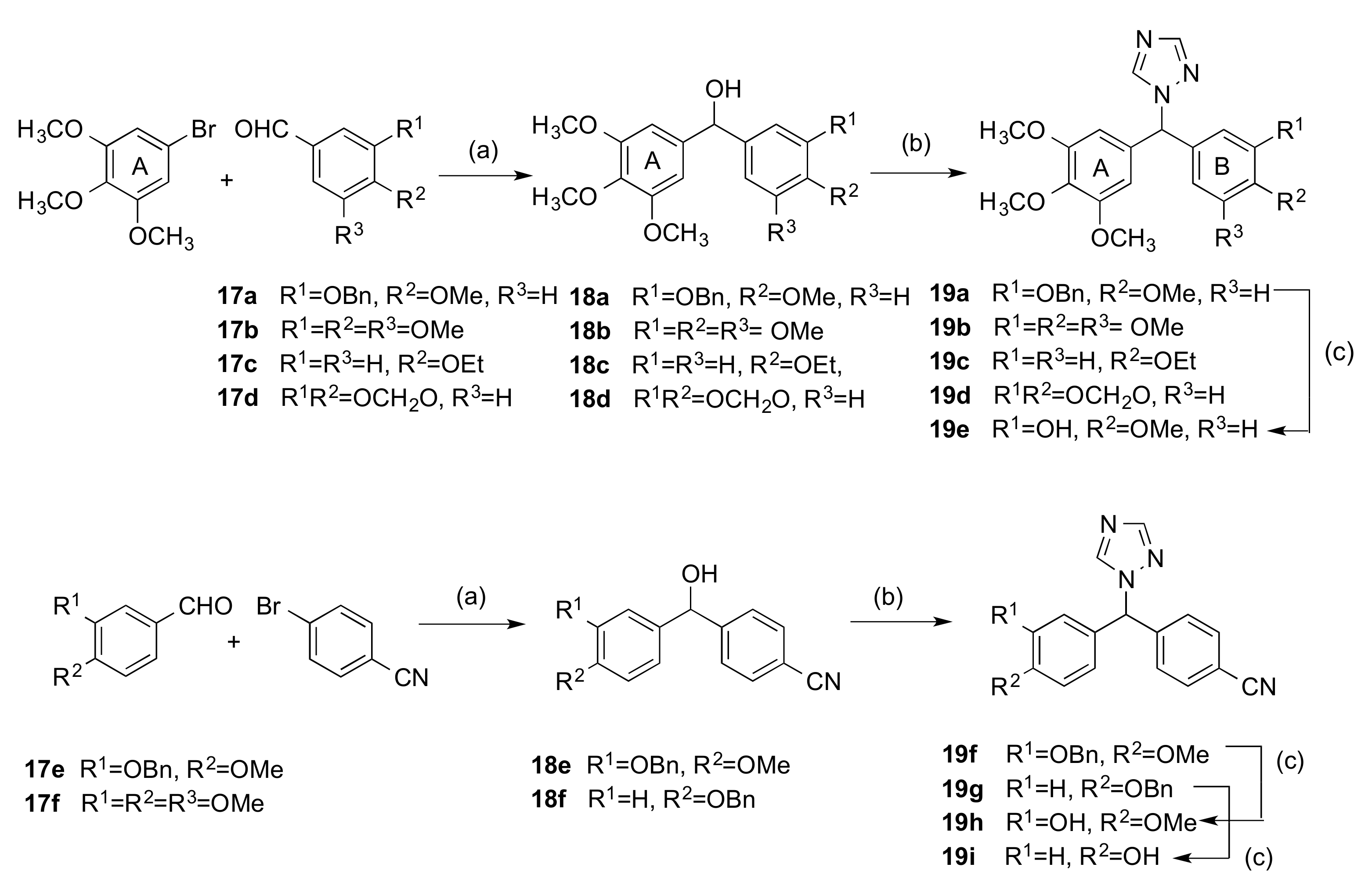
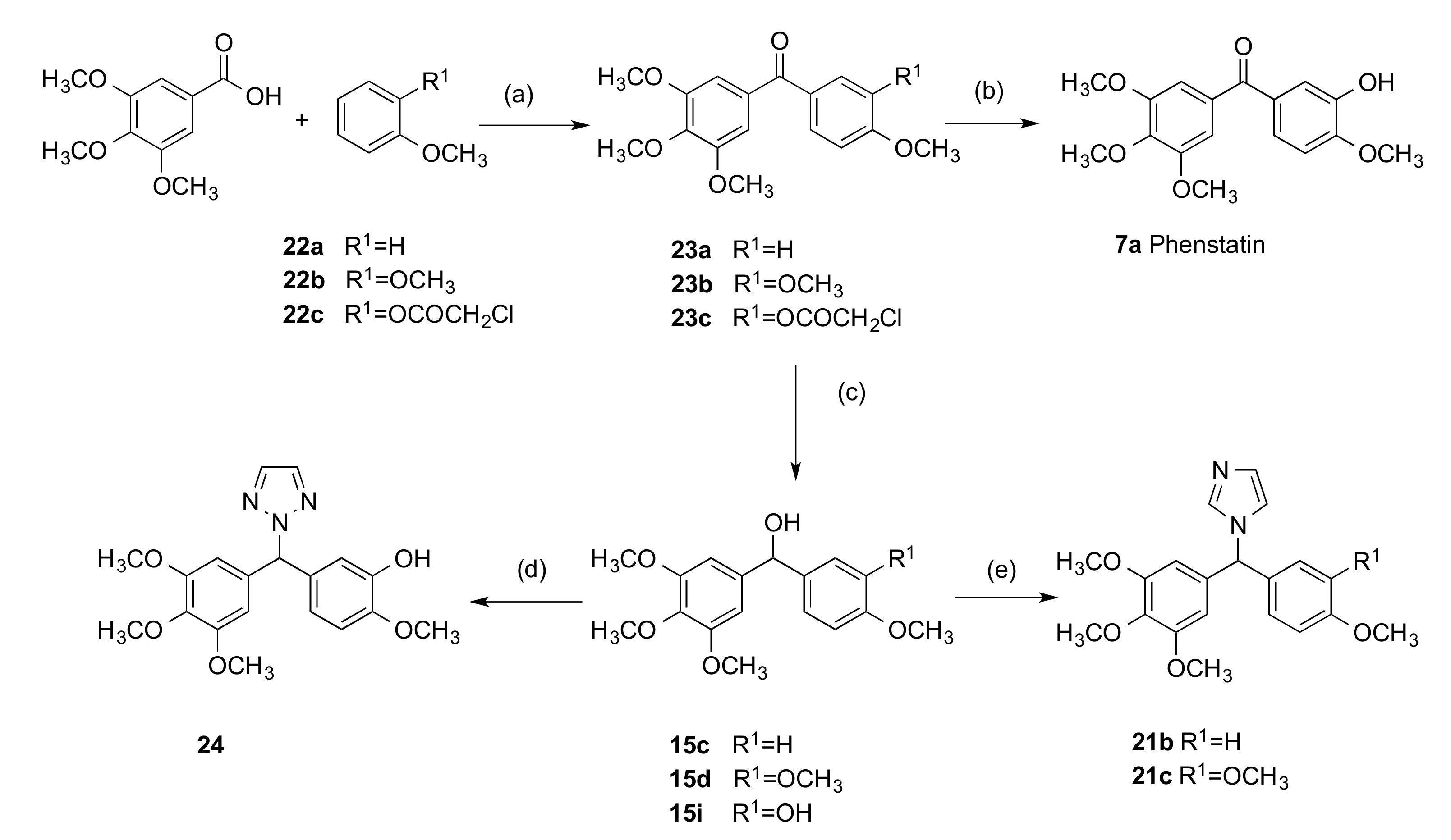
2.1.1. 1-(Diarylmethyl)-1H-1,2,4-triazoles (Series 1 and 2)
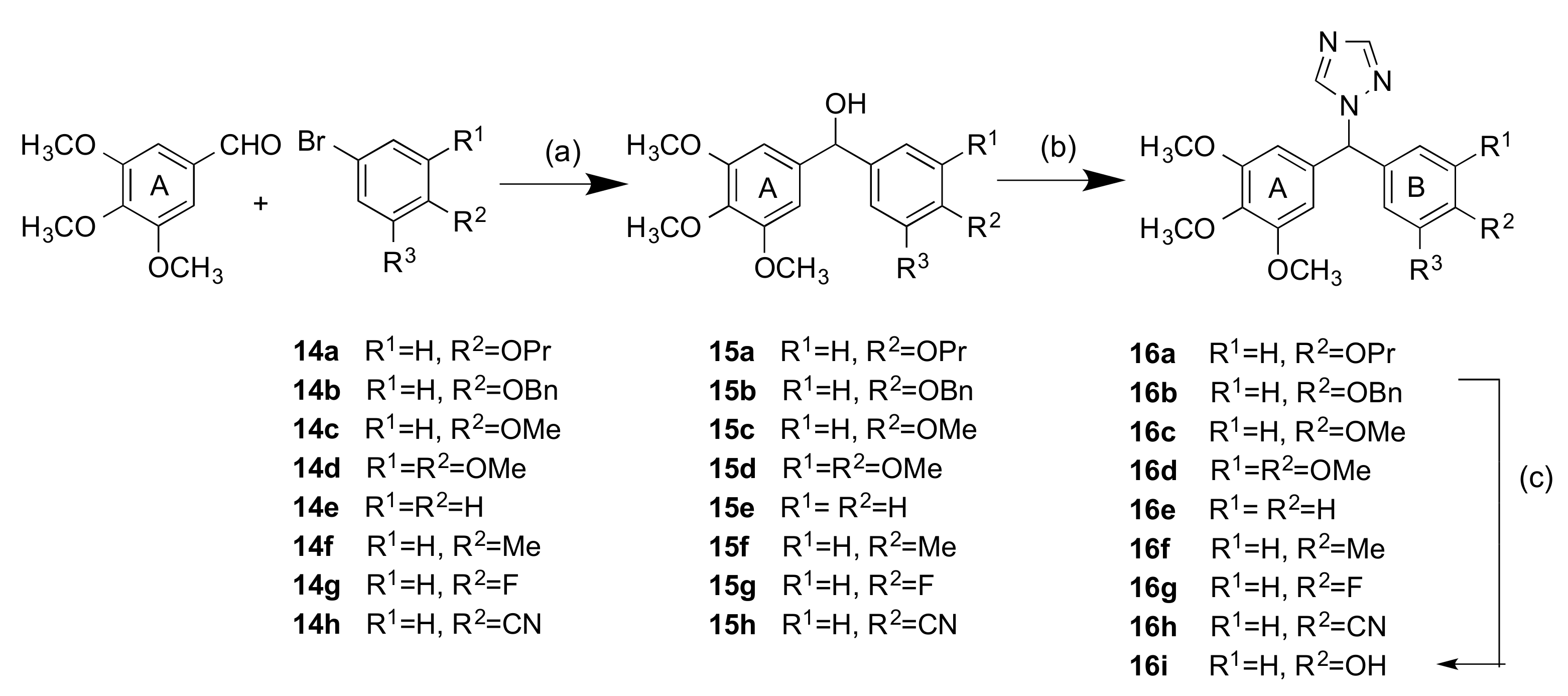
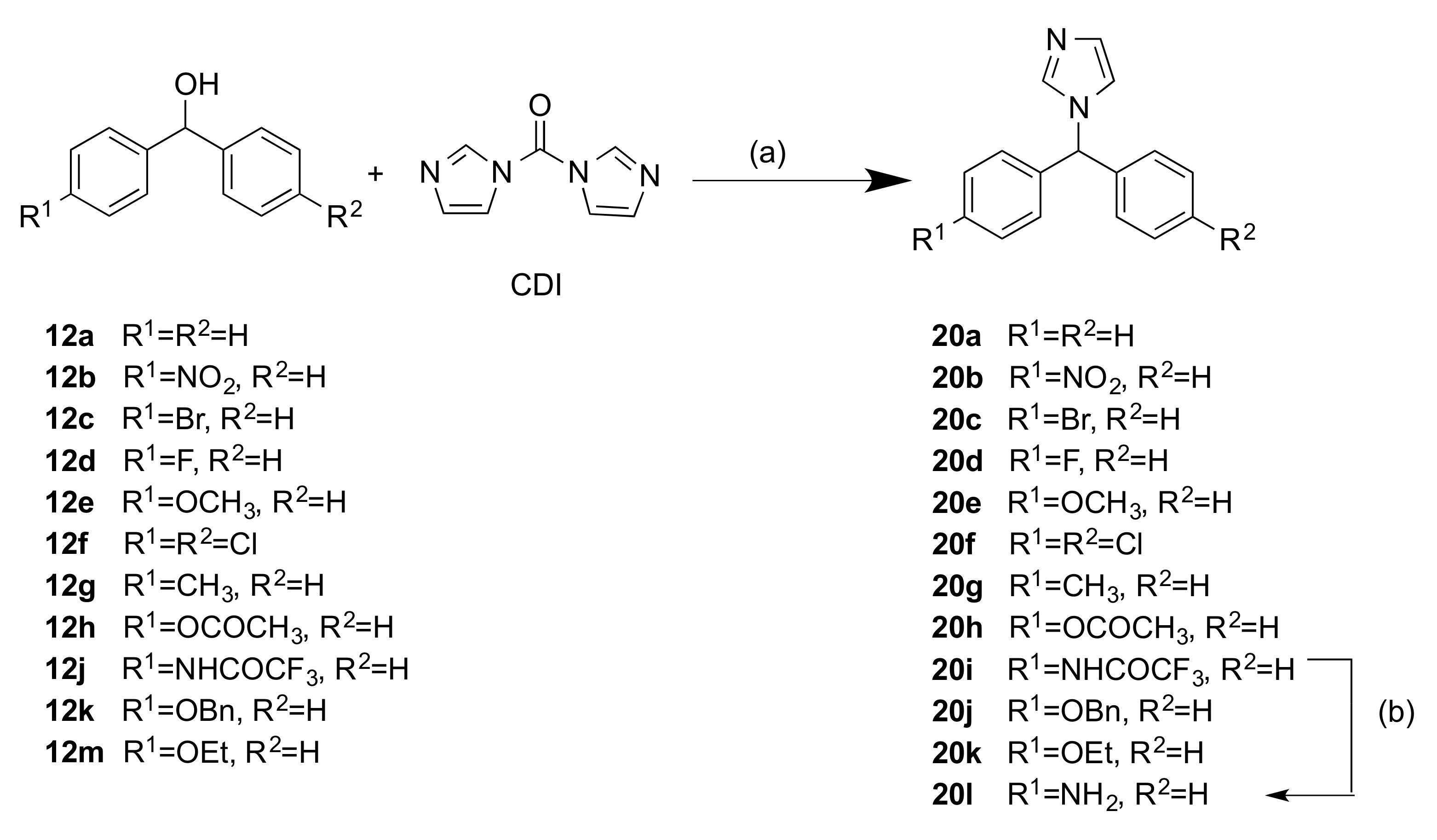
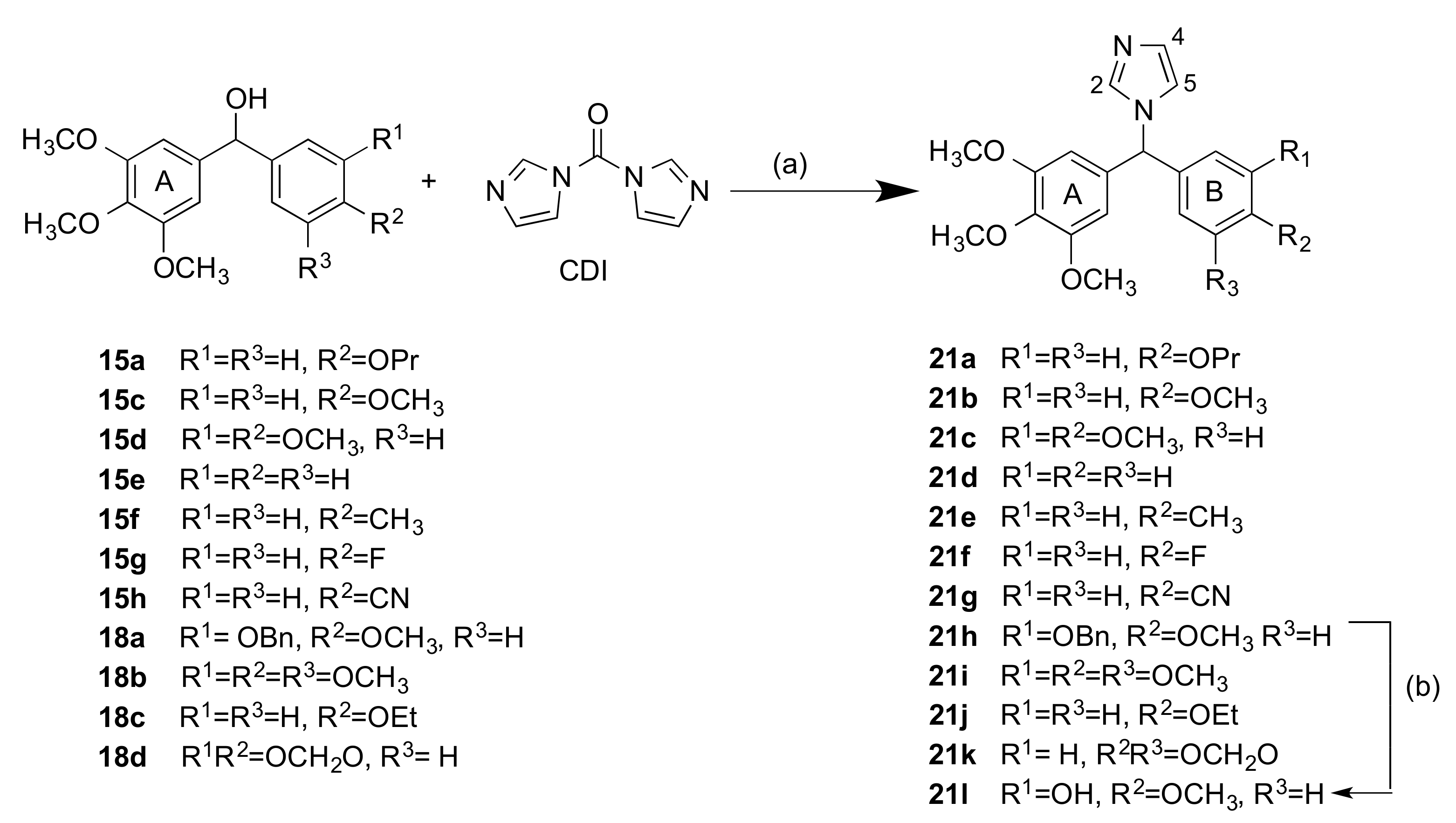
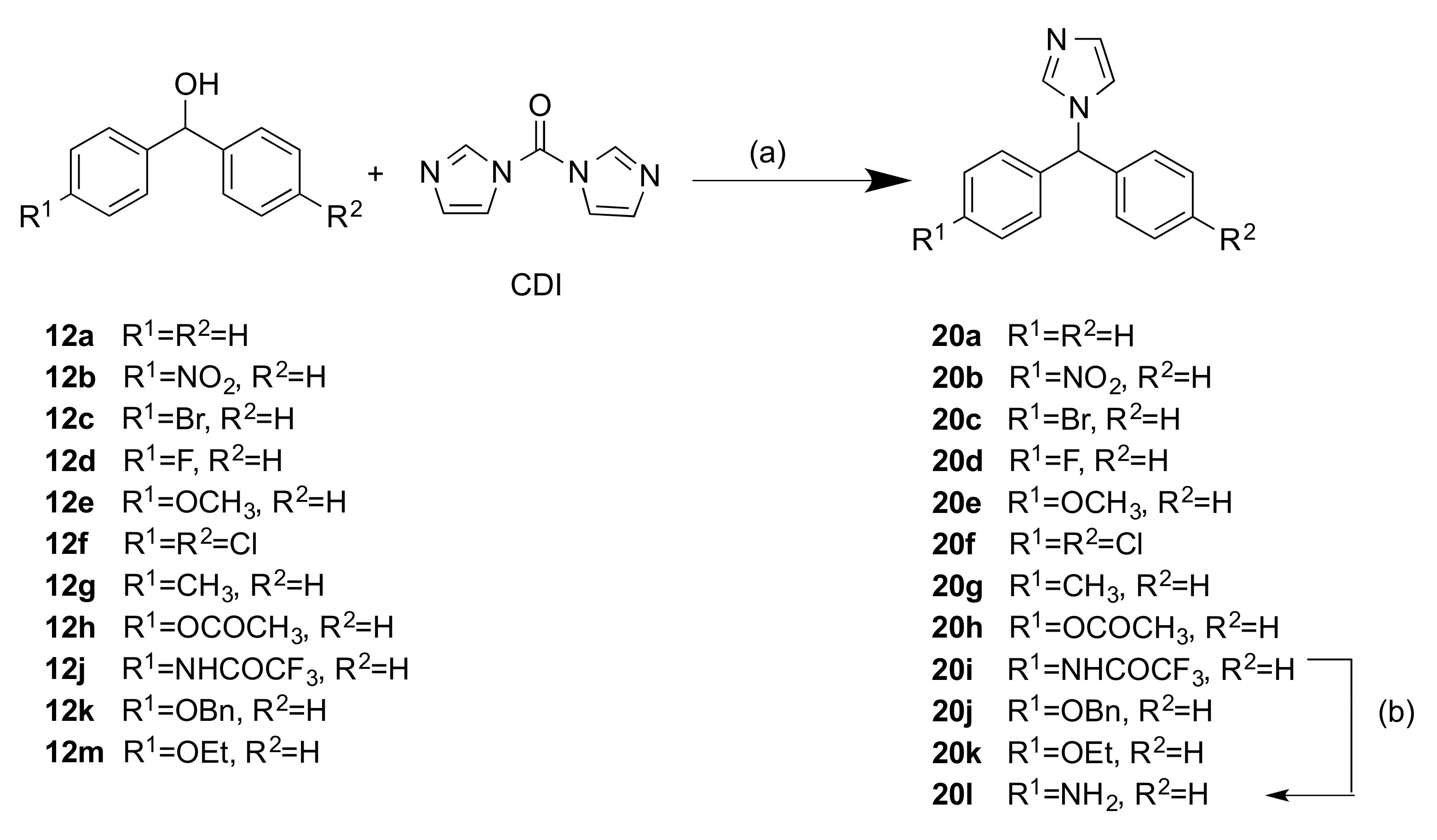
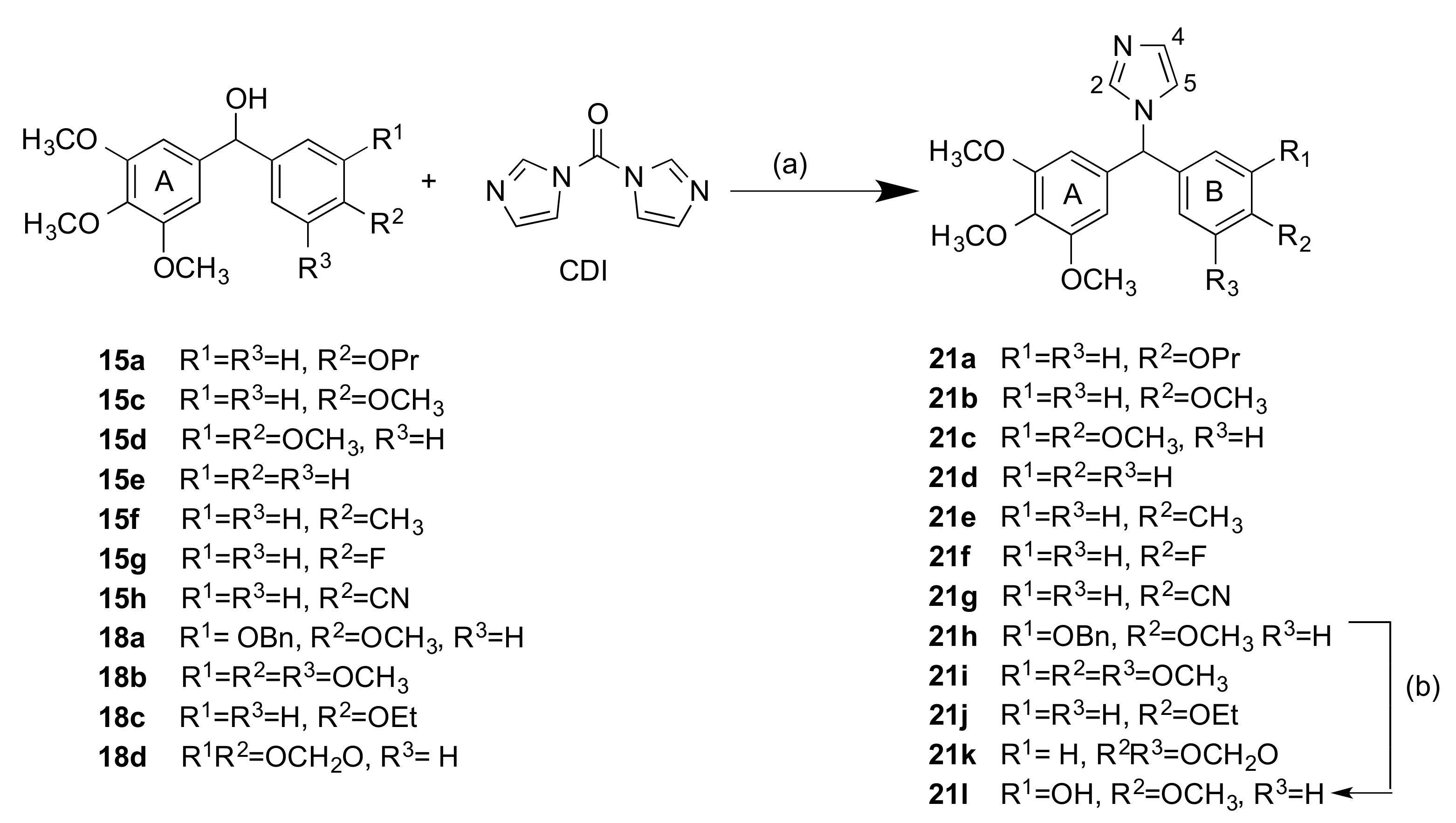
2.1.2. 1-(Diarylmethyl)-1H-Imidazoles (Series 3 and 4)
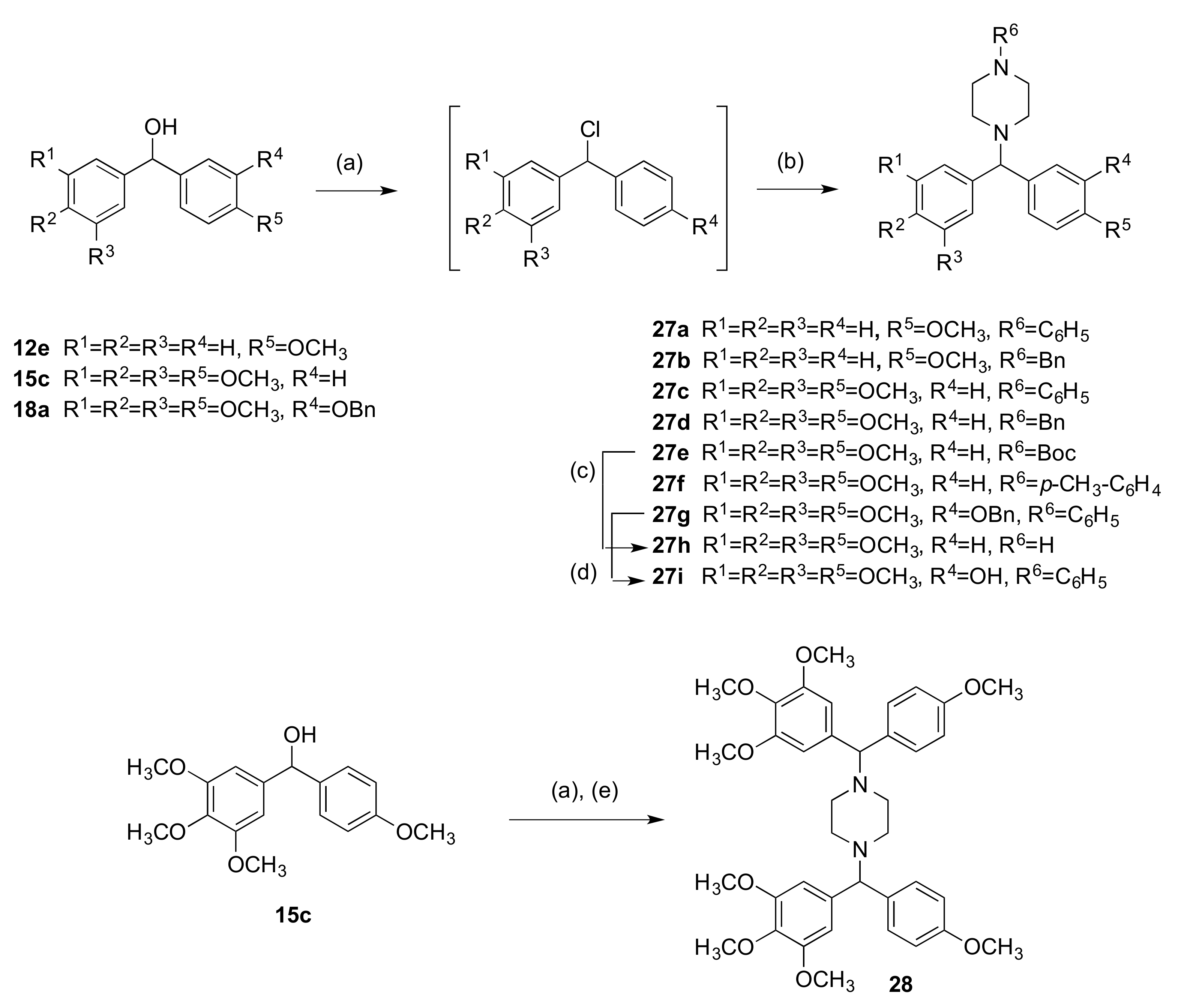
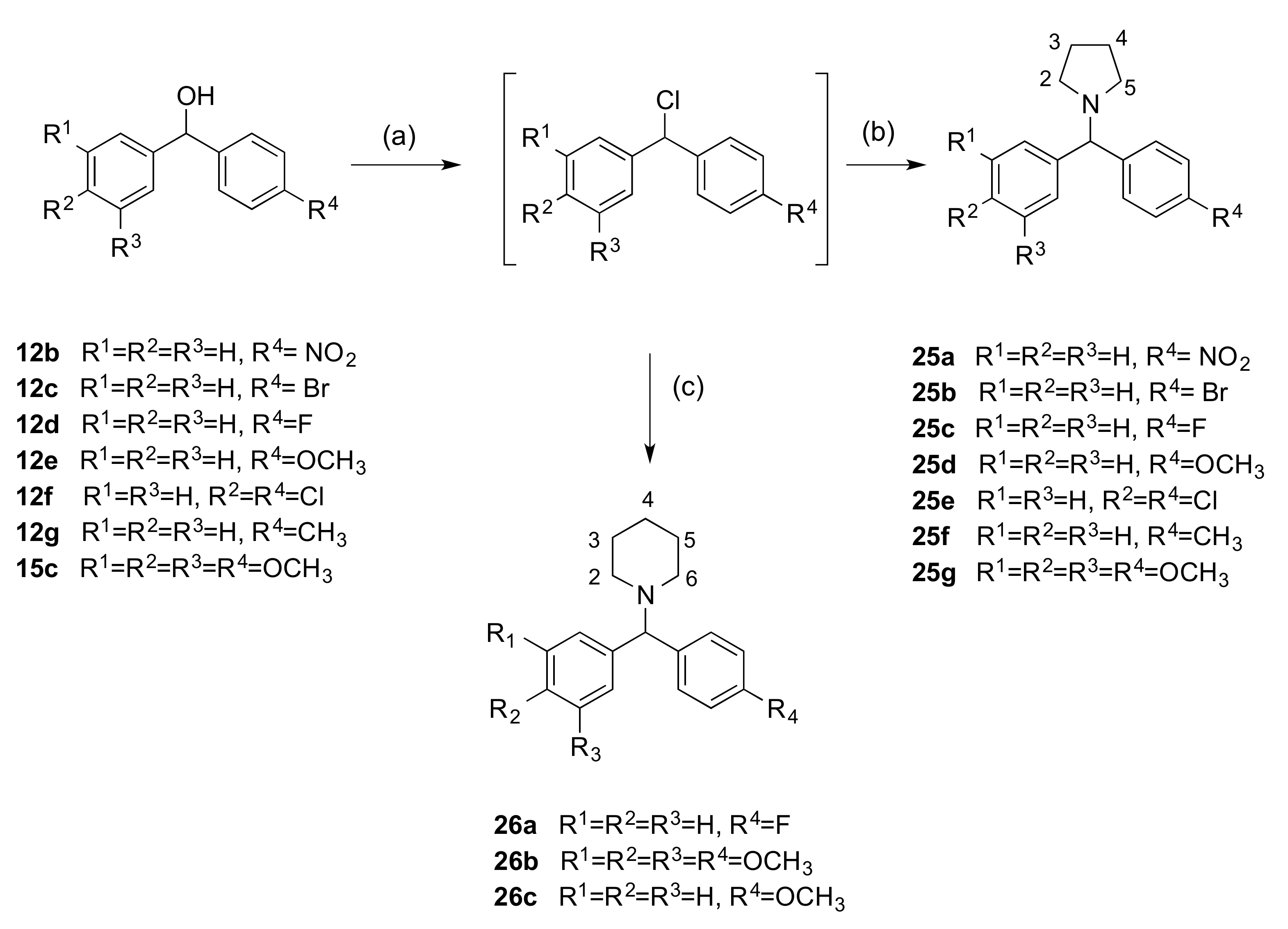
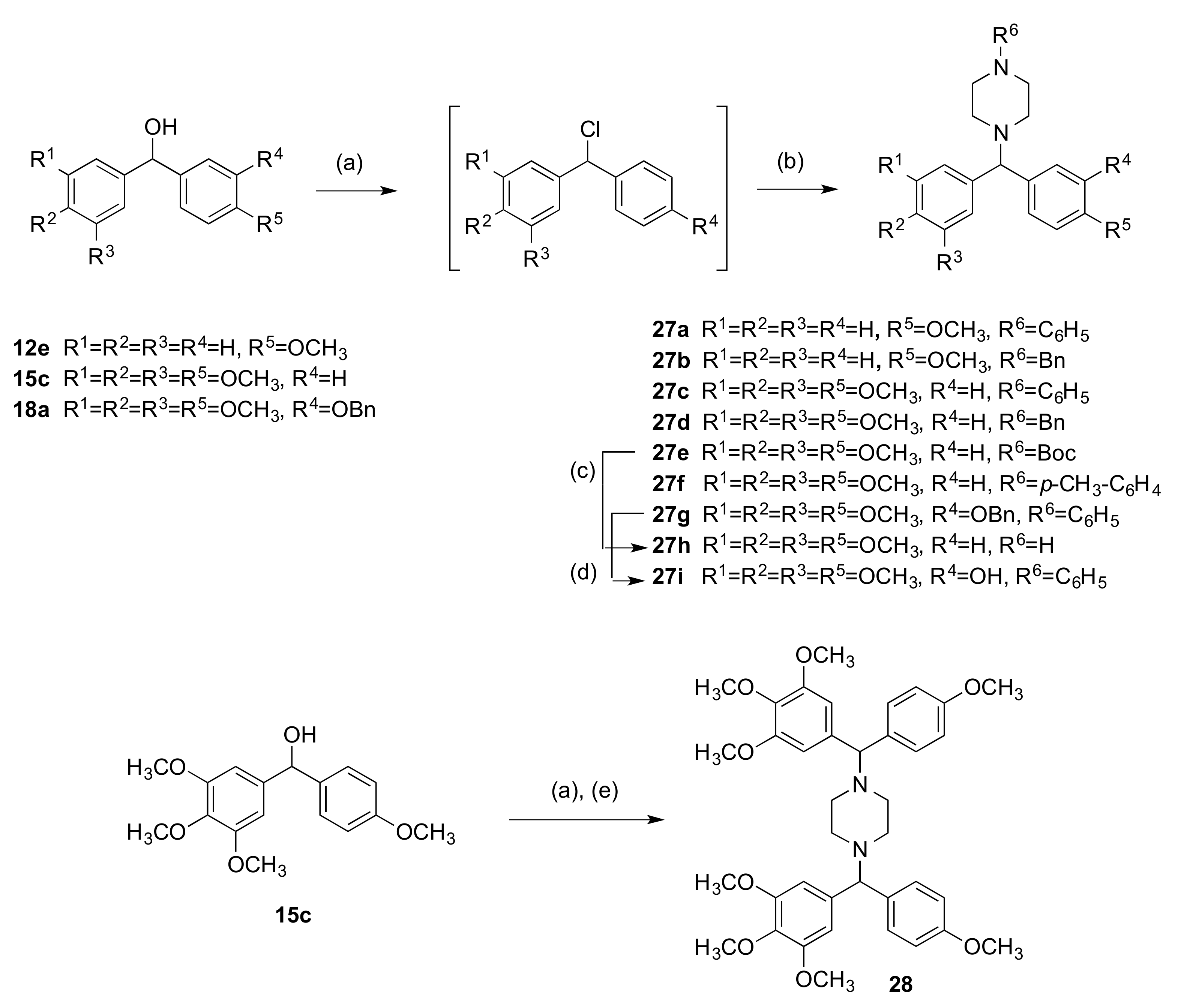
2.1.3. 1-(Diarylmethyl)Pyrrolidines, 1-(Diarylmethyl)Piperidines, and 1-(Diarylmethyl) Piperazines (Series 5)
2.2. Stability Studies
3. Biochemical Results and Discussion
3.1. In Vitro Antiproliferative Activity in MCF-7 Breast Cancer Cells
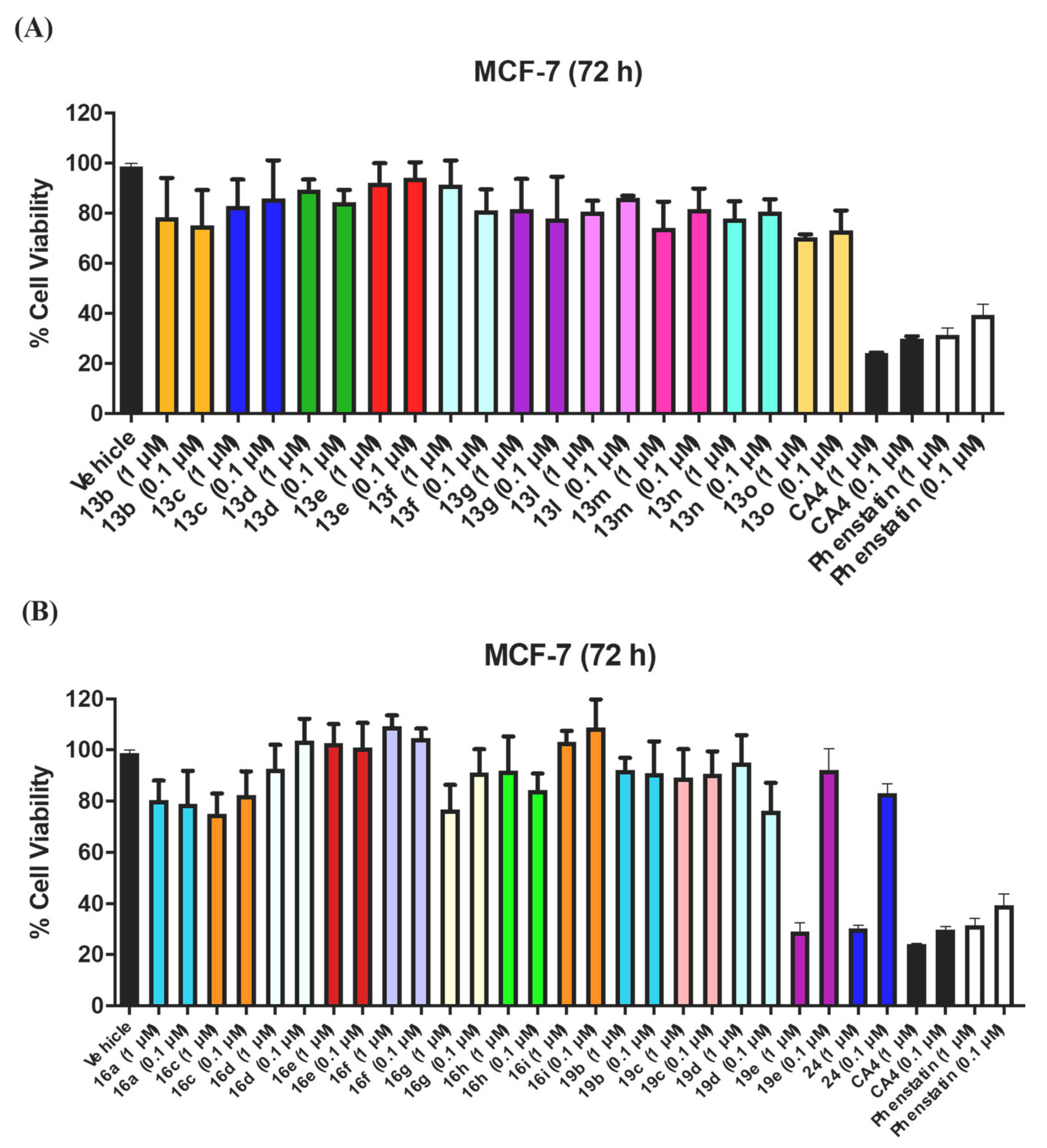
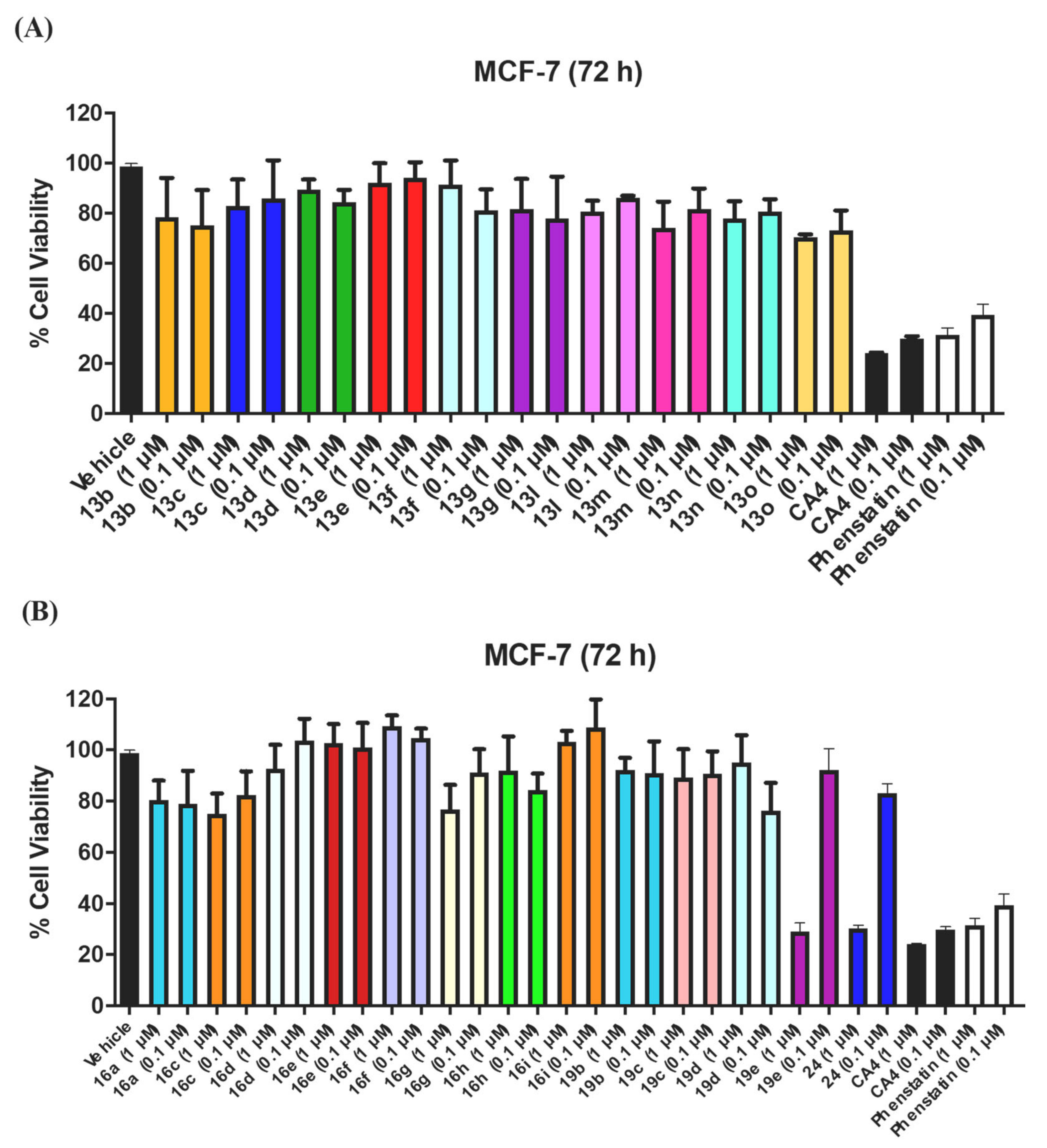
3.1.1. Series 1: 1-(Diarylmethyl)-1H-1,2,4-Triazoles 13b–g, l–o
3.1.2. Series 2: 1-(Aryl-(3,4,5-Trimethoxyphenyl)Methyl)-1H-1,2,4-Triazoles 16a,c–i, 19b–e, 19h, 19i and 1-(Phenyl(3,4,5-Trimethoxyphenyl)Methyl)-1H-1,2,3-Triazole 24
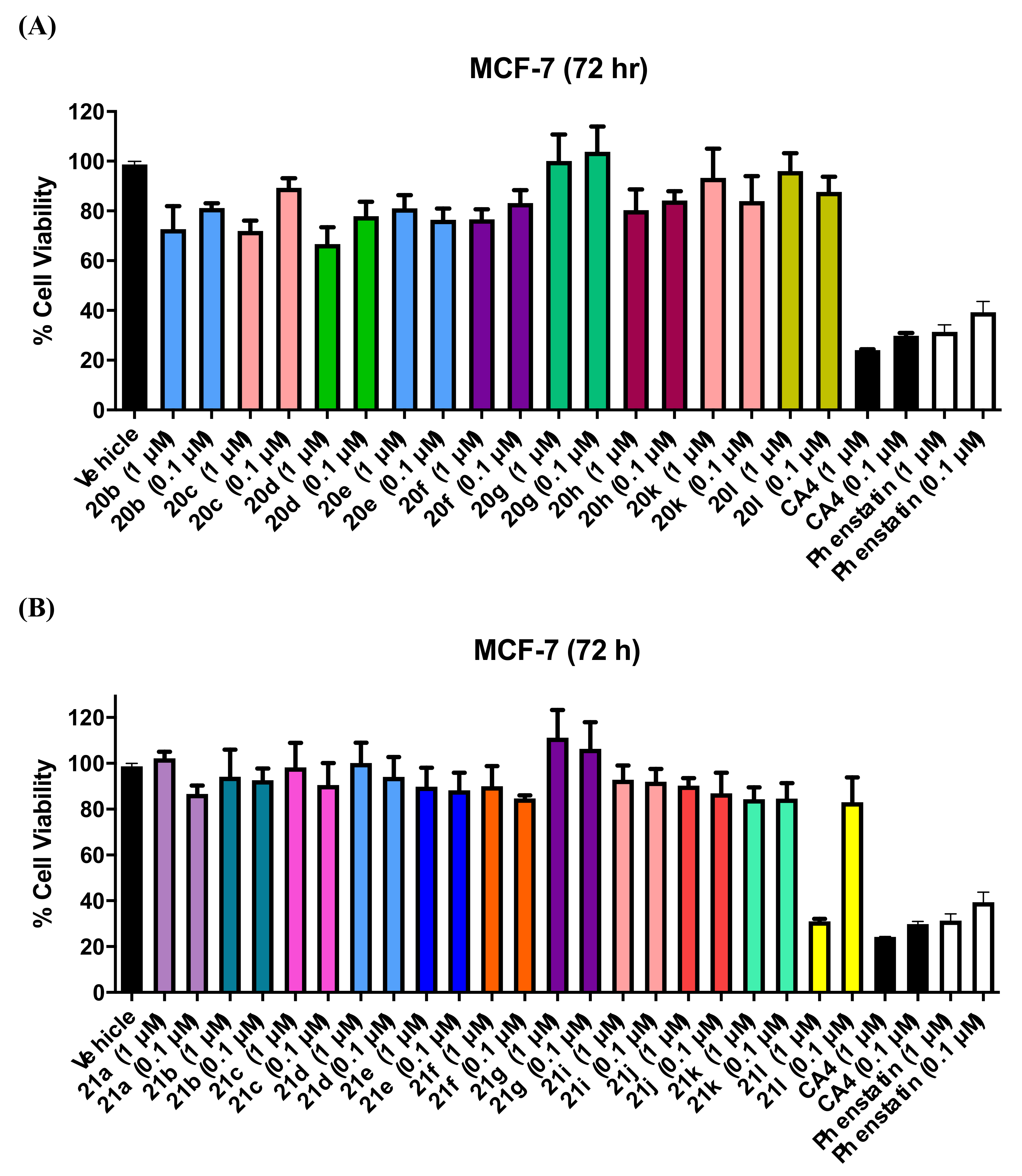
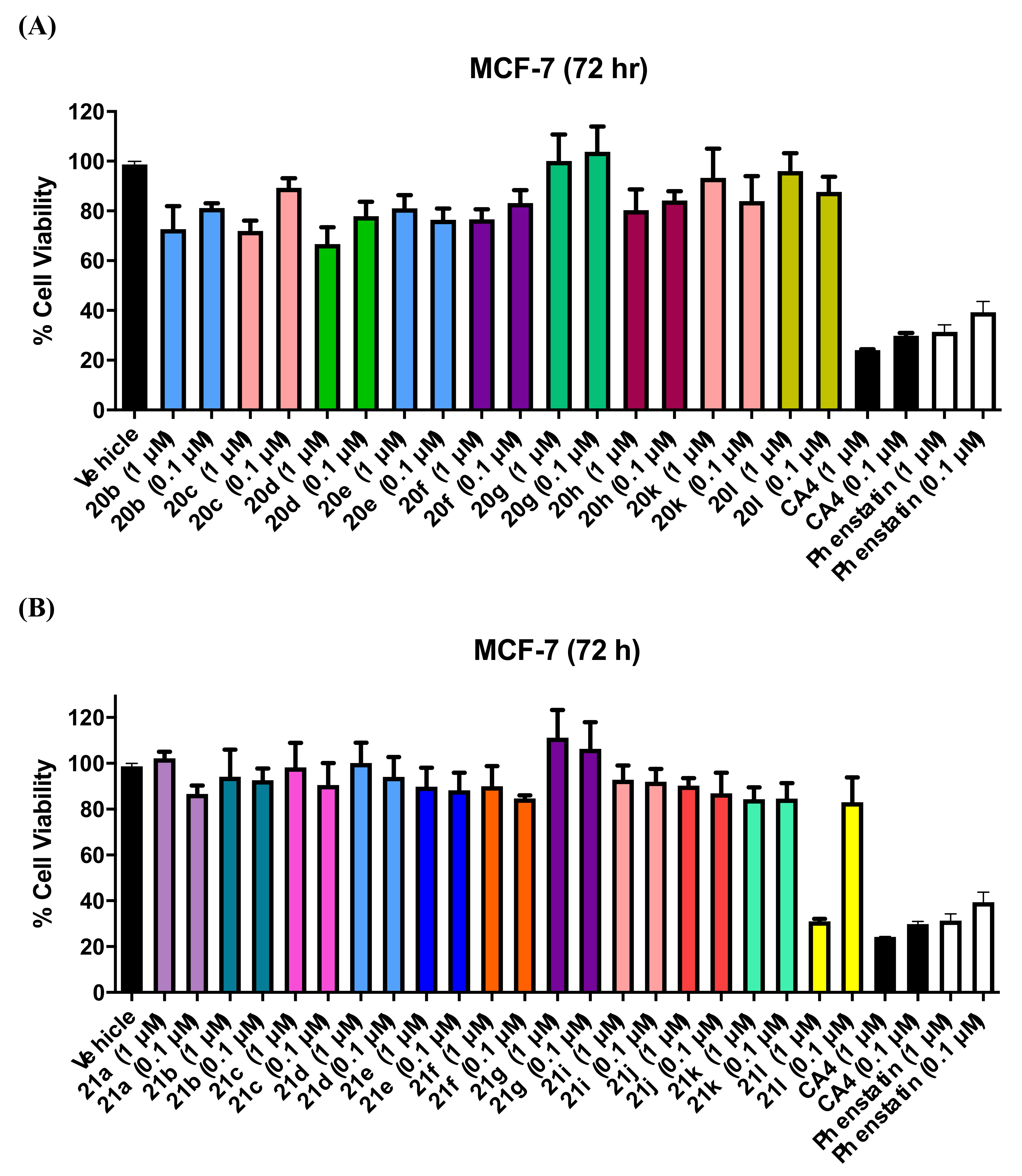
3.1.3. Series 3: 1-(Diarylmethyl)-1H-Imidazoles 20b–h,k,l
3.1.4. Series 4: 1-(Aryl-(3,4,5-Trimethoxyphenyl)Methyl)-1H-Imidazoles 21a-g, i-l
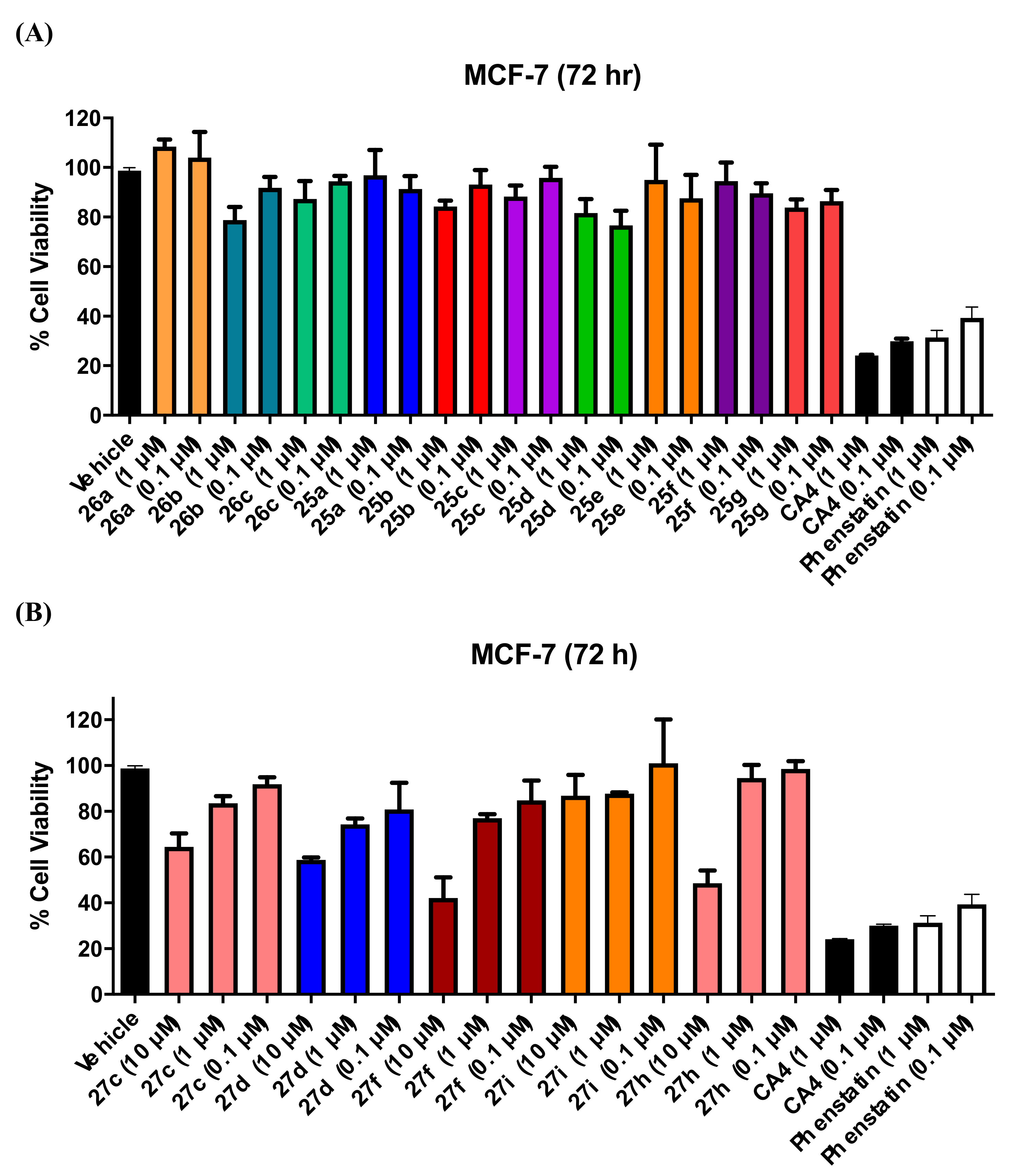
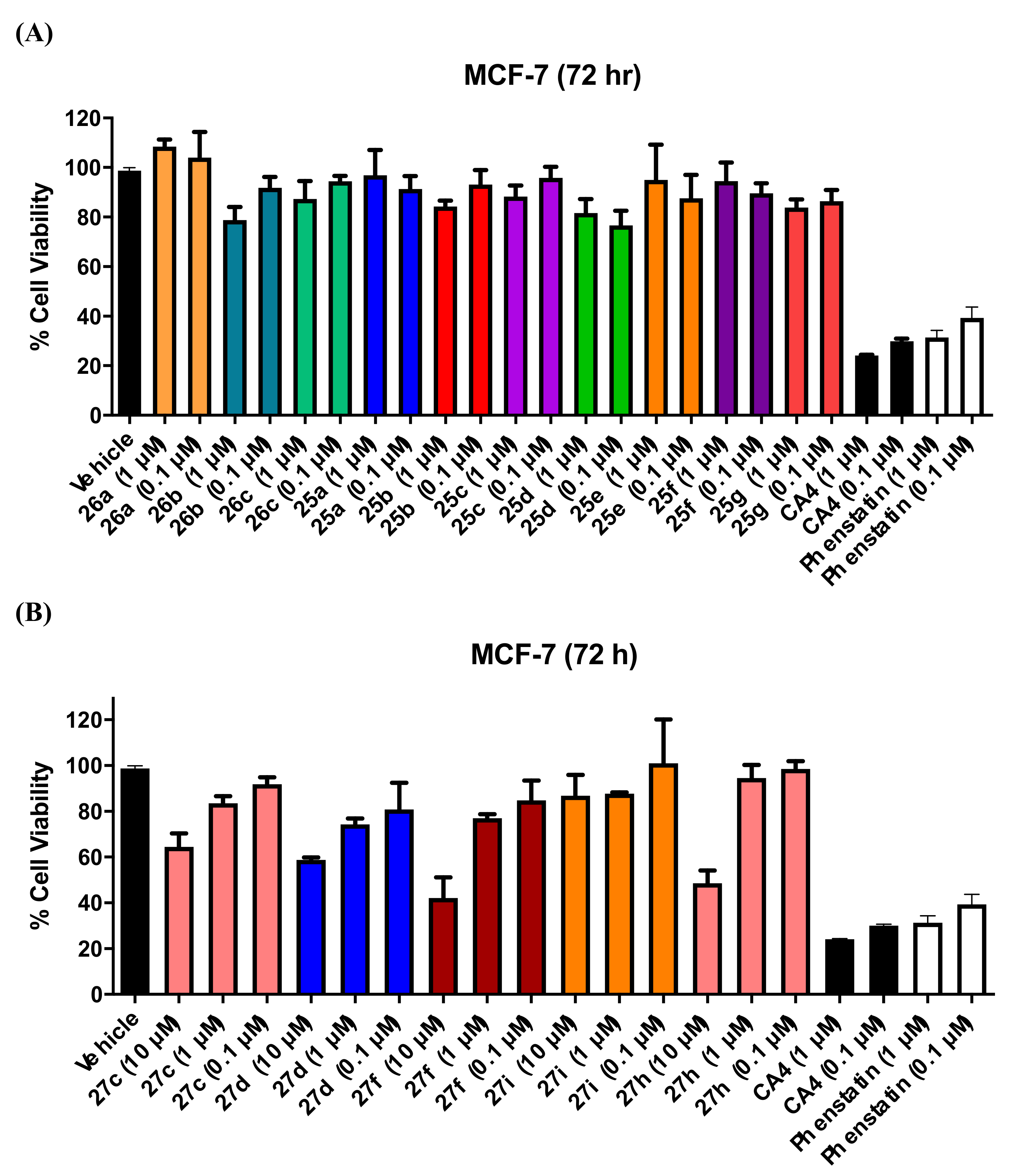
3.1.5. Series 5: 1-(Diarylmethyl)Pyrrolidines 25a–g and 1-(Diarylmethyl)Piperidines 26a–c, and 1-(Diarylmethyl)Piperazines 27a–g, 28
3.2. Antiproliferative Activity of Selected Analogues in MDA-MB-231 and HL60 Cell Lines
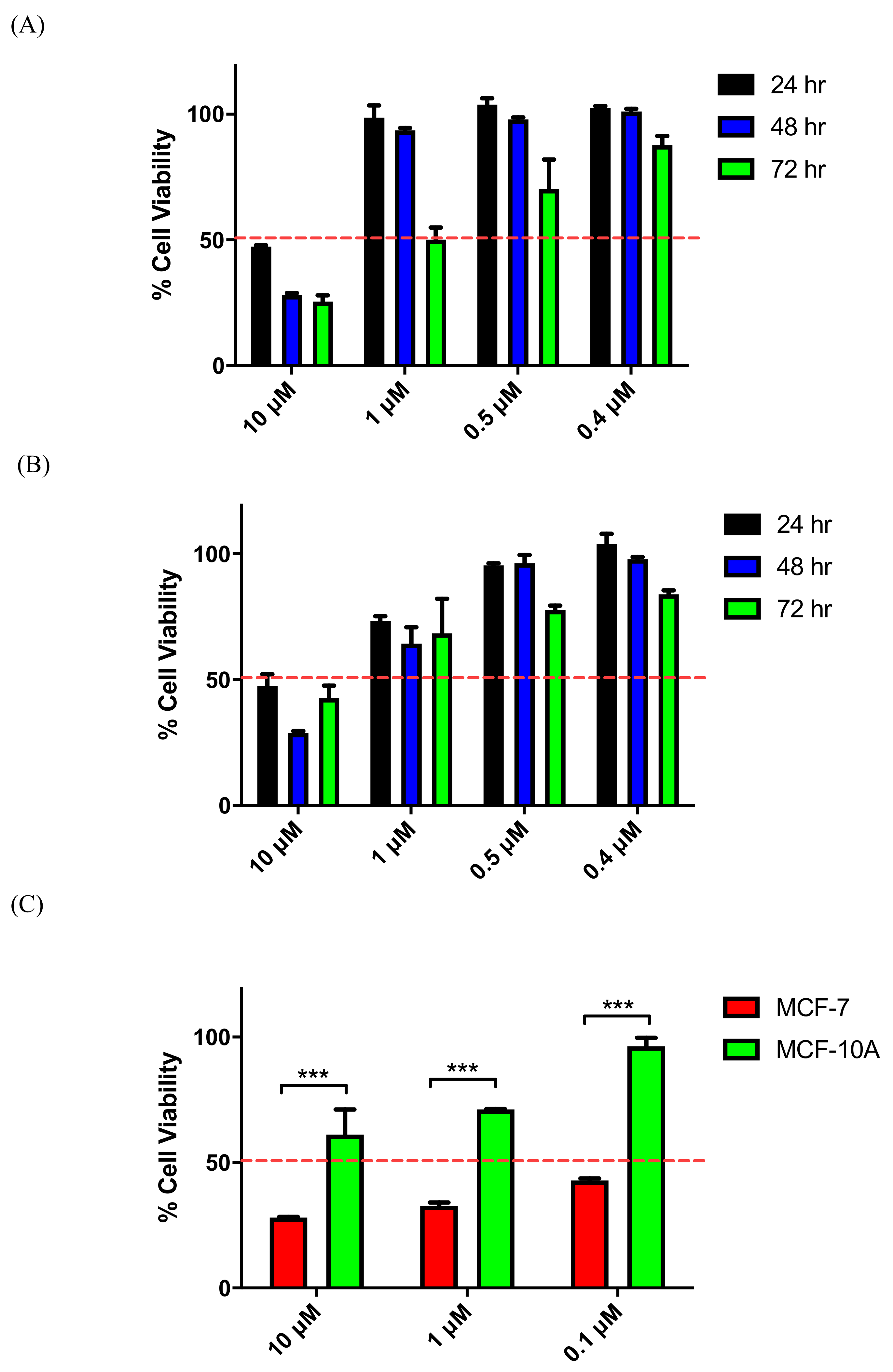
3.3. NCI Cell Line Screening for 19e, 21l, 25g, 26b, and 27d
3.4. Evaluation of Toxicity in MCF-10A Cells
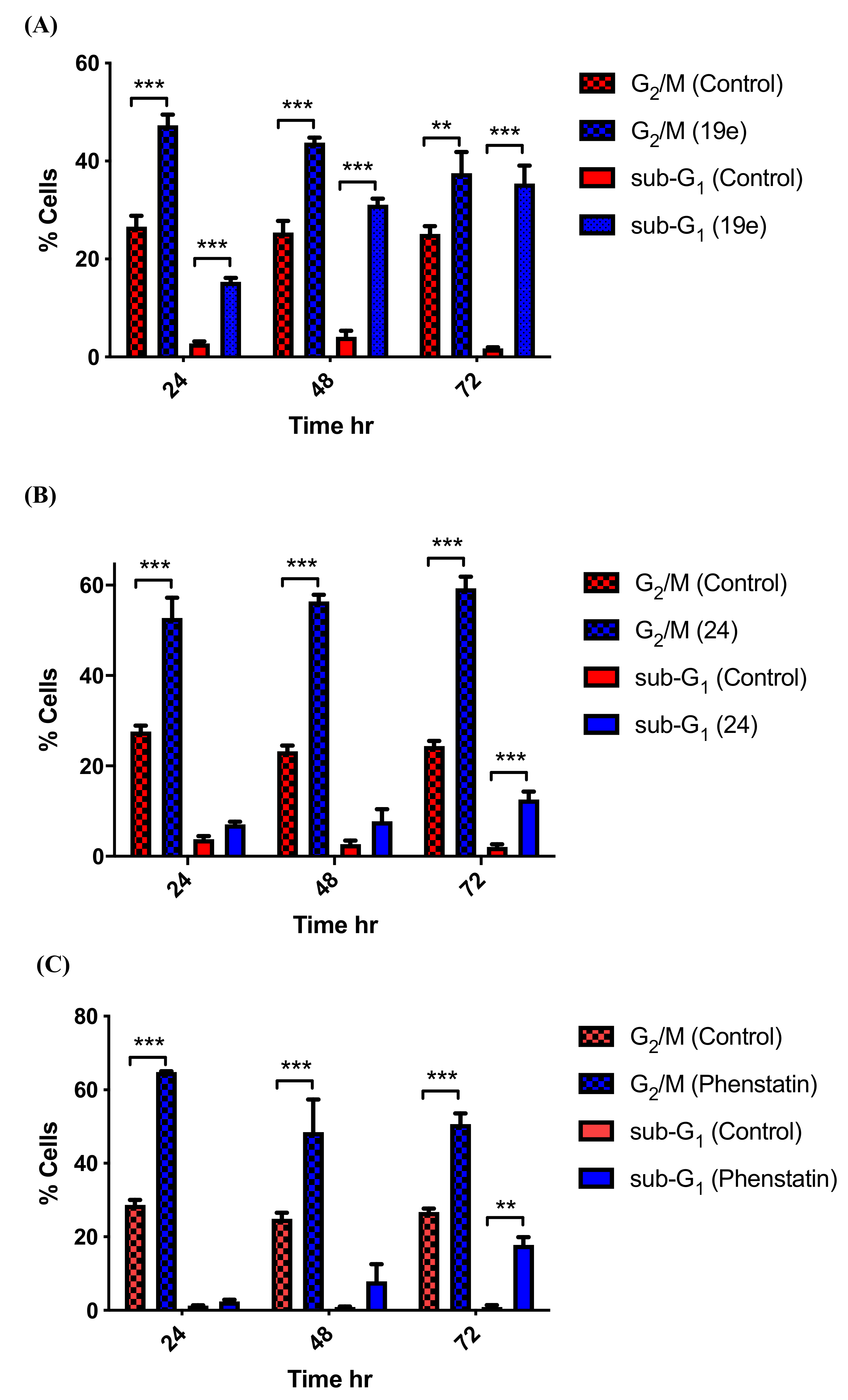
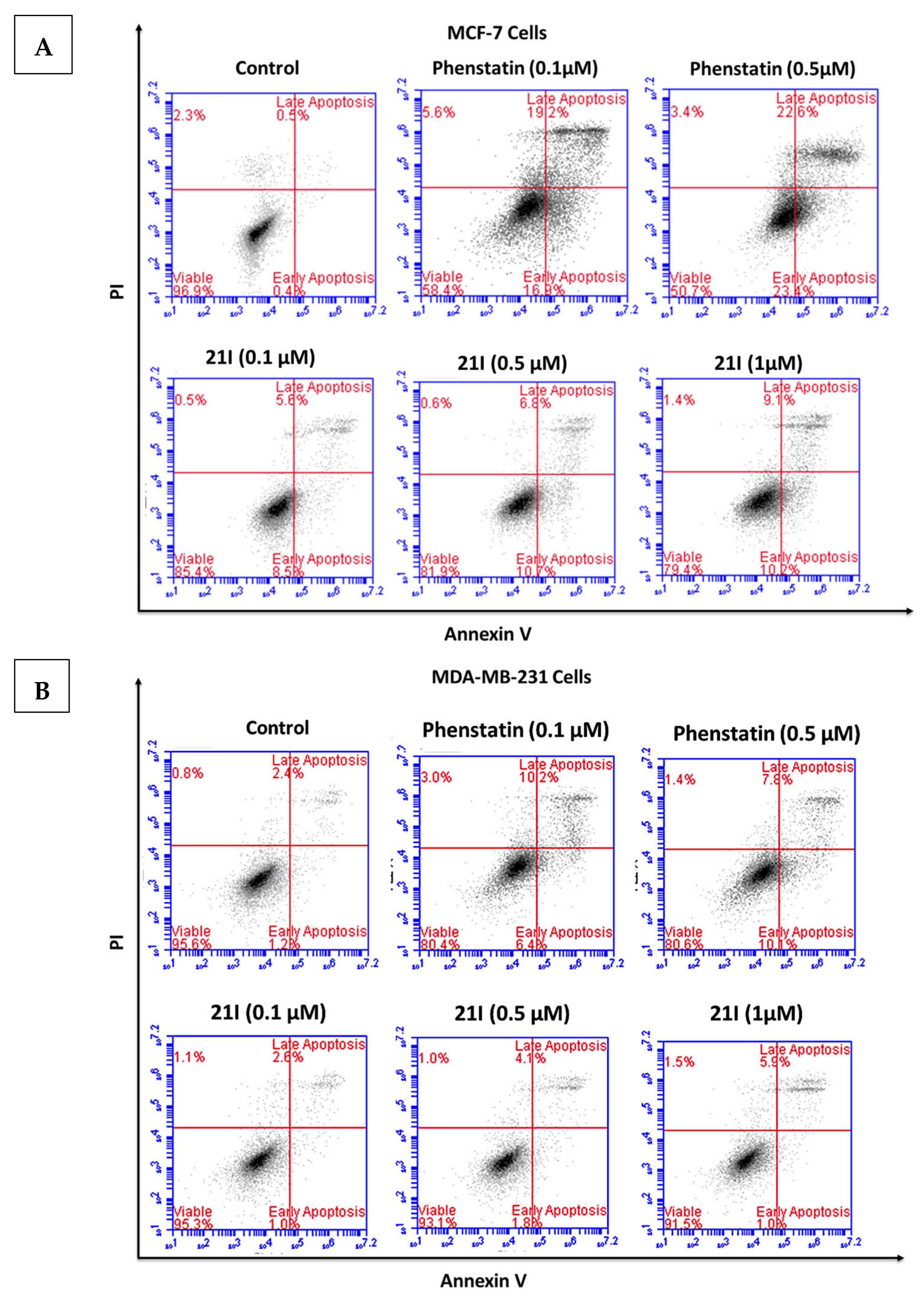
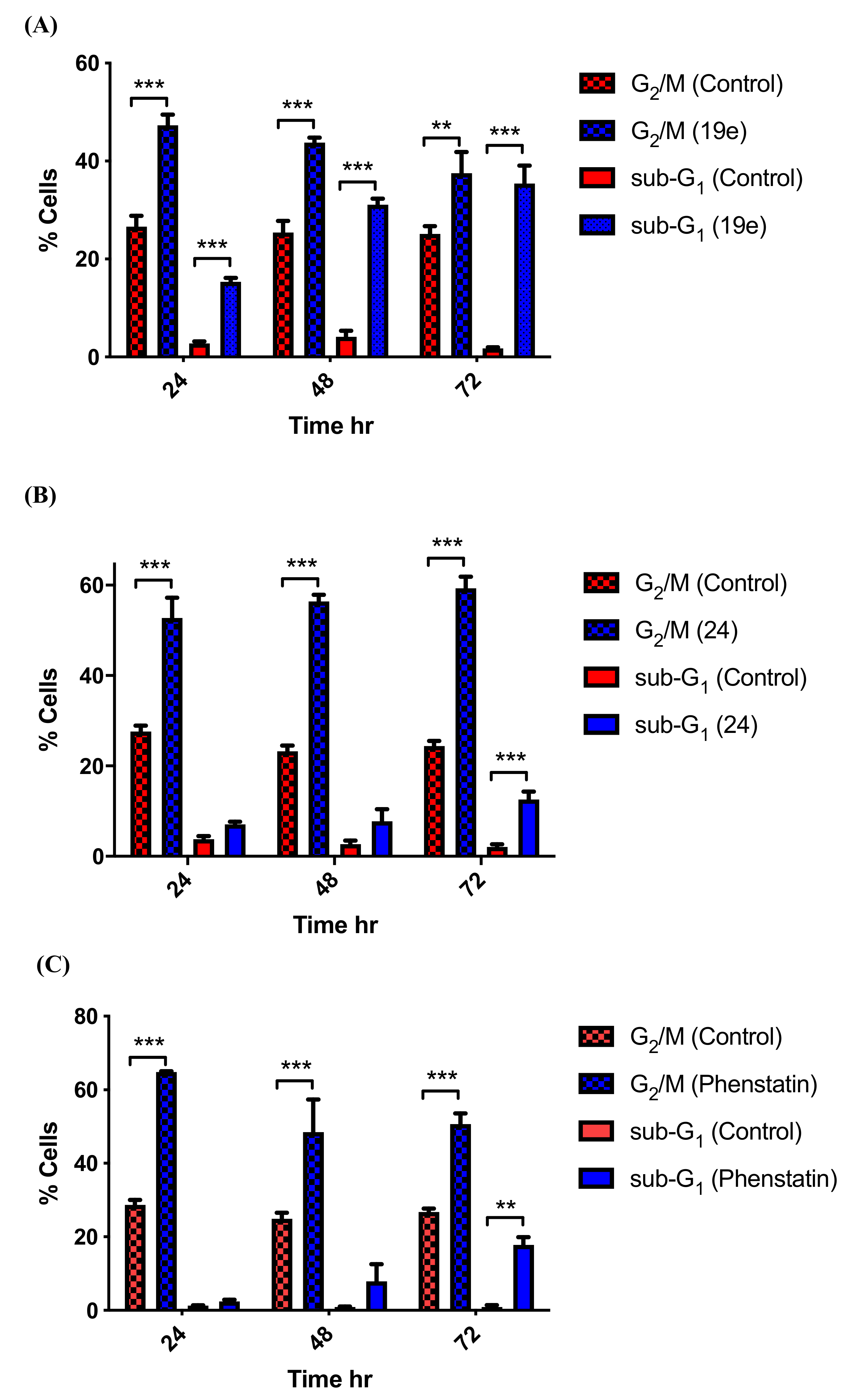
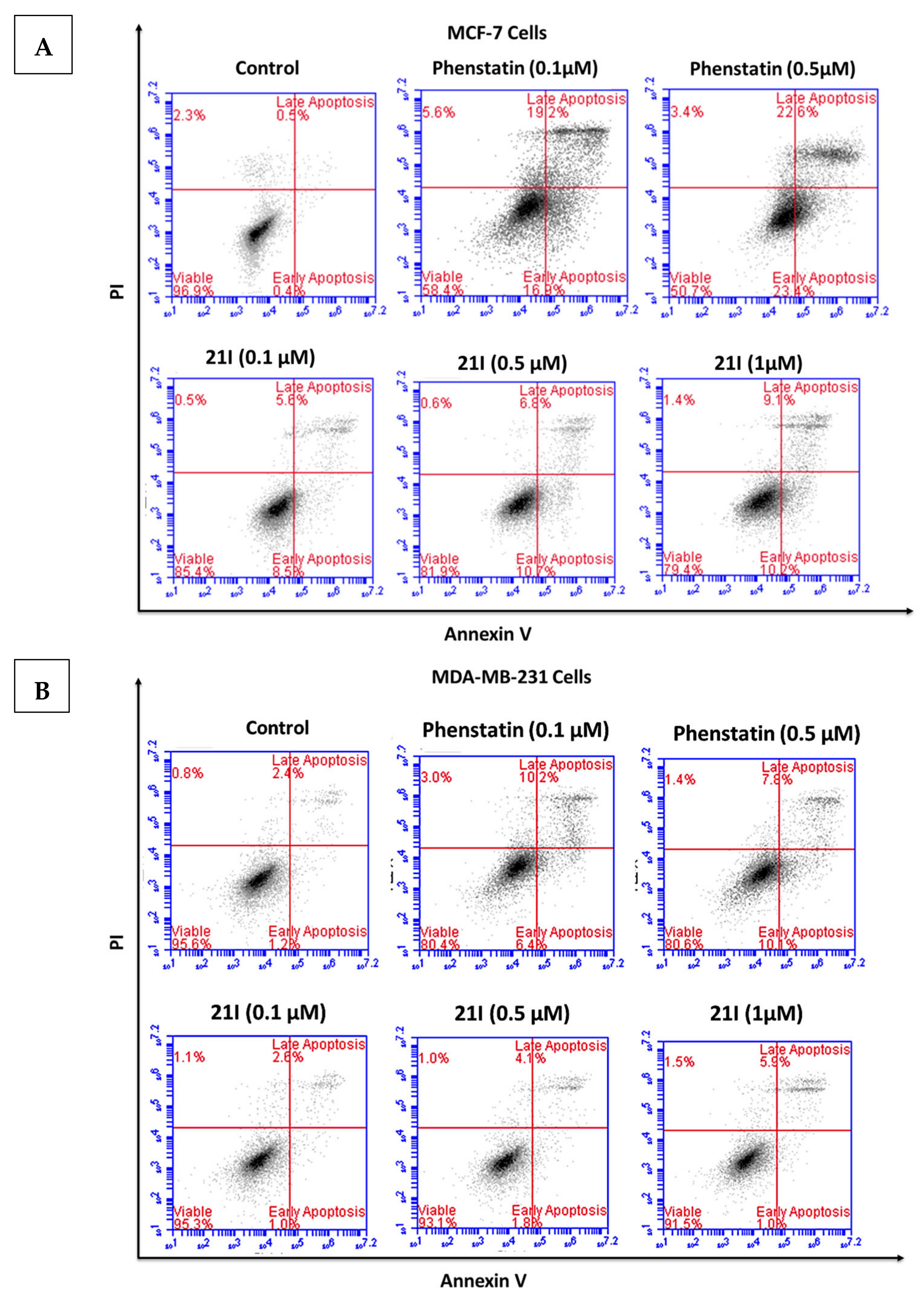
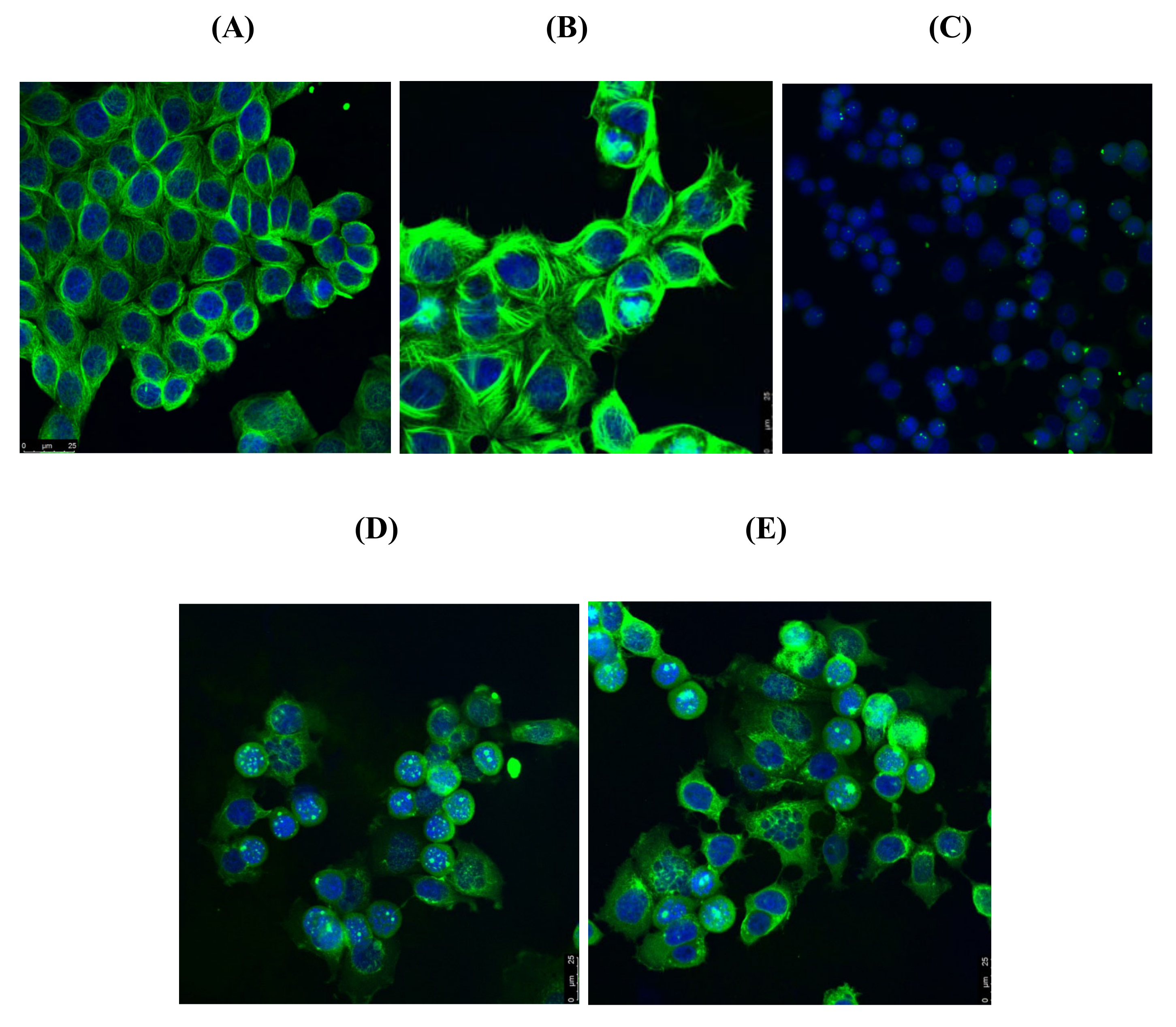
3.5. Effects of Compounds 21l and 24 on Cell Cycle Arrest and Apoptosis
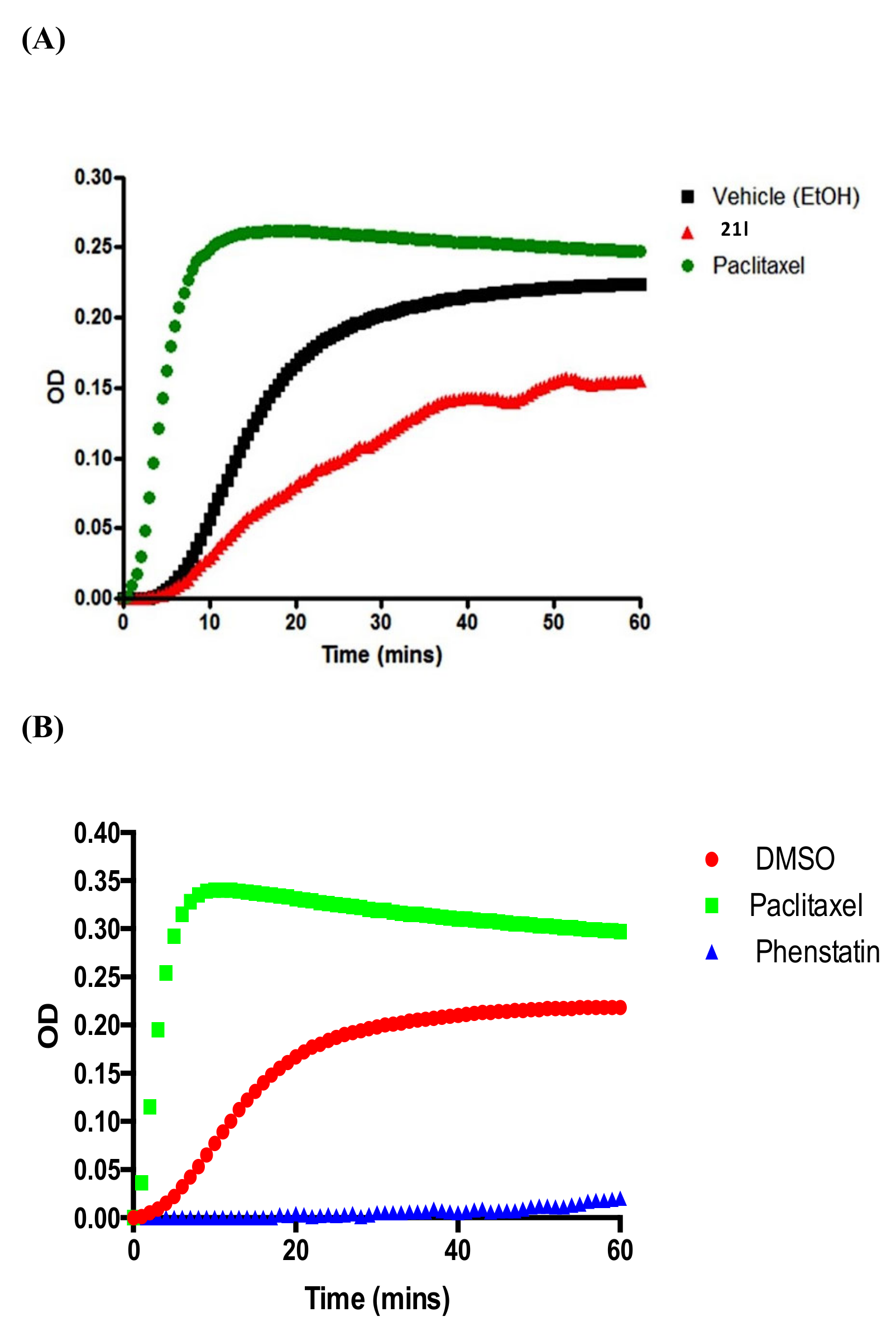
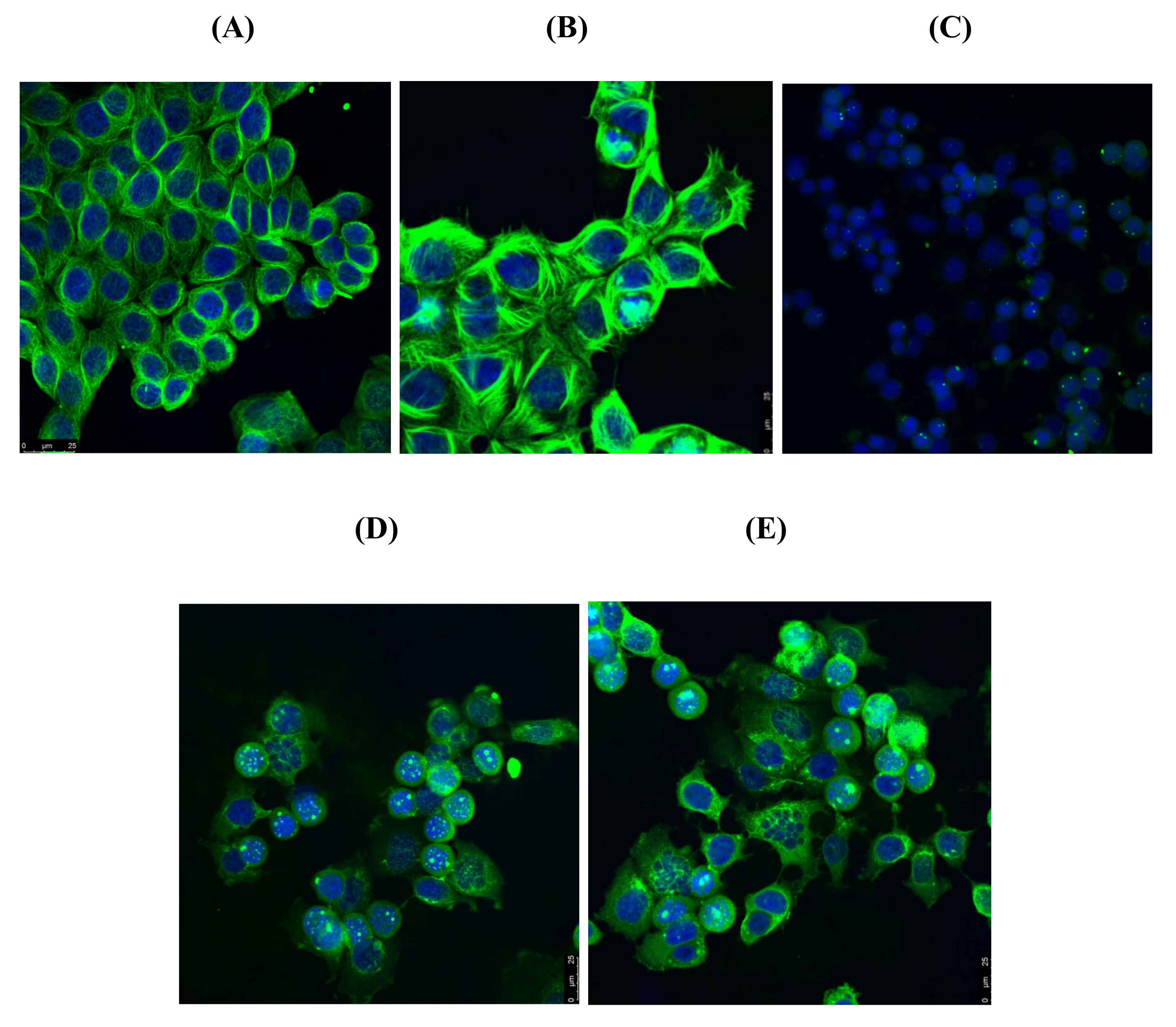
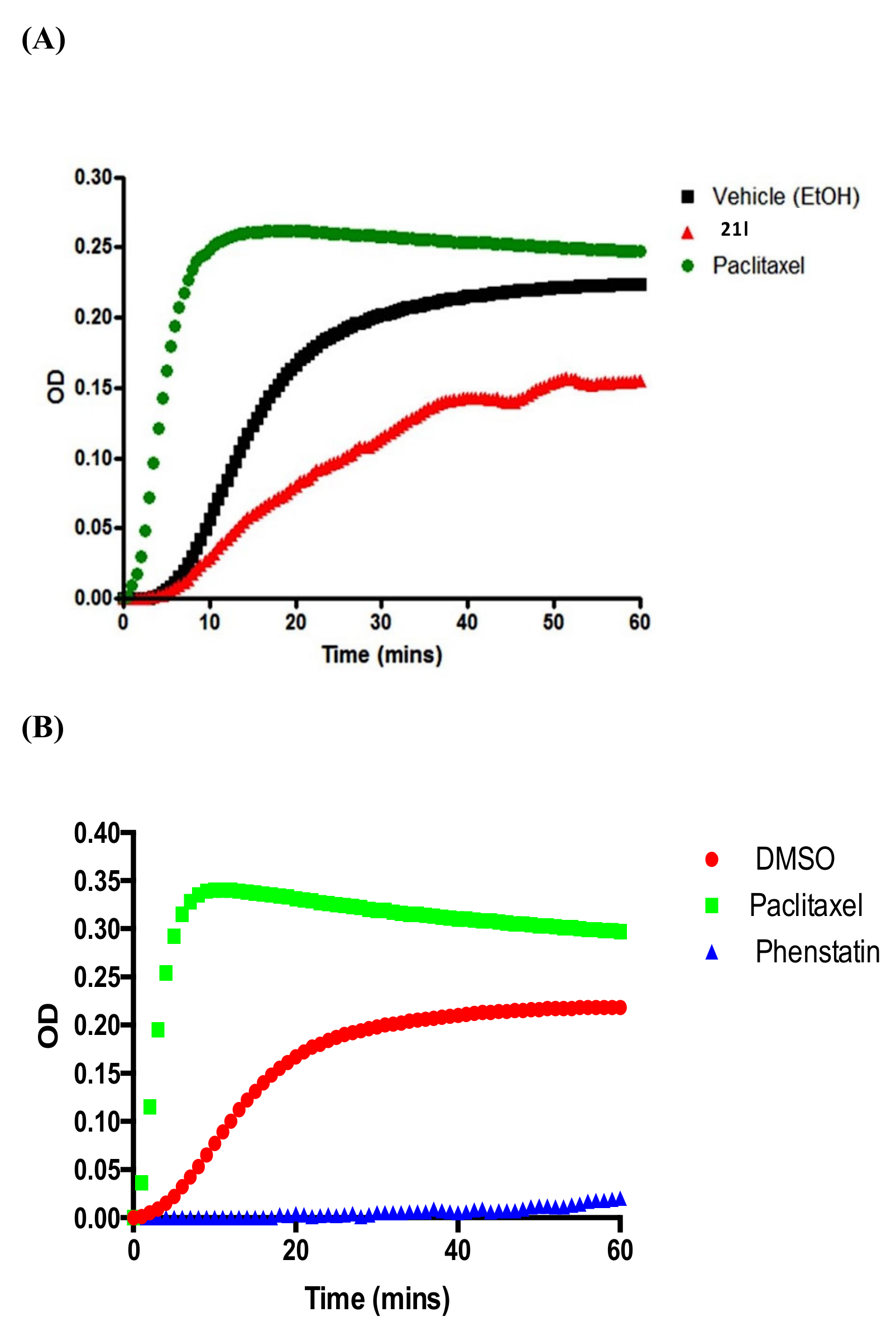
3.6. Tubulin Polymerisation
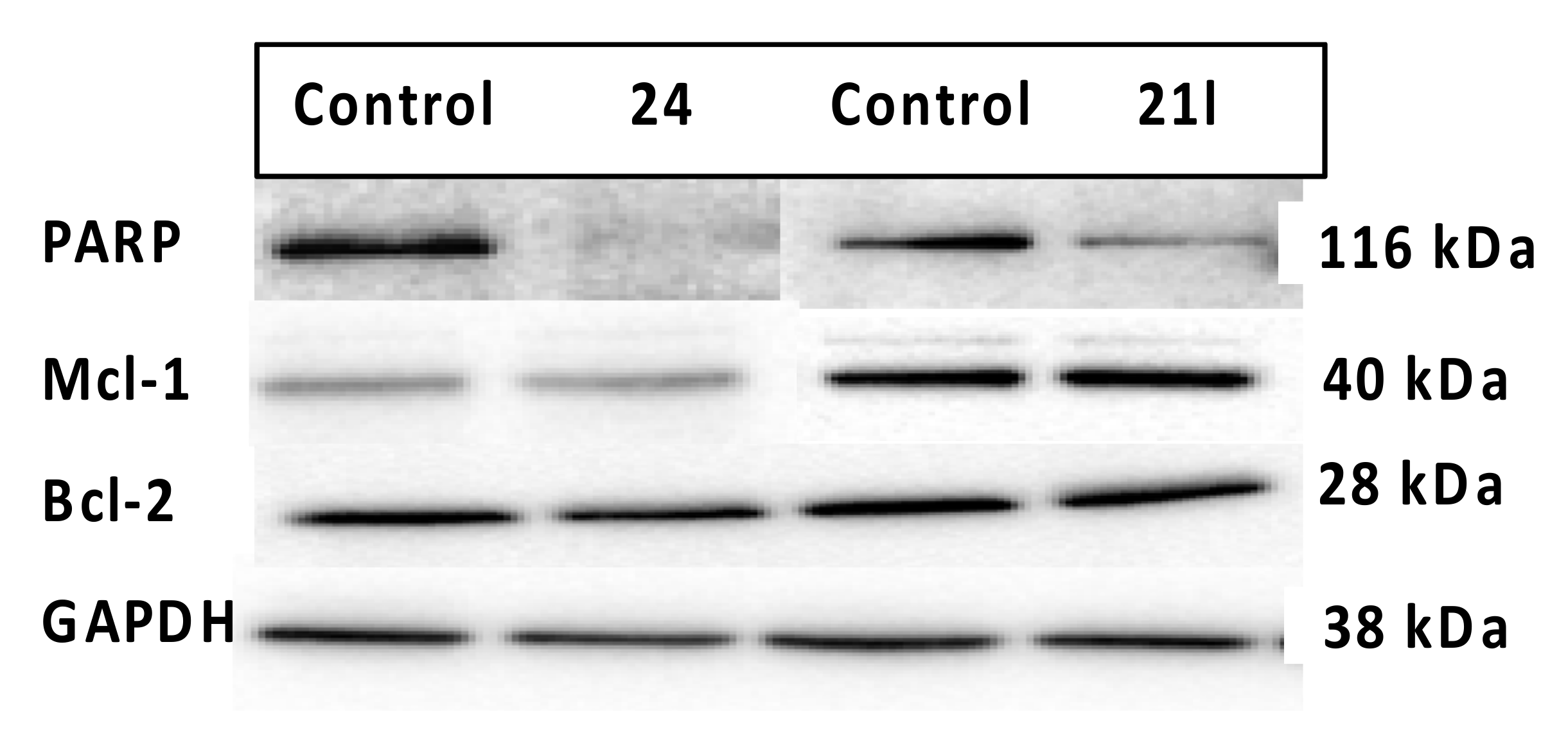
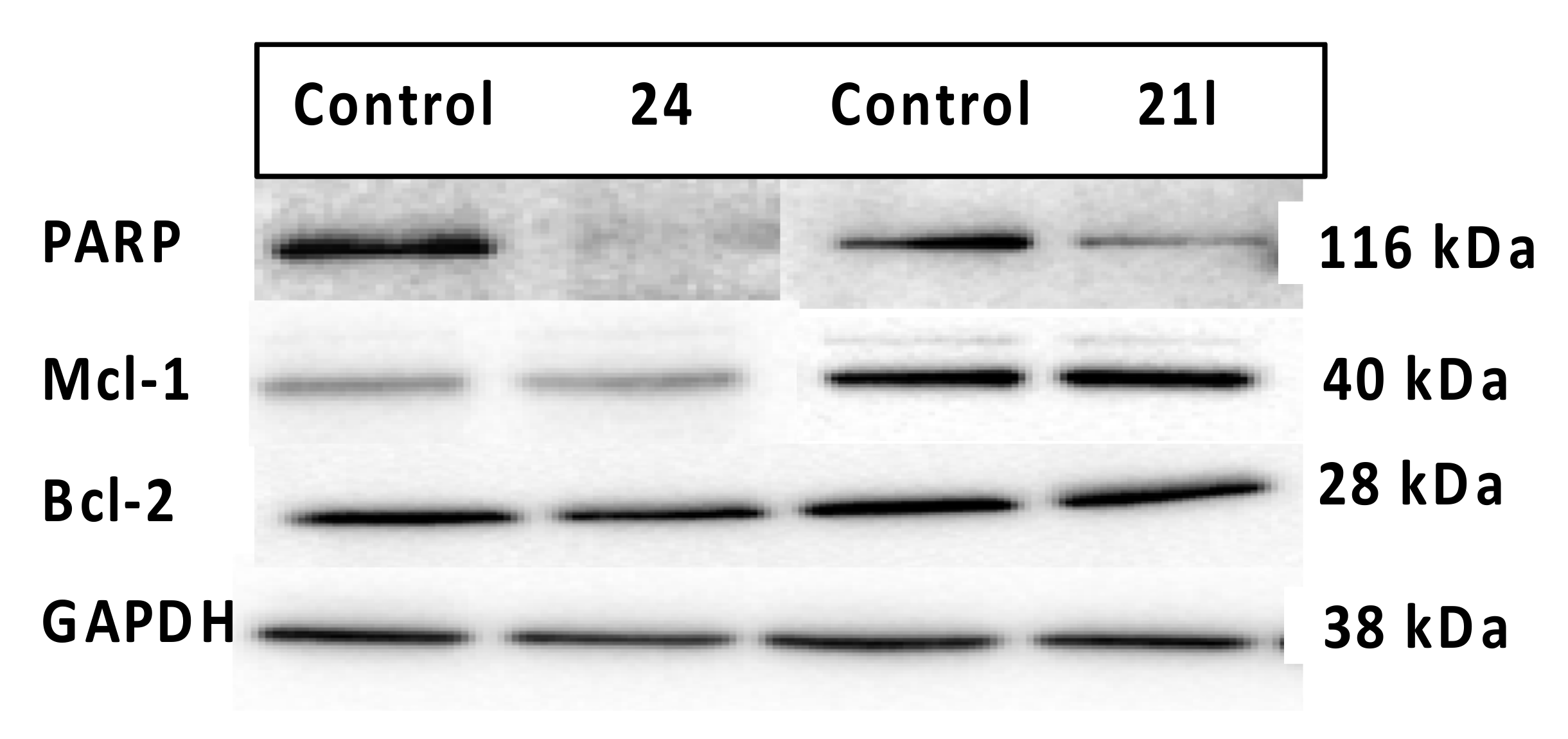
3.7. Effects of Compounds 21l and 24 on Expression Levels of Apoptosis-Associated Proteins
3.8. Aromatase Inhibition
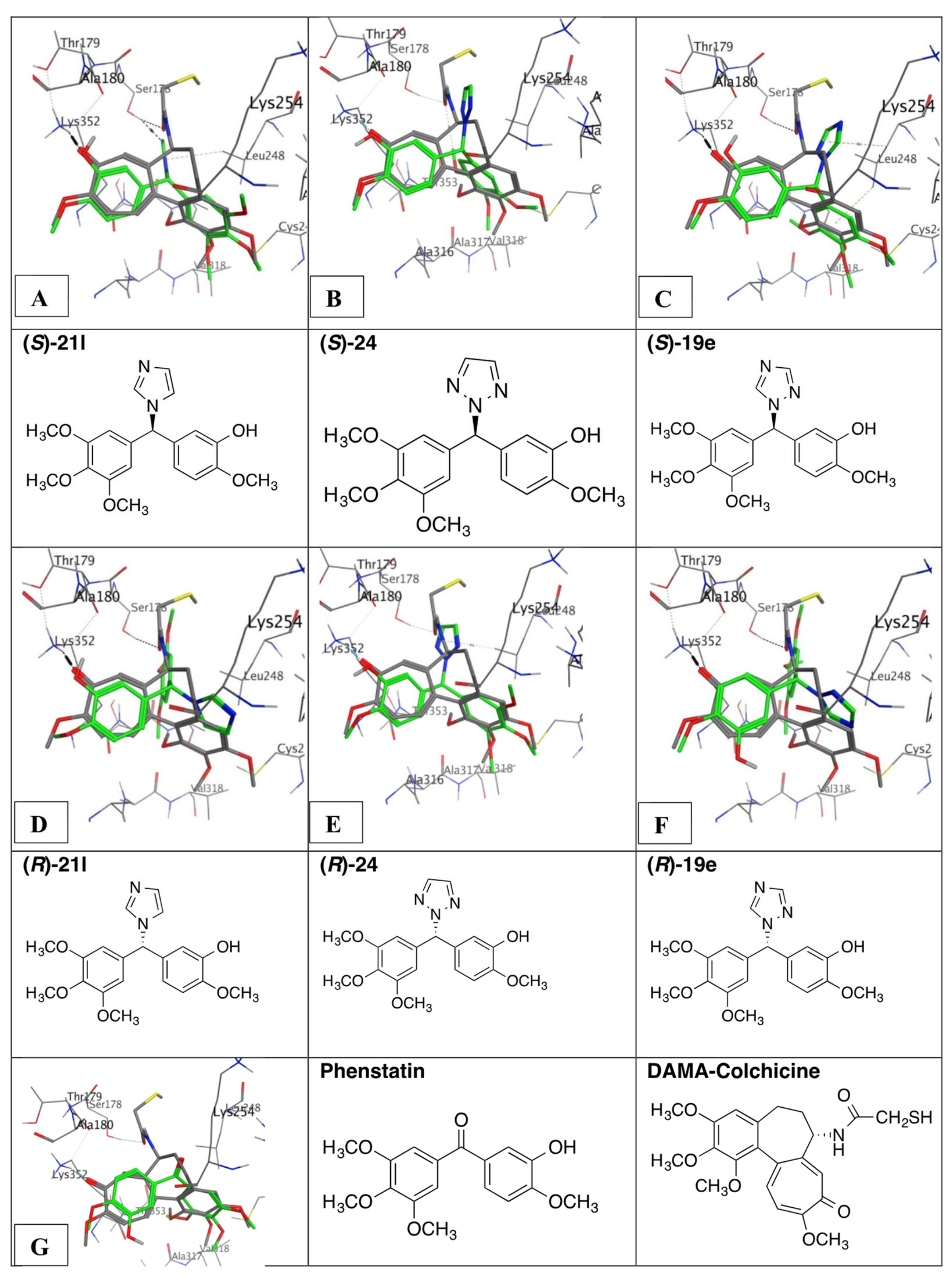
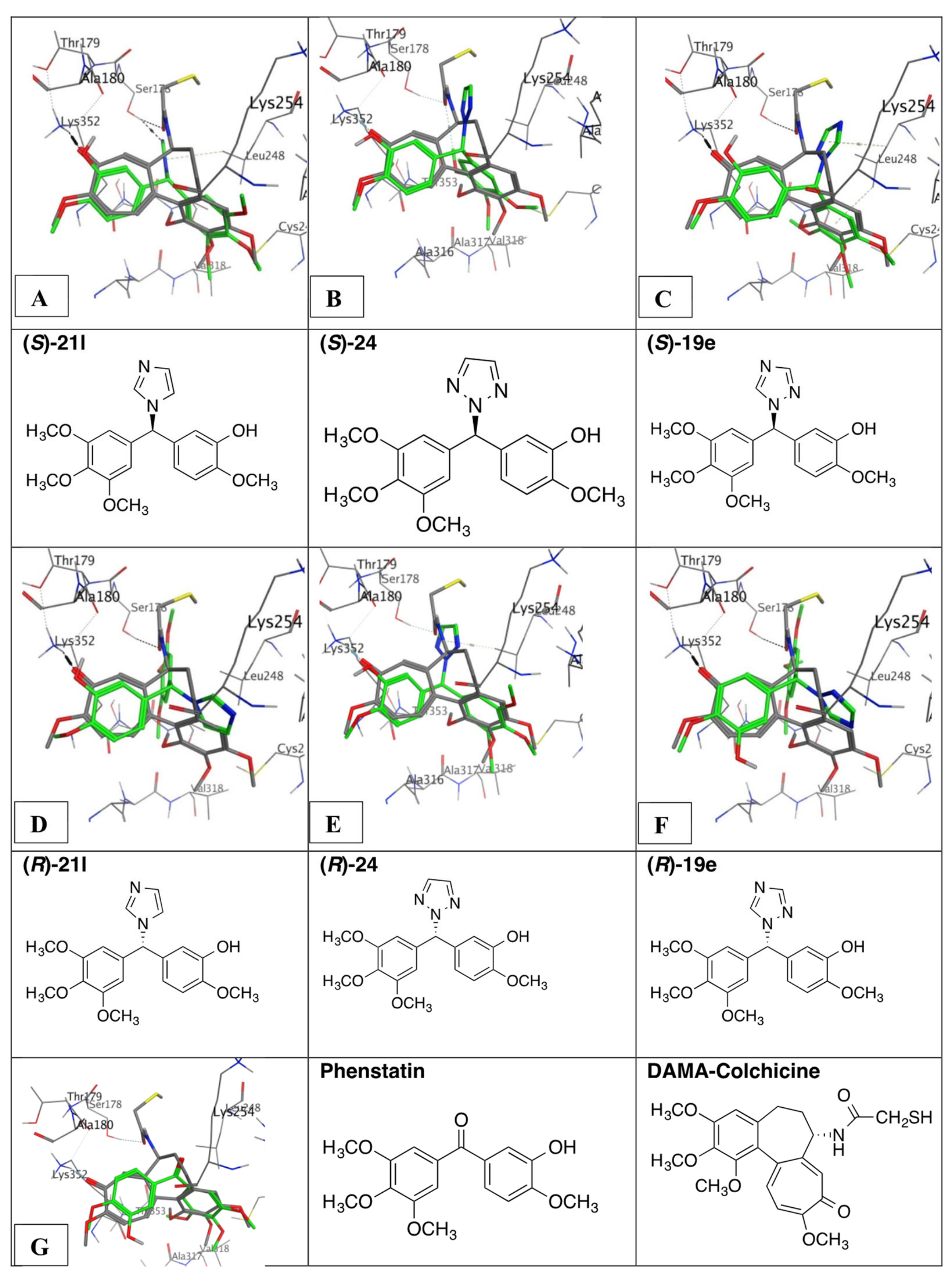
3.9. Molecular Docking of Phenstatin Hybrids 19e, 21l, and 24
4. Materials and Methods
4.1. Chemistry
4.1.1. (3-Hydroxy-4-Methoxyphenyl)(3,4,5-Trimethoxyphenyl)Methanone, Phenstatin (7a)
4.1.2. General Method A: Preparation of Alcohols
N-(4-(Hydroxy(Phenyl)Methyl)Phenyl)Acetamide (12i)
4-(Hydroxy(Phenyl)Methyl)Phenyl 2,2,2-Trifluoroacetate (12j)
4.1.3. General Method B: Preparation of 1-(Diarylmethyl)-1H-1,2,4-Triazoles
1-((4-Fluorophenyl)(Phenyl)Methyl)-1H-1,2,4-Triazole (13c)
2,2,2-Trifluoro-N-(4-(Phenyl(1H-1,2,4-Triazol-1-yl)Methyl)Phenyl)Acetamide (13i)
1-((4-(Benzyloxy)Phenyl)(Phenyl)Methyl)-1H-1,2,4-Triazole (13j)
1-((4-Ethoxyphenyl)(Phenyl)Methyl)-1H-1,2,4-Triazole (13l)
1-((4-(Benzyloxy)phenyl)(3,4,5-trimethoxyphenyl)methyl)-1H-1,2,4-triazole (16b)
1-(Phenyl(3,4,5-Trimethoxyphenyl)Methyl)-1H-1,2,4-Triazole (16e)
1-((3-(Benzyloxy)-4-Methoxyphenyl)(3,4,5-Trimethoxyphenyl)Methyl)-1H-1,2,4-Triazole (19a)
4-((3-(Benzyloxy)-4-Methoxyphenyl)(1H-1,2,4-Triazol-1-yl)Methyl)Benzonitrile (19f)
4-((4-(Benzyloxy)Phenyl)(1H-1,2,4-Triazol-1-yl)Methyl)Benzonitrile (19g)
5-((2H-1,2,3-Triazol-1-yl)(3,4,5-Trimethoxyphenyl)Methyl)-2-Methoxyphenol (24)
4.1.4. 4-(Phenyl(1H-1,2,4-Triazol-1-yl)Methyl)Aniline (13m)
4.1.5. 4-(Phenyl(1H-1,2,4-Triazol-1-yl)Methyl)Phenol (13n)
4.1.6. General Method C: Preparation of Secondary Alcohols 15b, 15c, 15d, 15g, 15h
(4-(Benzyloxy)Phenyl)(3,4,5-Trimethoxyphenyl)Methanol (15b)
(4-Methoxyphenyl)(3,4,5-Trimethoxyphenyl)Methanol (15c)
(3,4-Dimethoxyphenyl)(3,4,5-Trimethoxyphenyl)Methanol (15d)
4.1.7. 4-((1H-1,2,4-Triazol-1-yl)(3,4,5-Trimethoxyphenyl)Methyl)Phenol (16i)
4.1.8. 5-((1H-1,2,4-Triazol-1-yl)(3,4,5-Trimethoxyphenyl)methyl)-2-Methoxyphenol (19e)
4.1.9. 4-((3-Hydroxy-4-Methoxyphenyl)(1H-1,2,4-Triazol-1-yl)Methyl)Benzonitrile (19h)
4.1.10. 4-((4-Hydroxyphenyl)(1H-1,2,4-Triazol-1-yl)Methyl)Benzonitrile (19i)
4.1.11. General Method D: Preparation of 1-(Diarylmethyl)-1H-Imidazoles
1-((4-Bromophenyl)(Phenyl)methyl)-1H-Imidazole (20c)
1-((3-(Benzyloxy)-4-Methoxyphenyl)(3,4,5-Trimethoxyphenyl)Methyl)-1H-Imidazole (21h)
1-(Bis(3,4,5-Trimethoxyphenyl)Methyl)-1H-Imidazole (21i)
4.1.12. 4-((1H-Imidazol-1-yl)(Phenyl)Methyl)Aniline (20l)
4.1.13. 5-((1H-Imidazol-1-yl)(3,4,5-Trimethoxyphenyl)Methyl)-2-Methoxyphenol (21l)
4.1.14. (4-Methoxyphenyl)(3,4,5-trimethoxyphenyl)methanone (23a)
4.1.15. (3,4-Dimethoxyphenyl)(3,4,5-Trimethoxyphenyl)Methanone (23b)
4.1.16. General Method E for the Preparation of Diarylmethylpyrrolidines, Diarylmethylpiperidines and Diarylmethylpiperazines
1-((4-Bromophenyl)(Phenyl)methyl)Pyrrolidine (25b)
1-((4-Methoxyphenyl)(3,4,5-Trimethoxyphenyl)Methyl)Piperidine (26b)
1-((4-Methoxyphenyl)(3,4,5-Trimethoxyphenyl)Methyl)-4-Phenylpiperazine (27c)
Tert-Butyl 4-((4-Methoxyphenyl)(3,4,5-Trimethoxyphenyl)Methyl)Piperazine-1-Carboxylate (27e)
1-((3-(Benzyloxy)-4-Methoxyphenyl)(3,4,5-Trimethoxyphenyl)Methyl)-4-Phenylpiperazine (27g)
1,4-bis((4-Methoxyphenyl)(3,4,5-Trimethoxyphenyl)Methyl)Piperazine (28)
4.1.17. 1-((4-Methoxyphenyl)(3,4,5-Trimethoxyphenyl)Methyl)Piperazine (27h)
4.1.18. 2-Methoxy-5-((4-Phenylpiperazin-1-yl)(3,4,5-Trimethoxyphenyl)Methyl)Phenol (27i)
4.2. Stability Study of Compounds 21l and 24
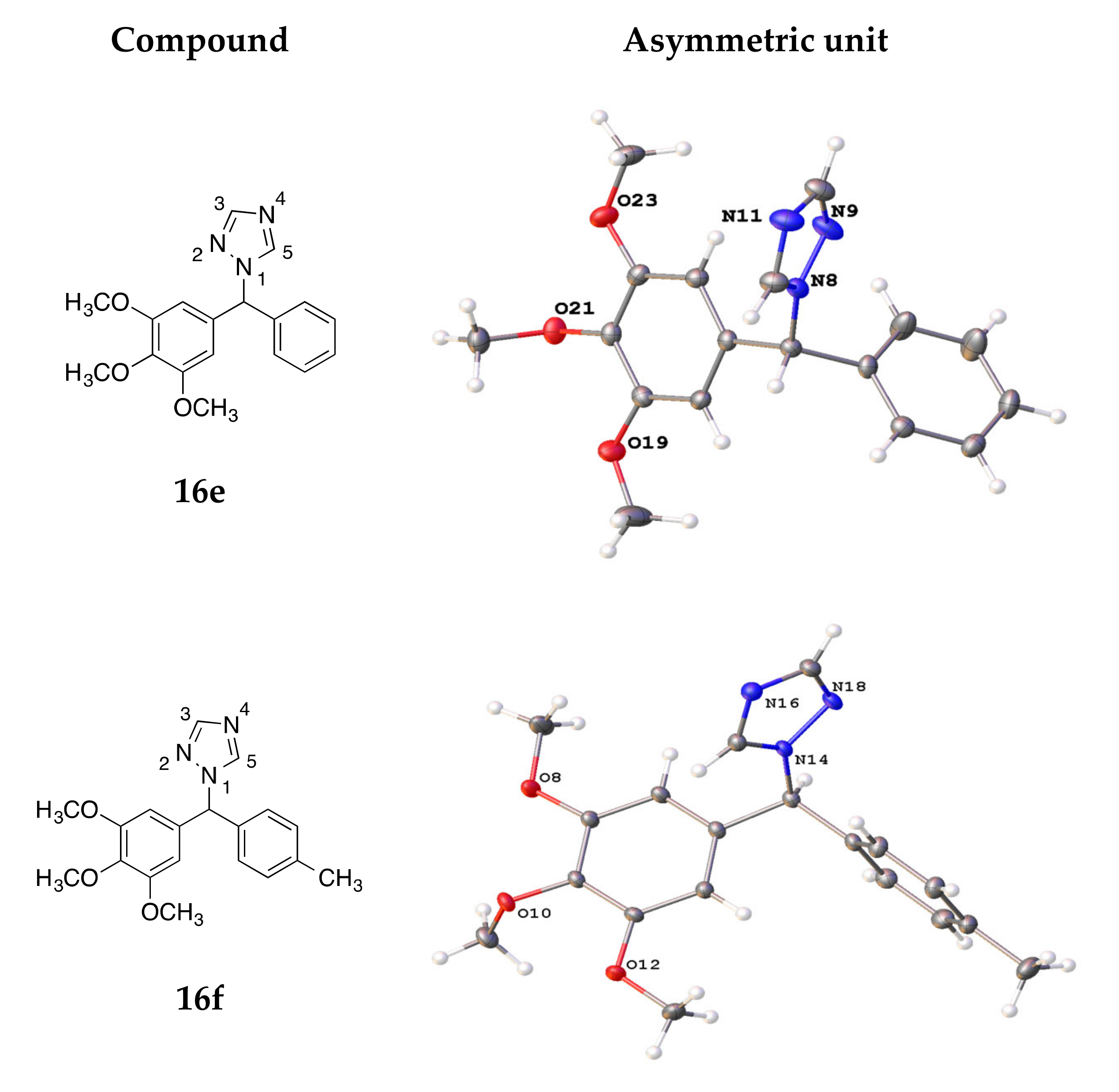
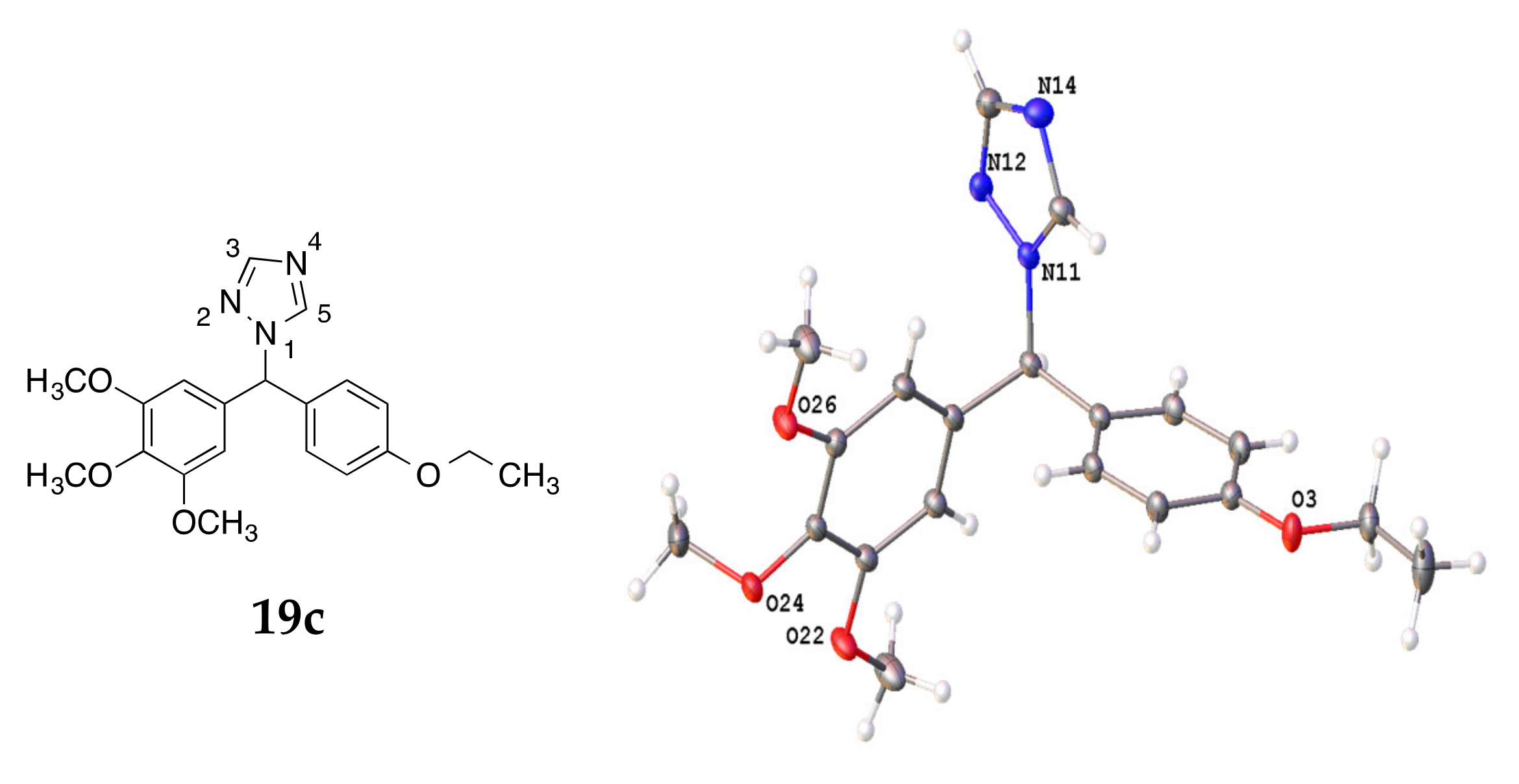
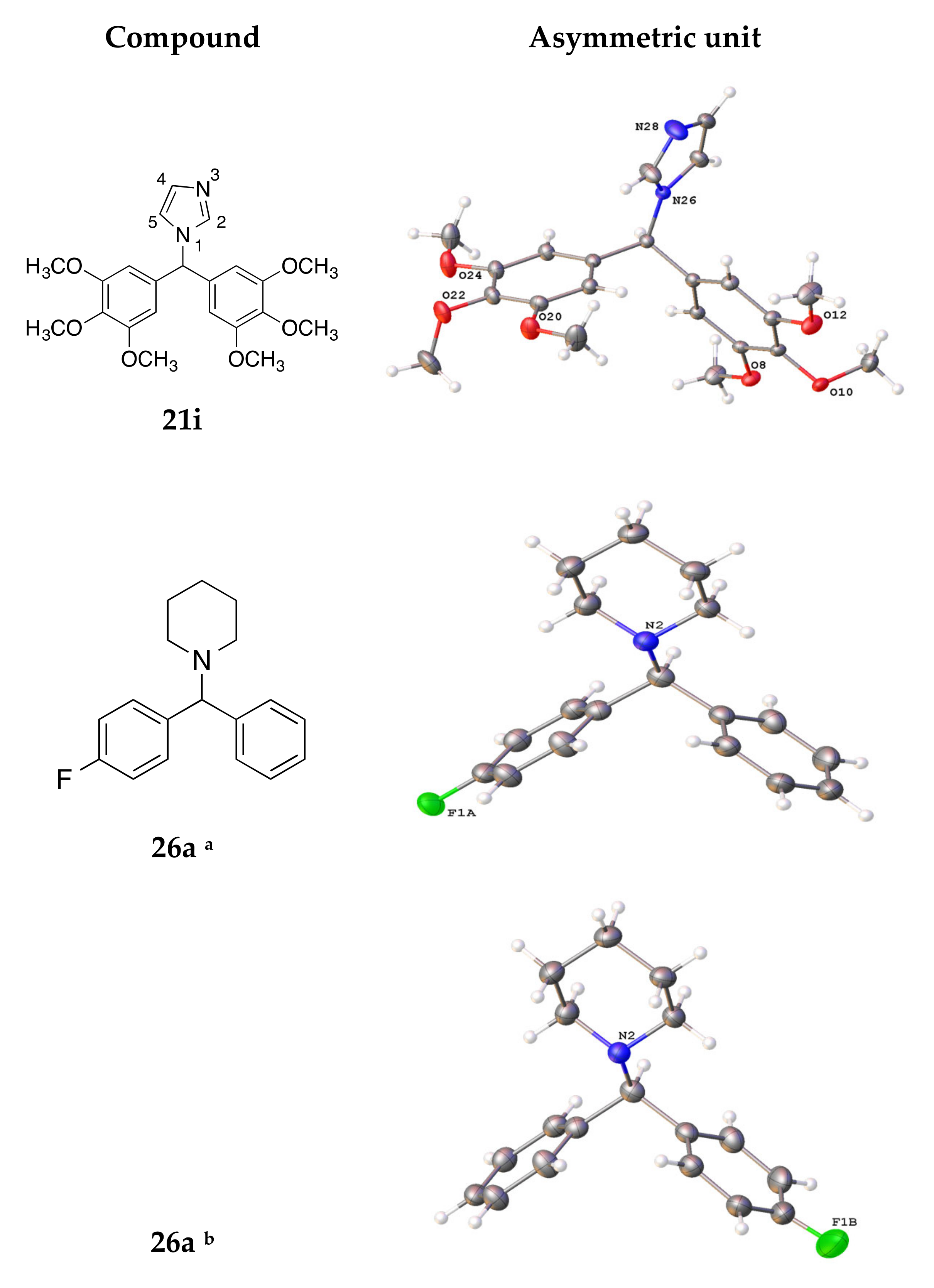
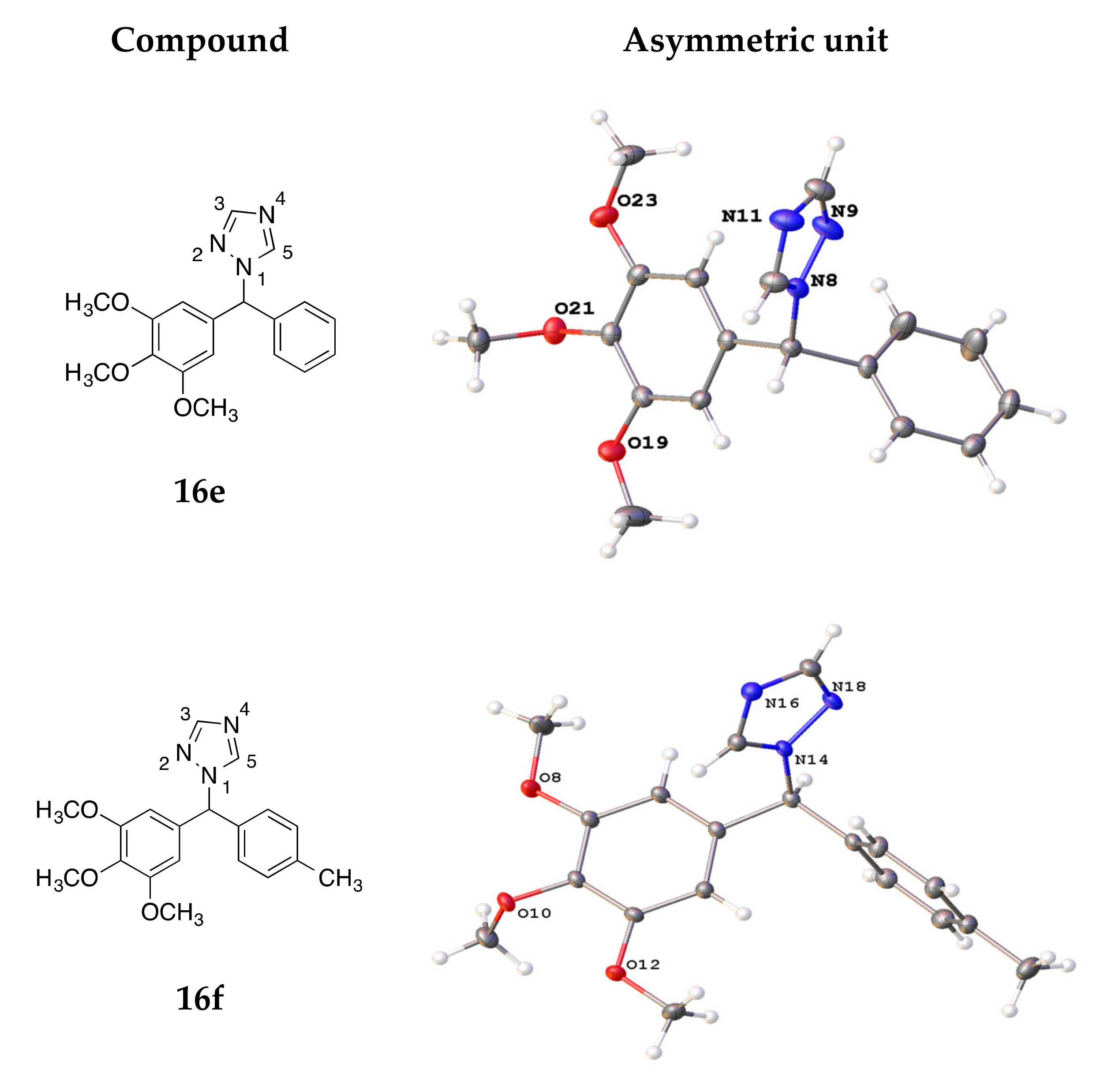
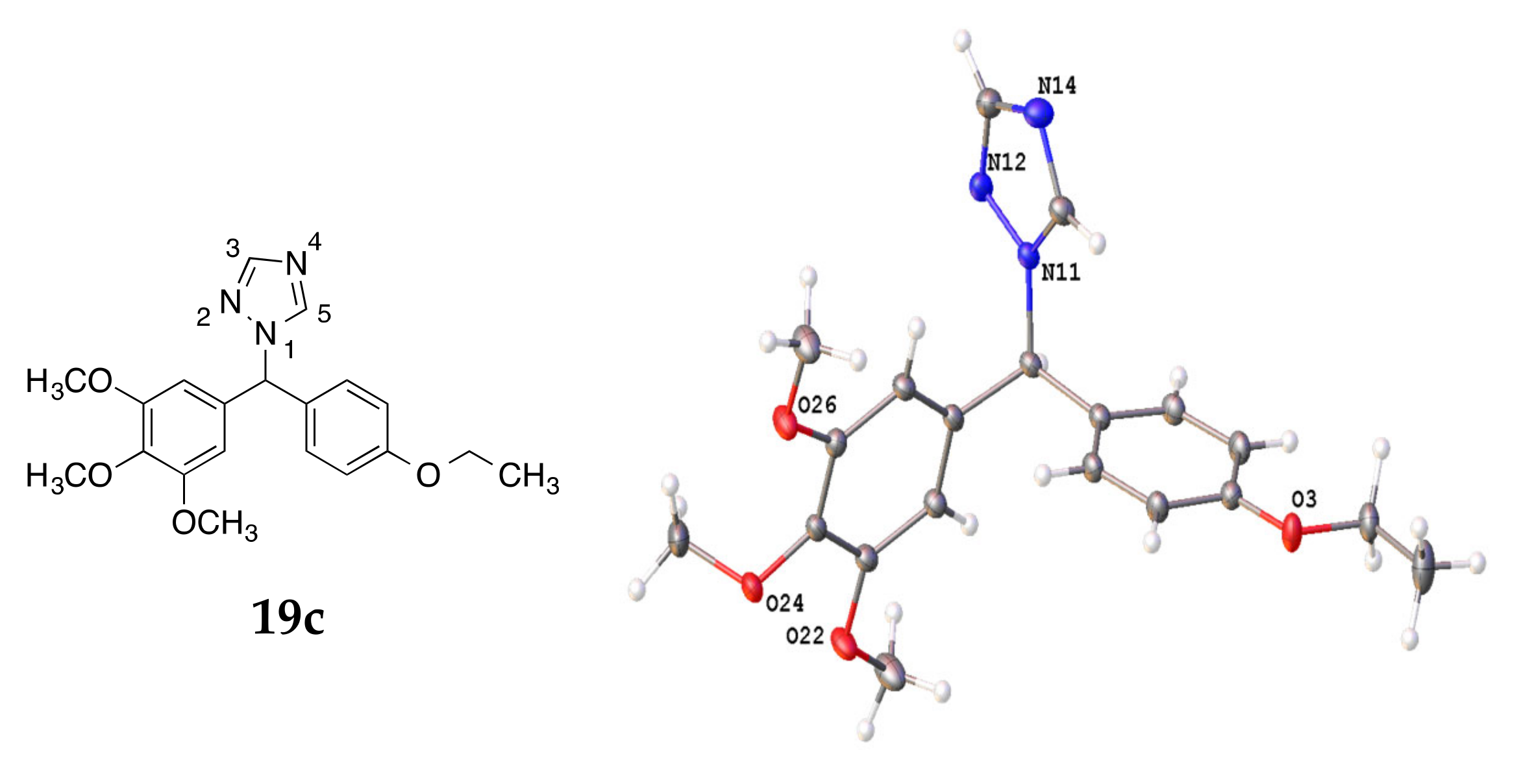
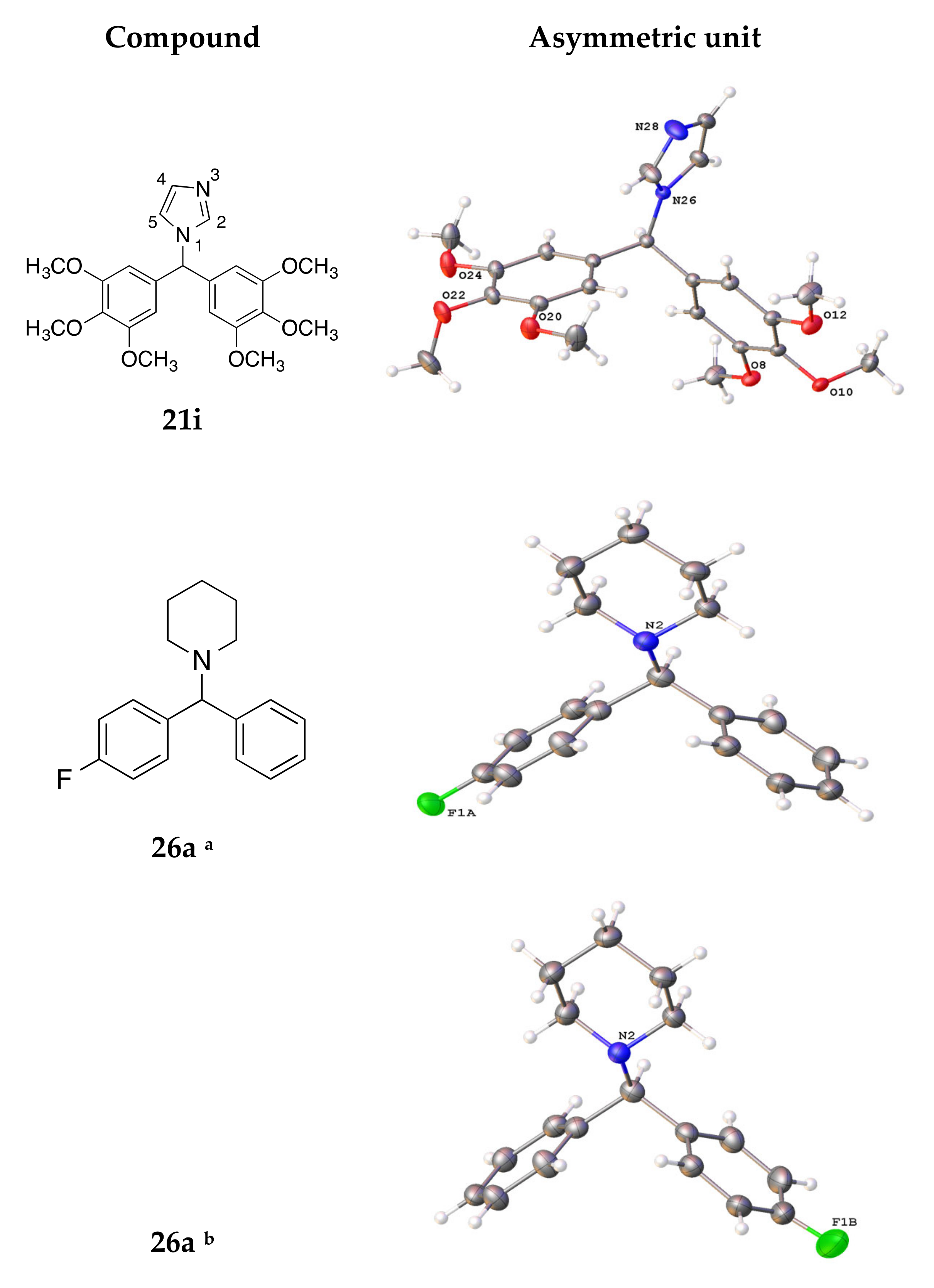
4.3. X-ray Crystallography
4.4. Biochemical Evaluation of Activity
4.4.1. Cell Culture
4.4.2. Cell Viability Assay
4.4.3. Cell Cycle Analysis
4.4.4. Annexin V/PI Apoptotic Assay
4.4.5. Immunofluorescence Microscopy
4.4.6. Evaluation of Expression Levels of Anti-Apoptotic Proteins Mcl-1, Bcl-2 and PARP Cleavage
4.4.7. Tubulin Polymerisation Assay
4.4.8. Cytochrome P450 Assays (CYP19 (Aromatase) and CYP1A1)
4.5. Molecular Modelling and Docking Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AI | Aromatase inhibitor |
| ADC | Antibody–drug conjugate |
| ATR | Attenuated total reflection |
| CDI | 1,1’-Carbonyldiimidazole |
| DEPT | Distortionless Enhancement by Polarization Transfer |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl sulfoxide |
| ECACC | European Collection of Animal Cell Cultures |
| EGFR | Epidermal growth factor receptor |
| ER | Estrogen receptor |
| FACS | Fluorescence activated cell sorting |
| FBS | Foetal bovine serum |
| GI50 | 50% Growth inhibitory concentration |
| HER2 | Human epidermal growth factor receptor 2 |
| HER/neu | Receptor tyrosine-protein kinase erbB-2, CD340 |
| HDBC | Hormone-dependent breast cancer |
| HR | Hormone receptor |
| LC50 | Median lethal concentration |
| MBC | Metastatic breast cancer |
| MDR | Multidrug resistance |
| MEM | Minimum essential media |
| NCI | National Cancer Institute |
| NMR | Nuclear magnetic resonance |
| PARP | Poly (ADP-ribose) polymerase |
| PBS | Phosphate buffered saline |
| PI | Propidium iodide |
| PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha |
| PR | Progesterone receptor |
| RIPA | Radioimmunoprecipitation assay |
| SERM | Selective estrogen receptor modulator |
| STS | Steroid sulfatase |
| TGI | Total growth inhibitory concentration |
| TLC | Thin layer chromatography |
| TNBC | Triple negative breast cancer |
References
- O’Boyle, N.M.; Meegan, M.J. Designed multiple ligands for cancer therapy. Curr. Med. Chem. 2011, 18, 4722–4737. [Google Scholar] [PubMed]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Gediya, L.K.; Njar, V.C.O. Promise and challenges in drug discovery and development of hybrid anticancer drugs. Expert Opin. Drug Dis. 2009, 4, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Fortin, S.; Berube, G. Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2013, 8, 1029–1047. [Google Scholar] [CrossRef]
- Ji, X.; Lu, Y.; Tian, H.; Meng, X.; Wei, M.; Cho, W.C. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed. Pharm. 2019, 114, 108800. [Google Scholar] [CrossRef] [PubMed]
- Jelovac, D.; Macedo, L.; Goloubeva, O.G.; Handratta, V.; Brodie, A.M. Additive antitumor effect of aromatase inhibitor letrozole and antiestrogen fulvestrant in a postmenopausal breast cancer model. Cancer Res. 2005, 65, 5439–5444. [Google Scholar] [CrossRef] [Green Version]
- Howell, A.; Cuzick, J.; Baum, M.; Buzdar, A.; Dowsett, M.; Forbes, J.F.; Hoctin-Boes, G.; Houghton, J.; Locker, G.Y.; Tobias, J.S.; et al. Results of the ATAC (arimidex, tamoxifen, alone or in combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet 2005, 365, 60–62. [Google Scholar]
- American Cancer Society. Breast Cancer Facts & Figures 2019–2020; American Cancer Society Inc.: Atlanta, GA, USA, 2019. [Google Scholar]
- World Cancer Research Fund, Breast Cancer Statistics. Available online: https://www.Wcrf.Org/dietandcancer/cancer-trends/breast-cancer-statistics (accessed on 4 November 2020).
- Cancer Trends 37—Breast Cancer, 1994–2016. Available online: https://www.Ncri.Ie/publications/cancer-trends-and-projections/cancer-trends-37-breast-cancer-1994-2016 (accessed on 4 November 2020).
- Cancer Incidence Projections for Ireland, 2020–2045. Available online: https://www.Ncri.Ie/publications/cancer-trends-and-projections/cancer-incidence-projections-ireland-2020-2045 (accessed on 4 November 2020).
- Early Breast Cancer Trialists’ Collaborative Group. Tamoxifen for early breast cancer: An overview of the randomised trials. Lancet 1998, 351, 1451–1467. [Google Scholar] [CrossRef]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates eralpha action in breast cancer. Nature 2015, 523, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, J.; Naftolin, F. Aromatase: Contributions to physiology and disease in women and men. Physiology 2016, 31, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Spinello, A.; Ritacco, I.; Magistrato, A. Recent advances in computational design of potent aromatase inhibitors: Open-eye on endocrine-resistant breast cancers. Expert Opin. Drug Discov. 2019, 14, 1065–1076. [Google Scholar] [CrossRef]
- Khodarahmi, G.; Asadi, P.; Farrokhpour, H.; Hassanzadeh, F.; Dinari, M. Design of novel potential aromatase inhibitors via hybrid pharmacophore approach: Docking improvement using the qm/mm method. RSC Adv. 2015, 5, 58055–58064. [Google Scholar] [CrossRef]
- Bhatnagar, A.S. The discovery and mechanism of action of letrozole. Breast Cancer Res. Treat. 2007, 105 (Suppl. 1), 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, C.J. The what, why and how of aromatase inhibitors: Hormonal agents for treatment and prevention of breast cancer. Int. J. Clin. Pract. 2007, 61, 2051–2063. [Google Scholar] [CrossRef] [Green Version]
- Deeks, E.D.; Scott, L.J. Exemestane: A review of its use in postmenopausal women with breast cancer. Drugs 2009, 69, 889–918. [Google Scholar] [CrossRef] [PubMed]
- Kumler, I.; Knoop, A.S.; Jessing, C.A.; Ejlertsen, B.; Nielsen, D.L. Review of hormone-based treatments in postmenopausal patients with advanced breast cancer focusing on aromatase inhibitors and fulvestrant. ESMO Open 2016, 1, e000062. [Google Scholar] [CrossRef] [Green Version]
- Pistelli, M.; Mora, A.D.; Ballatore, Z.; Berardi, R. Aromatase inhibitors in premenopausal women with breast cancer: The state of the art and future prospects. Curr. Oncol. 2018, 25, e168–e175. [Google Scholar] [CrossRef] [Green Version]
- Needleman, S.J.; Tobias, J.S. Review of the ATAC study: Tamoxifen versus anastrozole in early-stage breast cancer. Expert Rev. Anticancer Ther. 2008, 8, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.R.; Larionov, A.A. Understanding the mechanisms of aromatase inhibitor resistance. Breast Cancer Res. 2012, 14, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming endocrine resistance in breast cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.; Coombes, R.C. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2002, 2, 101–112. [Google Scholar] [CrossRef]
- Fleming, C.A.; Heneghan, H.M.; O’Brien, D.; McCartan, D.P.; McDermott, E.W.; Prichard, R.S. Meta-analysis of the cumulative risk of endometrial malignancy and systematic review of endometrial surveillance in extended tamoxifen therapy. Br. J. Surg. 2018, 105, 1098–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, M.P.; Suman, V.J.; Reid, J.M.; Northfelt, D.W.; Mahr, M.A.; Ralya, A.T.; Kuffel, M.; Buhrow, S.A.; Safgren, S.L.; McGovern, R.M.; et al. First-in-human phase I study of the tamoxifen metabolite z-endoxifen in women with endocrine-refractory metastatic breast cancer. J. Clin. Oncol. 2017, 35, 3391–3400. [Google Scholar] [CrossRef] [PubMed]
- Sestak, I. Preventative therapies for healthy women at high risk of breast cancer. Cancer Manag. Res. 2014, 6, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Cuzick, J.; Sestak, I.; Cawthorn, S.; Hamed, H.; Holli, K.; Howell, A.; Forbes, J.F.; Investigators, I.-I. Tamoxifen for prevention of breast cancer: Extended long-term follow-up of the ibis-i breast cancer prevention trial. Lancet Oncol. 2015, 16, 67–75. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Czerniecki, B.J. Clinical development of immunotherapies for her2(+) breast cancer: A review of HER2-directed monoclonal antibodies and beyond. NPJ Breast Cancer 2020, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Li, B.T.; Shen, R.; Buonocore, D.; Olah, Z.T.; Ni, A.; Ginsberg, M.S.; Ulaner, G.A.; Offin, M.; Feldman, D.; Hembrough, T.; et al. Ado-trastuzumab emtansine for patients with her2-mutant lung cancers: Results from a phase ii basket trial. J. Clin. Oncol. 2018, 36, 2532–2537. [Google Scholar] [CrossRef]
- Tsang, R.Y.; Sadeghi, S.; Finn, R.S. Lapatinib, a dual-targeted small molecule inhibitor of egfr and her2, in her2-amplified breast cancer: From bench to bedside. Clin. Med. Insights Ther. 2011, 3, 1–13. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Jubair, S.A.; Alkhateeb, A.; Tabl, A.A.; Rueda, L.; Ngom, A. A novel approach to identify subtype-specific network biomarkers of breast cancer survivability. Netw. Modeling Anal. Health Inform. Bioinform. 2020, 9, 43. [Google Scholar] [CrossRef]
- Iwata, T.N.; Ishii, C.; Ishida, S.; Ogitani, Y.; Wada, T.; Agatsuma, T. A HER2-targeting antibody-drug conjugate, trastuzumab deruxtecan (ds-8201a), enhances antitumor immunity in a mouse model. Mol. Cancer 2018, 17, 1494–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enhertu (Trastuzumab Deruxtecan) Approved in the US for HER2-Positive Unresectable or Metastatic Breast Cancer Following Two or More Prior Anti-HER2 Based Regimens. Available online: https://www.astrazeneca.com/media-centre/press-releases/2019/enhertu-trastuzumab-deruxtecan-approved-in-the-us-for-her2-positive-unresectable-or-metastatic-breast-cancer-following-2-or-more-prior-anti-her2-based-regimens.html (accessed on 4 November 2020).
- FDA Approves Alpelisib for Metastatic Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-alpelisib-metastatic-breast-cancer (accessed on 4 November 2020).
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for pik3ca-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- A Study of Tucatinib, vs. Placebo in Combination with Capecitabine & Trastuzumab in Patients with Advanced her2+ Breast Cancer (her2climb). Available online: https://www.Clinicaltrials.Gov/ct2/show/nct02614794 (accessed on 4 November 2020).
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Reviews Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Van Vuuren, R.J.; Visagie, M.H.; Theron, A.E.; Joubert, A.M. Antimitotic drugs in the treatment of cancer. Cancer Chemother. Pharm. 2015, 76, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- FDA Grants Accelerated Approval to Sacituzumab Govitecan-Hziy for Metastatic Triple Negative Breast Cancer. Available online: https://www.Fda.Gov/drugs/drug-approvals-and-databases/fda-grants-accelerated-approval-sacituzumab-govitecan-hziy-metastatic-triple-negative-breast-cancer (accessed on 4 November 2020).
- Safety and Efficacy of Sgn-Liv1a Plus Pembrolizumab for Patients with Locally-Advanced or Metastatic Triple-Negative Breast Cancer. Available online: https://www.Clinicaltrials.Gov/ct2/show/nct03310957 (accessed on 4 November 2020).
- Potter, B.V.L. Sulfation pathways: Steroid sulphatase inhibition via aryl sulphamates: Clinical progress, mechanism and future prospects. J. Mol. Endocrinol. 2018, 61, T233–T252. [Google Scholar] [CrossRef] [Green Version]
- Synnott, N.C.; Murray, A.; McGowan, P.M.; Kiely, M.; Kiely, P.A.; O’Donovan, N.; O’Connor, D.P.; Gallagher, W.M.; Crown, J.; Duffy, M.J. Mutant p53: A novel target for the treatment of patients with triple-negative breast cancer? Int. J. Cancer 2017, 140, 234–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettit, G.R.; Singh, S.B.; Boyd, M.R.; Hamel, E.; Pettit, R.K.; Schmidt, J.M.; Hogan, F. Antineoplastic agents. 291. Isolation and synthesis of combretastatins A-4, A-5, and A-6(1A). J. Med. Chem. 1995, 38, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Perez-Perez, M.J.; Priego, E.M.; Bueno, O.; Martins, M.S.; Canela, M.D.; Liekens, S. Blocking blood flow to solid tumors by destabilizing tubulin: An approach to targeting tumor growth. J. Med. Chem. 2016, 59, 8685–8711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, M.; Huang, J.; Liu, S.; Xiao, Y.; Qin, X.; Liu, J.; Pi, C.; Luo, T.; Li, J.; Chen, X.; et al. The anti-angiogenic effect and novel mechanisms of action of combretastatin A-4. Sci. Rep. 2016, 6, 28139. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [Green Version]
- McLoughlin, E.C.; O’Boyle, N.M. Colchicine-binding site inhibitors from chemistry to clinic: A review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Greene, L.M.; Meegan, M.J.; Zisterer, D.M. Combretastatins: More than just vascular targeting agents? J. Pharmacol. Exp. Ther. 2015, 355, 212–227. [Google Scholar] [CrossRef]
- Gaspari, R.; Prota, A.E.; Bargsten, K.; Cavalli, A.; Steinmetz, M.O. Structural basis of cis- and trans-combretastatin binding to tubulin. Chem 2017, 2, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Pettit, G.R.; Toki, B.E.; Herald, D.L.; Boyd, M.R.; Hamel, E.; Pettit, R.K.; Chapuis, J.C. Antineoplastic agents. 410. Asymmetric hydroxylation of trans-combretastatin a-4. J. Med. Chem. 1999, 42, 1459–1465. [Google Scholar] [CrossRef]
- Malebari, A.M.; Fayne, D.; Nathwani, S.M.; O’Connell, F.; Noorani, S.; Twamley, B.; O’Boyle, N.M.; O’Sullivan, J.; Zisterer, D.M.; Meegan, M.J. Beta-lactams with antiproliferative and antiapoptotic activity in breast and chemoresistant colon cancer cells. Eur. J. Med. Chem. 2020, 189, 112050. [Google Scholar] [CrossRef] [PubMed]
- Odlo, K.; Hentzen, J.; dit Chabert, J.F.; Ducki, S.; Gani, O.A.; Sylte, I.; Skrede, M.; Florenes, V.A.; Hansen, T.V. 1,5-disubstituted 1,2,3-triazoles as cis-restricted analogues of combretastatin A-4: Synthesis, molecular modeling and evaluation as cytotoxic agents and inhibitors of tubulin. Bioorganic Med. Chem. 2008, 16, 4829–4838. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.; Anwar, S.; Elgamal, F.; Ahmed, E.R.; Aly, O.M. Potent combretastatin A-4 analogs containing 1,2,4-triazole: Synthesis, antiproliferative, anti-tubulin activity, and docking study. Eur. J. Med. Chem. 2019, 183, 111697. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Prencipe, F.; Oliva, P.; Baraldi, S.; Tabrizi, M.A.; Lopez-Cara, L.C.; Ferla, S.; Brancale, A.; Hamel, E.; et al. Design and synthesis of potent in vitro and in vivo anticancer agents based on 1-(3’,4’,5’-trimethoxyphenyl)-2-aryl-1h-imidazole. Sci. Rep. 2016, 6, 26602. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Xu, F.; Shuai, W.; Sun, H.; Yao, H.; Ma, C.; Xu, S.; Yao, H.; Zhu, Z.; Yang, D.H.; et al. Discovery of novel quinoline-chalcone derivatives as potent antitumor agents with microtubule polymerization inhibitory activity. J. Med. Chem. 2019, 62, 993–1013. [Google Scholar] [CrossRef]
- He, J.; Zhang, M.; Tang, L.; Liu, J.; Zhong, J.; Wang, W.; Xu, J.P.; Wang, H.T.; Li, X.F.; Zhou, Z.Z. Synthesis, biological evaluation, and molecular docking of arylpyridines as antiproliferative agent targeting tubulin. ACS Med. Chem. Lett. 2020, 11, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, S.; Treguier, B.; Hamze, A.; Provot, O.; Peyrat, J.F.; De Losada, J.R.; Liu, J.M.; Bignon, J.; Wdzieczak-Bakala, J.; Thoret, S.; et al. Isocombretastatins a versus combretastatins a: The forgotten isoca-4 isomer as a highly promising cytotoxic and antitubulin agent. J. Med. Chem. 2009, 52, 4538–4542. [Google Scholar] [CrossRef]
- La Regina, G.; Bai, R.; Rensen, W.M.; Di Cesare, E.; Coluccia, A.; Piscitelli, F.; Famiglini, V.; Reggio, A.; Nalli, M.; Pelliccia, S.; et al. Toward highly potent cancer agents by modulating the c-2 group of the arylthioindole class of tubulin polymerization inhibitors. J. Med. Chem. 2013, 56, 123–149. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.B.; Nayak, V.L.; Sayeed, I.B.; Reddy, V.S.; Shaik, A.B.; Mahesh, R.; Baig, M.F.; Shareef, M.A.; Ravikumar, A.; Kamal, A. Design, synthesis of phenstatin/isocombretastatin-oxindole conjugates as antimitotic agents. Bioorganic Med. Chem. 2016, 24, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Naret, T.; Khelifi, I.; Provot, O.; Bignon, J.; Levaique, H.; Dubois, J.; Souce, M.; Kasselouri, A.; Deroussent, A.; Paci, A.; et al. 1,1-diheterocyclic ethylenes derived from quinaldine and carbazole as new tubulin-polymerization inhibitors: Synthesis, metabolism, and biological evaluation. J. Med. Chem. 2019, 62, 1902–1916. [Google Scholar] [CrossRef]
- Pettit, G.R.; Toki, B.; Herald, D.L.; Verdier-Pinard, P.; Boyd, M.R.; Hamel, E.; Pettit, R.K. Antineoplastic agents. 379. Synthesis of phenstatin phosphate. J. Med. Chem. 1998, 41, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Ghinet, A.; Rigo, B.; Henichart, J.P.; Le Broc-Ryckewaert, D.; Pommery, J.; Pommery, N.; Thuru, X.; Quesnel, B.; Gautret, P. Synthesis and biological evaluation of phenstatin metabolites. Bioorganic Med. Chem. 2011, 19, 6042–6054. [Google Scholar] [CrossRef]
- Cascioferro, S.; Attanzio, A.; Di Sarno, V.; Musella, S.; Tesoriere, L.; Cirrincione, G.; Diana, P.; Parrino, B. New 1,2,4-oxadiazole nortopsentin derivatives with cytotoxic activity. Mar. Drugs 2019, 17, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, D.; Parrino, B.; Cascioferro, S.; Pecoraro, C.; Giovannetti, E.; Di Sarno, V.; Musella, S.; Auriemma, G.; Cirrincione, G.; Diana, P. 1,2,4-oxadiazole topsentin analogs with antiproliferative activity against pancreatic cancer cells, targeting GSK3b kinase. ChemMedChem 2021, 16, 537–554. [Google Scholar] [CrossRef]
- Cascioferro, S.; Petri, G.L.; Parrino, B.; Carbone, D.; Funel, N.; Bergonzini, C.; Mantini, G.; Dekker, H.; Geerke, D.; Peters, G.J.; et al. Imidazo[2,1-b] [1,3,4]thiadiazoles with antiproliferative activity against primary and gemcitabine-resistant pancreatic cancer cells. Eur. J. Med. Chem. 2020, 189, 112088. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Chuang, H.Y.; Lee, H.Y.; Yeh, T.K.; Kuo, C.C.; Chang, C.Y.; Chang, J.Y.; Liou, J.P. Antimitotic and vascular disrupting agents: 2-hydroxy-3,4,5-trimethoxybenzophenones. Eur. J. Med. Chem. 2014, 77, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Brancale, A.; Silvestri, R. Indole, a core nucleus for potent inhibitors of tubulin polymerization. Med. Res. Rev. 2007, 27, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Hadimani, M.B.; Macdonough, M.T.; Ghatak, A.; Strecker, T.E.; Lopez, R.; Sriram, M.; Nguyen, B.L.; Hall, J.J.; Kessler, R.J.; Shirali, A.R.; et al. Synthesis of a 2-aryl-3-aroyl indole salt (Oxi8007) resembling combretastatin A-4 with application as a vascular disrupting agent. J. Nat. Prod. 2013, 76, 1668–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, Y.S.; Coumar, M.S.; Wu, Y.S.; Shiao, H.Y.; Chang, J.Y.; Liou, J.P.; Shukla, P.; Chang, C.W.; Chang, C.Y.; Kuo, C.C.; et al. Scaffold-hopping strategy: Synthesis and biological evaluation of 5,6-fused bicyclic heteroaromatics to identify orally bioavailable anticancer agents. J. Med. Chem. 2011, 54, 3076–3080. [Google Scholar] [CrossRef]
- Chen, J.; Ahn, S.; Wang, J.; Lu, Y.; Dalton, J.T.; Miller, D.D.; Li, W. Discovery of novel 2-aryl-4-benzoyl-imidazole (ABI-III) analogues targeting tubulin polymerization as antiproliferative agents. J. Med. Chem. 2012, 55, 7285–7289. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Arnst, K.E.; Wang, Y.; Kumar, G.; Ma, D.; White, S.W.; Miller, D.D.; Li, W.; Li, W. Structure-guided design, synthesis, and biological evaluation of (2-(1H-indol-3-yl)-1H-imidazol-4-yl)(3,4,5-trimethoxyphenyl) methanone (ABI-231) analogues targeting the colchicine binding site in tubulin. J. Med. Chem. 2019, 62, 6734–6750. [Google Scholar] [CrossRef]
- Bai, Z.; Gao, M.; Zhang, H.; Guan, Q.; Xu, J.; Li, Y.; Qi, H.; Li, Z.; Zuo, D.; Zhang, W.; et al. BZML, a novel colchicine binding site inhibitor, overcomes multidrug resistance in a549/taxol cells by inhibiting P-gp function and inducing mitotic catastrophe. Cancer Lett. 2017, 402, 81–92. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Pollock, J.K.; Carr, M.; Knox, A.J.; Nathwani, S.M.; Wang, S.; Caboni, L.; Zisterer, D.M.; Meegan, M.J. Beta-lactam estrogen receptor antagonists and a dual-targeting estrogen receptor/tubulin ligand. J. Med. Chem. 2014, 57, 9370–9382. [Google Scholar] [CrossRef] [PubMed]
- Knox, A.J.; Price, T.; Pawlak, M.; Golfis, G.; Flood, C.T.; Fayne, D.; Williams, D.C.; Meegan, M.J.; Lloyd, D.G. Integration of ligand and structure-based virtual screening for the identification of the first dual targeting agent for heat shock protein 90 (Hsp90) and tubulin. J. Med. Chem. 2009, 52, 2177–2180. [Google Scholar] [CrossRef]
- Zhong, B.; Chennamaneni, S.; Lama, R.; Yi, X.; Geldenhuys, W.J.; Pink, J.J.; Dowlati, A.; Xu, Y.; Zhou, A.; Su, B. Synthesis and anticancer mechanism investigation of dual Hsp27 and tubulin inhibitors. J. Med. Chem. 2013, 56, 5306–5320. [Google Scholar] [CrossRef] [Green Version]
- Lv, W.; Liu, J.; Skaar, T.C.; Flockhart, D.A.; Cushman, M. Design and synthesis of norendoxifen analogues with dual aromatase inhibitory and estrogen receptor modulatory activities. J. Med. Chem. 2015, 58, 2623–2648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, W.; Liu, J.; Lu, D.; Flockhart, D.A.; Cushman, M. Synthesis of mixed (E,Z)-, (E)-, and (Z)-norendoxifen with dual aromatase inhibitory and estrogen receptor modulatory activities. J. Med. Chem. 2013, 56, 4611–4618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.J.; Desta, Z.; Flockhart, D.A. Tamoxifen metabolites as active inhibitors of aromatase in the treatment of breast cancer. Breast Cancer Res. Treat. 2012, 131, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Woo, L.W.; Bubert, C.; Purohit, A.; Potter, B.V. Hybrid dual aromatase-steroid sulfatase inhibitors with exquisite picomolar inhibitory activity. ACS Med. Chem. Lett. 2011, 2, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohle, W.; Jourdan, F.L.; Menchon, G.; Prota, A.E.; Foster, P.A.; Mannion, P.; Hamel, E.; Thomas, M.P.; Kasprzyk, P.G.; Ferrandis, E.; et al. Quinazolinone-based anticancer agents: Synthesis, antiproliferative sar, antitubulin activity, and tubulin co-crystal structure. J. Med. Chem. 2018, 61, 1031–1044. [Google Scholar] [CrossRef]
- Dohle, W.; Prota, A.E.; Menchon, G.; Hamel, E.; Steinmetz, M.O.; Potter, B.V.L. Tetrahydroisoquinoline sulfamates as potent microtubule disruptors: Synthesis, antiproliferative and antitubulin activity of dichlorobenzyl-based derivatives, and a tubulin cocrystal structure. ACS Omega 2019, 4, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Gangjee, A.; Pavana, R.K.; Ihnat, M.A.; Thorpe, J.E.; Disch, B.C.; Bastian, A.; Bailey-Downs, L.C.; Hamel, E.; Bai, R. Discovery of antitubulin agents with antiangiogenic activity as single entities with multitarget chemotherapy potential. ACS Med. Chem. Lett. 2014, 5, 480–484. [Google Scholar] [CrossRef]
- Doiron, J.; Soultan, A.H.; Richard, R.; Toure, M.M.; Picot, N.; Richard, R.; Cuperlovic-Culf, M.; Robichaud, G.A.; Touaibia, M. Synthesis and structure-activity relationship of 1- and 2-substituted-1,2,3-triazole letrozole-based analogues as aromatase inhibitors. Eur. J. Med. Chem. 2011, 46, 4010–4024. [Google Scholar] [CrossRef]
- Wood, P.M.; Woo, L.W.; Labrosse, J.R.; Trusselle, M.N.; Abbate, S.; Longhi, G.; Castiglioni, E.; Lebon, F.; Purohit, A.; Reed, M.J.; et al. Chiral aromatase and dual aromatase-steroid sulfatase inhibitors from the letrozole template: Synthesis, absolute configuration, and in vitro activity. J. Med. Chem. 2008, 51, 4226–4238. [Google Scholar] [CrossRef]
- Negi, A.S.; Gautam, Y.; Alam, S.; Chanda, D.; Luqman, S.; Sarkar, J.; Khan, F.; Konwar, R. Natural antitubulin agents: Importance of 3,4,5-trimethoxyphenyl fragment. Bioorganic Med. Chem. 2015, 23, 373–389. [Google Scholar] [CrossRef]
- Misawa, T.; Aoyama, H.; Furuyama, T.; Dodo, K.; Sagawa, M.; Miyachi, H.; Kizaki, M.; Hashimoto, Y. Structural development of benzhydrol-type 1′-acetoxychavicol acetate (aca) analogs as human leukemia cell-growth inhibitors based on quantitative structure-activity relationship (QSAR) analysis. Chem. Pharm Bull. 2008, 56, 1490–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.B.; Bei, F.L.; Li, R.Y.; Yang, X.J.; Wang, X. Synthesis, characterization and single crystal structure of 4,4′-(1H-1,2,4-triazol-1-methylene)-bisbenzonitrile. Chin. J. Org. Chem. 2004, 24, 550. [Google Scholar]
- Plobeck, N.; Delorme, D.; Wei, Z.Y.; Yang, H.; Zhou, F.; Schwarz, P.; Gawell, L.; Gagnon, H.; Pelcman, B.; Schmidt, R.; et al. New diarylmethylpiperazines as potent and selective nonpeptidic delta opioid receptor agonists with increased in vitro metabolic stability. J. Med. Chem. 2000, 43, 3878–3894. [Google Scholar] [CrossRef]
- Tang, Y.; Dong, Y.; Vennerstrom, J.L. The reaction of carbonyldiimidazole with alcohols to form carbamates and N-alkylimidazoles. Synthesis 2004, 15, 2540–2544. [Google Scholar] [CrossRef]
- Ohta, S.; Kawasaki, I.; Uemura, T.; Yamashita, M.; Yoshioka, T.; Yamaguchi, S. Alkylation and acylation of the 1,2,3-triazole ring. Chem. Pharm. Bull. 1997, 45, 1140–1145. [Google Scholar] [CrossRef]
- Belskaya, N.P.; Subbotina, J.; Lesogorova, S. Synthesis of 2H-1,2,3-Triazoles; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Martins, P.; Jesus, J.; Santos, S.; Raposo, L.R.; Roma-Rodrigues, C.; Baptista, P.V.; Fernandes, A.R. Heterocyclic anticancer compounds: Recent advances and the paradigm shift towards the use of nanomedicine’s tool box. Molecules 2015, 20, 16852–16891. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Sakai, N.; Hori, H.; Yoshida, Y.; Konakahara, T.; Ogiwara, Y. Copper(I)-catalyzed coupling reaction of aryl boronic acids with N,O-acetals and N,N-aminals under atmosphere leading to alpha-aryl glycine derivatives and diarylmethylamine derivatives. Tetrahedron 2015, 71, 4722–4729. [Google Scholar] [CrossRef]
- LeGall, E.; Troupel, M.; Nedelec, J. One-step three-component coupling of aromatic organozinc reagents, secondary amines, and aromatic aldehydes into functionalized diarylmethylamines. Tetrahedron 2006, 62, 9953–9965. [Google Scholar] [CrossRef]
- Sengmany, S.L.; LeGall, E.; LeJean, C.; Troupel, M.; Nedelec, J. Straightforward three-component synthesis of diarylmethylpiperazines and 1,2-diarylethylpiperazines. Tetrahedron 2007, 63, 3672–3681. [Google Scholar] [CrossRef]
- Malebari, A.M.; Greene, L.M.; Nathwani, S.M.; Fayne, D.; O’Boyle, N.M.; Wang, S.; Twamley, B.; Zisterer, D.M.; Meegan, M.J. Beta-lactam analogues of combretastatin A-4 prevent metabolic inactivation by glucuronidation in chemoresistant HT-29 colon cancer cells. Eur. J. Med. Chem. 2017, 130, 261–285. [Google Scholar] [CrossRef]
- Cushman, M.; Nagarathnam, D.; Gopal, D.; He, H.M.; Lin, C.M.; Hamel, E. Synthesis and evaluation of analogues of (z)-1-(4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)ethene as potential cytotoxic and antimitotic agents. J. Med. Chem. 1992, 35, 2293–2306. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, E.G.; Bega, L.A.; Beatriz, A.; Sarkar, T.; Hamel, E.; do Amaral, M.S.; de Lima, D.P. A diaryl sulfide, sulfoxide, and sulfone bearing structural similarities to combretastatin a-4. Eur. J. Med. Chem. 2009, 44, 2685–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, A.; Kumar, G.B.; Vishnuvardhan, M.V.; Shaik, A.B.; Reddy, V.S.; Mahesh, R.; Sayeeda, I.B.; Kapure, J.S. Synthesis of phenstatin/isocombretastatin-chalcone conjugates as potent tubulin polymerization inhibitors and mitochondrial apoptotic inducers. Org. Biomol. Chem. 2015, 13, 3963–3981. [Google Scholar] [CrossRef]
- Alvarez, R.; Alvarez, C.; Mollinedo, F.; Sierra, B.G.; Medarde, M.; Pelaez, R. Isocombretastatins A: 1,1-diarylethenes as potent inhibitors of tubulin polymerization and cytotoxic compounds. Bioorganic Med. Chem. 2009, 17, 6422–6431. [Google Scholar] [CrossRef]
- National Cancer Institute. DCTD Division of Cancer Treatment and Diagnostics, DTP Development Therapeutics Programme; National Cancer Institute: Bethesda, MD, USA; Available online: https://dtp.cancer.gov/organization/btb/default.htm (accessed on 20 February 2020).
- Qu, Y.; Han, B.; Yu, Y.; Yao, W.; Bose, S.; Karlan, B.Y.; Giuliano, A.E.; Cui, X. Evaluation of MCF10a as a reliable model for normal human mammary epithelial cells. PLoS ONE 2015, 10, e0131285. [Google Scholar] [CrossRef] [Green Version]
- Visconti, R.; Grieco, D. Fighting tubulin-targeting anticancer drug toxicity and resistance. Endocr. Relat. Cancer 2017, 24, T107–T117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mc Gee, M.M. Targeting the mitotic catastrophe signaling pathway in cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, I.; Antoccia, A.; Cenciarelli, C.; Crateri, P.; Meschini, S.; Arancia, G.; Pisano, C.; Tanzarella, C. Combretastatin CA-4 and combretastatin derivative induce mitotic catastrophe dependent on spindle checkpoint and caspase-3 activation in non-small cell lung cancer cells. Apoptosis Int. J. Program. Cell Death 2007, 12, 155–166. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Ana, G.; Kelly, P.M.; Nathwani, S.M.; Noorani, S.; Fayne, D.; Bright, S.A.; Twamley, B.; Zisterer, D.M.; Meegan, M.J. Synthesis and evaluation of antiproliferative microtubule-destabilising combretastatin A-4 piperazine conjugates. Org. Biomol. Chem. 2019, 17, 6184–6200. [Google Scholar] [CrossRef]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y. Role of BCL-2 family proteins in apoptosis: Apoptosomes or mitochondria? Genes Cells 1998, 3, 697–707. [Google Scholar] [CrossRef]
- Michels, J.; Johnson, P.W.; Packham, G. Mcl-1. Int. J. Biochem. Cell Biol. 2005, 37, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Beumer, J.H.; Hamieh, L.; Tawbi, H. Role of PARP inhibitors in cancer biology and therapy. Curr. Med. Chem. 2012, 19, 3907–3921. [Google Scholar] [CrossRef]
- Stresser, D.M.; Turner, S.D.; McNamara, J.; Stocker, P.; Miller, V.P.; Crespi, C.L.; Patten, C.J. A high-throughput screen to identify inhibitors of aromatase (CYP19). Anal. Biochem. 2000, 284, 427–430. [Google Scholar] [CrossRef]
- Maiti, A.; Cuendet, M.; Croy, V.L.; Endringer, D.C.; Pezzuto, J.M.; Cushman, M. Synthesis and biological evaluation of (+/-)-abyssinone II and its analogues as aromatase inhibitors for chemoprevention of breast cancer. J. Med. Chem. 2007, 50, 2799–2806. [Google Scholar] [CrossRef]
- Endringer, D.C.; Guimaraes, K.G.; Kondratyuk, T.P.; Pezzuto, J.M.; Braga, F.C. Selective inhibition of aromatase by a dihydroisocoumarin from xyris pterygoblephara. J. Nat. Prod. 2008, 71, 1082–1084. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Shin, Y.G.; Kosmeder, J.W.; Pezzuto, J.M.; van Breemen, R.B. Liquid chromatography/tandem mass spectrometric determination of inhibition of human cytochrome p450 isozymes by resveratrol and resveratrol-3-sulfate. Rapid Commun. Mass Spectrom. 2003, 17, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, R.B.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Tafi, A.; Anastassopoulou, J.; Theophanides, T.; Botta, M.; Corelli, F.; Massa, S.; Artico, M.; Costi, R.; Di Santo, R.; Ragno, R. Molecular modeling of azole antifungal agents active against Candida albicans. 1. A comparative molecular field analysis study. J. Med. Chem. 1996, 39, 1227–1235. [Google Scholar] [CrossRef]
- Bruker AXS Inc. Bruker APEX v2014; Bruker AXS Inc.: Madison, WI, USA, 2014. [Google Scholar]
- SADABS. Area Detector Absorption Correction Program, Sheldrick, G.M. University of Göttingen, Germany. 2014. Available online: https://journals.iucr.org/e/services/stdswrefs.html; (accessed on 20 February 2021).
- Sheldrick, G.M. Shelxt-integrated space-group and crystal-structure determination. Acta Cryst. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with Shelxl. Acta Cryst. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Cytoskeleton. Available online: https://www.Cytoskeleton.Com/tubulin-resources (accessed on 1 December 2020).
- Chemical Computing Group Inc. Molecular Operating Environment (MOE), 2015.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2015. [Google Scholar]
- Kendall, A.; Dowsett, M. Novel concepts for the chemoprevention of breast cancer through aromatase inhibition. Endocr. Relat. Cancer 2006, 13, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 16e | 16f | 19c | 21i | 26a |
|---|---|---|---|---|---|
| CCDC no. | 2015431 | 2015432 | 2015433 | 2015434 | 2015435 |
| Empirical formula | C18H19N3O3 | C19H21N3O3 | C20H23N3O4 | C22H26N2O6 | C18H20FN |
| M (g/mol) | 325.36 | 339.39 | 369.41 | 414.45 | 269.35 |
| T (K) | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) |
| Crystal System | monoclinic | monoclinic | monoclinic | monoclinic | monoclinic |
| SG | P21/n | P21/c | P21/c | P21/c | P21/c |
| a (Å) | 8.1796(5) | 9.2487(4) | 12.8748(6) | 14.1937(6) | 5.9228(2) |
| b (Å) | 14.1725(10) | 9.6620(4) | 9.8550(5) | 13.1553(5) | 14.0705(5) |
| c (Å) | 14.4742(10) | 20.0006(9) | 14.8091(7) | 12.4613(5) | 17.7920(6) |
| α (°) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 98.7882(11) | 97.8110(10) | 93.1792(8) | 113.2250(10) | 98.275(2) |
| γ (°) | 90 | 90 | 90 | 90 | 90 |
| V (Å3) | 1658.23(19) | 1770.69(13) | 1876.10(16) | 2138.25(15) | 1467.29(9) |
| Z | 4 | 4 | 4 | 4 | 4 |
| Dcalc (g/cm3) | 1.303 | 1.273 | 1.308 | 1.287 | 1.219 |
| μ (mm−1) | 0.09 | 0.088 | 0.092 | 0.094 | 0.628 |
| F(000) | 688 | 720 | 784 | 880 | 576 |
| Radiation | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | CuKα (λ = 1.54178) |
| Reflections collected | 61043 | 115661 | 95764 | 85391 | 20214 |
| Independent reflections | 4298 [Rint = 0.0532, Rsigma = 0.0237] | 5438 [Rint = 0.0264, Rsigma = 0.0097] | 5504 [Rint = 0.0364, Rsigma = 0.0143] | 6261 [Rint = 0.0385, Rsigma = 0.0188] | 2684 [Rint = 0.0634, Rsigma = 0.0356] |
| Data/restraints/parameters | 4298/0/217 | 5438/0/230 | 5504/0/248 | 6261/0/277 | 2684/1/191 |
| Goodness-of-fit on F2 (S) | 1.018 | 1.043 | 1.046 | 1.005 | 1.063 |
| Final R indexes [I ≥ 2σ (I)] * | R1 = 0.0407, wR2 = 0.0904 | R1 = 0.0374, wR2 = 0.1018 | R1 = 0.0381, wR2 = 0.0938 | R1 = 0.0405, wR2 = 0.0967 | R1 = 0.0479, wR2 = 0.1325 |
| Final R indexes [all data] | R1 = 0.0613, wR2 = 0.1005 | R1 = 0.0445, wR2 = 0.1083 | R1 = 0.0506, wR2 = 0.1025 | R1 = 0.0580, wR2 = 0.1079 | R1 = 0.0547, wR2 = 0.1369 |
| Largest diff. peak/hole/e Å-3 | 0.31/-0.21 | 0.42/-0.39 | 0.42/-0.21 | 0.38/-0.24 | 0.19/-0.21 |
| Compound | Antiproliferative Activity a MCF-7 Cells IC50 (μM) | Antiproliferative Activity a MDA-MB-231 Cells IC50 (μM) | Antiproliferative Activity a HL-60 Cells IC50 (μM) |
|---|---|---|---|
| 19e | 0.042 ± 0.070 | 0.978 ± 0.130 | 0.261b |
| 21l | 0.132 ± 0.007 | 0.237 ± 0.040 | 0.156 ± 0.052 |
| 24 | 0.052 ± 0.040 | 0.074 ± 0.030 | 0.173 ± 0.055 |
| CA-4 | 0.0039 ± 0.00032c | 0.0430c | 0.0019 ± 0.0005c |
| Cell Line | Compound 21l c | Compound 19e d | Cell Line | Compound 21l c | Compound 19e d |
|---|---|---|---|---|---|
| GI50 (μM) b | GI50 (μM) b | GI50 (μM) b | GI50 (μM) b | ||
| Leukemia | Melanoma | ||||
| CCRF-CEM | 0.289 | 0.402 | LOX IMV1 | 0.523 | 0.523 |
| HL-60 (TB) | 0.229 | 0.261 | MALME-3M | Nd e | Nd e |
| K-562 | 0.225 | 0.427 | M14 | 0.180 | 0.374 |
| MOLT-4 | 0.565 | 0.510 | MDA-MB-435 | 0.119 | 0.181 |
| RPMI-8226 | 0.385 | 0.452 | SK-MEL-2 | 0.324 | 0.361 |
| SR | 0.182 | 0.376 | UACC-62 | 0.668 | 0.550 |
| Non-Small Cell Lung Cancer | SK-MEL-28 | Nd e | Nd e | ||
| A549/ATCC | 0.836 | 0.969 | SK-MEL-5 | 0.367 | 0.440 |
| HOP-62 | 0.516 | 0.514 | UACC-257 | 1.44 | >100 |
| HOP-92 | 28.6 | 9.69 | Ovarian Cancer | ||
| EKVX | 0.702 | 2.24 | IGROV1 | 0.862 | 0.959 |
| NCI-H226 | 0.640 | 1.43 | OVCAR-3 | 0.264 | 0.323 |
| NCI-H23 | 0.882 | 0.977 | OVCAR-4 | 3.44 | 3.02 |
| NCI-H332M | 0.789 | 0.696 | OVCAR-5 | 0.648 | 1.99 |
| NCI-H460 | 0.377 | 0.414 | OVCAR-8 | 0.450 | 1.14 |
| NCI-H552 | 0.247 | 0.395 | NCI/ADR-RES | 1.92 | 1.18 |
| Colon Cancer | SK-OV-3 | 0.465 | 0.402 | ||
| COLO 205 | 0.361 | 0.332 | Renal Cancer | ||
| HCT-2998 | 1.72 | 1.35 | 786-0 | 0.369 | 0.475 |
| HCT-116 | 0.249 | 0.448 | A498 | 0.304 | 0.324 |
| HCT-15 | 0.264 | 0.438 | ACHN | 0.693 | 0.653 |
| HT29 | 0.312 | 0.330 | CAKI-1 | 0.681 | 0.567 |
| KM12 | 0.389 | 0.418 | RXF 393 | Nde | 0.321 |
| SW-620 | 0.387 | 0.431 | SN12C | 0.910 | 0.853 |
| CNS Cancer | TK-10 | Nde | >100 | ||
| SF-268 | 0.731 | 0.469 | UO-31 | 0.883 | 0.903 |
| SF295 | 0.307 | 0.325 | Breast Cancer | ||
| SF539 | 0.217 | 0.300 | MCF-7 | 0.339 | 0.347 |
| SNB-19 | 0.532 | 0.578 | MDA-MB-231/ATCC | 0.664 | 1.50 |
| SNB-75 | 0.192 | Nde | HS 578T | 0.414 | 0.548 |
| AC | 0.391 | 0.446 | BT-549 | 0.306 | 0.618 |
| Prostate cancer | T-47D | Nde | Nde | ||
| PC-3 | 0.384 | 0.431 | MDA-MB-468 | 0.316 | 0.371 |
| DU-145 | 0.407 | 0.530 | |||
| MG-MID (µM) f | 0.234 | 0.243 |
| Rank | 19e | r | 21l | r |
|---|---|---|---|---|
| Based on GI50 mean graph | Based on GI50 mean graph | |||
| 1 | Vinblastine sulphate, hiConc:-7.6 | 0.586 | Paclitaxel (Taxol) hiConc:-6.0 | 0.587 |
| 2 | S-Trityl-L-cysteine | 0.575 | S-Trityl-L-cysteine | 0.58 |
| 3 | Maytansine | 0.547 | Paclitaxel (Taxol) hiConc:-4.0 | 0.544 |
| 4 | Rhizoxin | 0.544 | Maytansine | 0.53 |
| 5 | Vinblastine sulphate, hiConc:-5.6 | 0.509 | Paclitaxel (Taxol) hiConc:-4.6 | 0.518 |
| Based on TGI mean graph | Based on TGI mean graph | |||
| 1 | Vinblastine sulfate | 0.737 | Maytansine hiConc: -9.0 | 0.775 |
| 2 | Rhizoxin | 0.726 | Vinblastine sulfate | 0.765 |
| 3 | Paclitaxel (Taxol) | 0.704 | Rhizoxin | 0.748 |
| 4 | Maytansine hiConc:-9.0 | 0.696 | Paclitaxel (Taxol) | 0.703 |
| 5 | Maytansine hiConc:-4.0 | 0.689 | Maytansine hiConc:-9.0 | 0.700 |
| Compound | % Inhibition at 20 µg/mLa | IC50 (µM) a,b | ||
|---|---|---|---|---|
| CYP19 | CYP1A1 | CYP19 | CYP1A1 | |
| 19e | 85.34 | 18.08 | 29.62 | >53.85 |
| 21l | 74.73 | 18.44 | >53.99 | >53.99 |
| 24 | 0.01 | 12.81 | >53.85 | >53.85 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ana, G.; Kelly, P.M.; Malebari, A.M.; Noorani, S.; Nathwani, S.M.; Twamley, B.; Fayne, D.; O’Boyle, N.M.; Zisterer, D.M.; Pimentel, E.F.; et al. Synthesis and Biological Evaluation of 1-(Diarylmethyl)-1H-1,2,4-triazoles and 1-(Diarylmethyl)-1H-imidazoles as a Novel Class of Anti-Mitotic Agent for Activity in Breast Cancer. Pharmaceuticals 2021, 14, 169. https://doi.org/10.3390/ph14020169
Ana G, Kelly PM, Malebari AM, Noorani S, Nathwani SM, Twamley B, Fayne D, O’Boyle NM, Zisterer DM, Pimentel EF, et al. Synthesis and Biological Evaluation of 1-(Diarylmethyl)-1H-1,2,4-triazoles and 1-(Diarylmethyl)-1H-imidazoles as a Novel Class of Anti-Mitotic Agent for Activity in Breast Cancer. Pharmaceuticals. 2021; 14(2):169. https://doi.org/10.3390/ph14020169
Chicago/Turabian StyleAna, Gloria, Patrick M. Kelly, Azizah M. Malebari, Sara Noorani, Seema M. Nathwani, Brendan Twamley, Darren Fayne, Niamh M. O’Boyle, Daniela M. Zisterer, Elisangela Flavia Pimentel, and et al. 2021. "Synthesis and Biological Evaluation of 1-(Diarylmethyl)-1H-1,2,4-triazoles and 1-(Diarylmethyl)-1H-imidazoles as a Novel Class of Anti-Mitotic Agent for Activity in Breast Cancer" Pharmaceuticals 14, no. 2: 169. https://doi.org/10.3390/ph14020169
APA StyleAna, G., Kelly, P. M., Malebari, A. M., Noorani, S., Nathwani, S. M., Twamley, B., Fayne, D., O’Boyle, N. M., Zisterer, D. M., Pimentel, E. F., Endringer, D. C., & Meegan, M. J. (2021). Synthesis and Biological Evaluation of 1-(Diarylmethyl)-1H-1,2,4-triazoles and 1-(Diarylmethyl)-1H-imidazoles as a Novel Class of Anti-Mitotic Agent for Activity in Breast Cancer. Pharmaceuticals, 14(2), 169. https://doi.org/10.3390/ph14020169