
Phenotypical Characterization of the Nuclear Egress of Recombinant Cytomegaloviruses Reveals Defective Replication upon ORF-UL50 Deletion but Not pUL50 Phosphosite Mutation


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Virus Infection and Transient Transfection
2.2. Plasmids
2.3. Generation of Recombinant HCMV
2.4. Antibodies
2.5. CoIP, Phosphate Affinity (Phos-Tag) SDS-PAGE and Western Blot (Wb) Analyses
2.6. Indirect Immunofluorescence Assay and Confocal Laser-Scanning Microscopy
2.7. Cytomegalovirus Multistep Replication Curve Analysis
2.8. Transmission Electron Microscopy (TEM)
3. Results
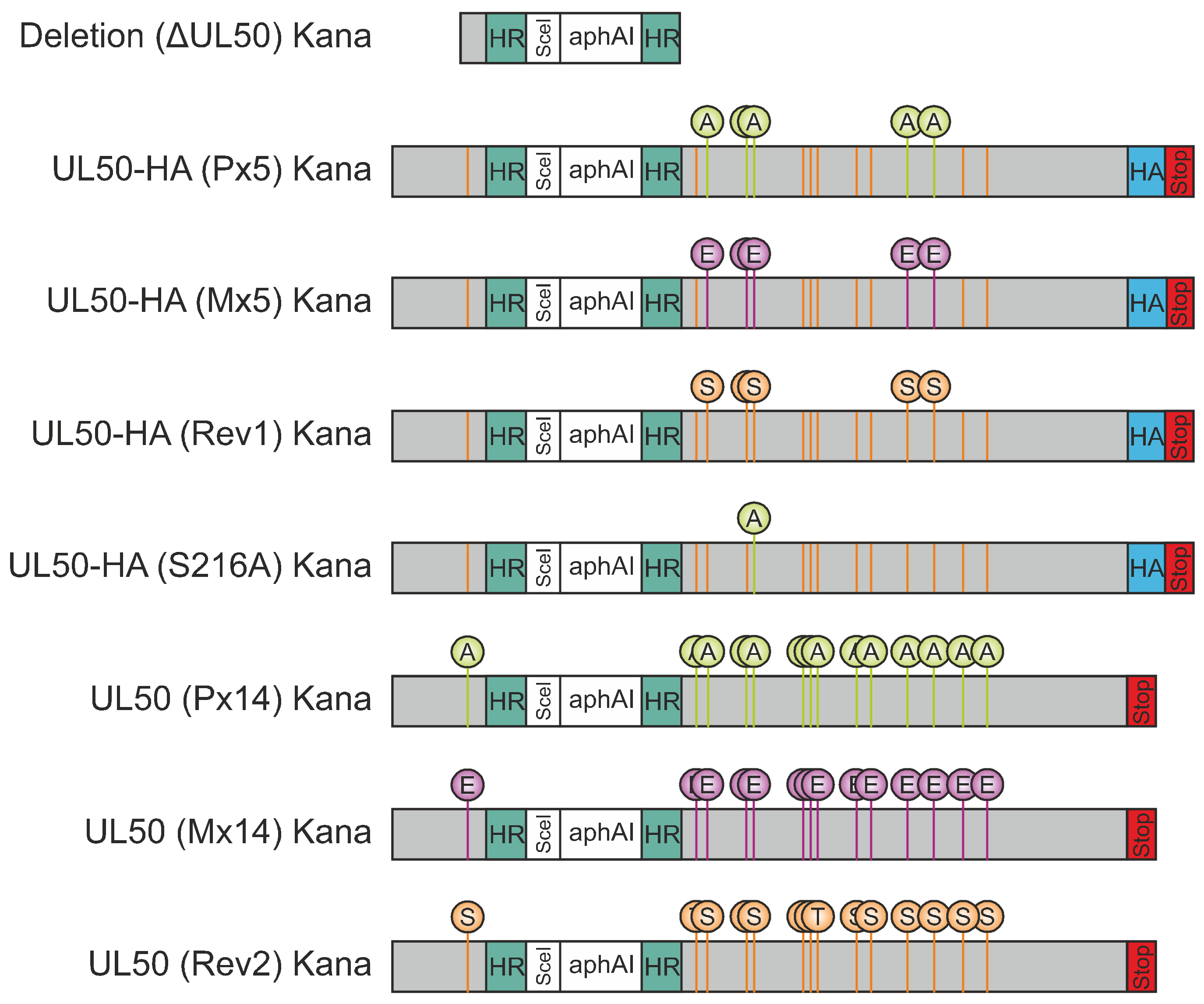
3.1. Construction of Expression Plasmids and Recombinant Viruses Encoding Mutant Versions of ORF-UL50
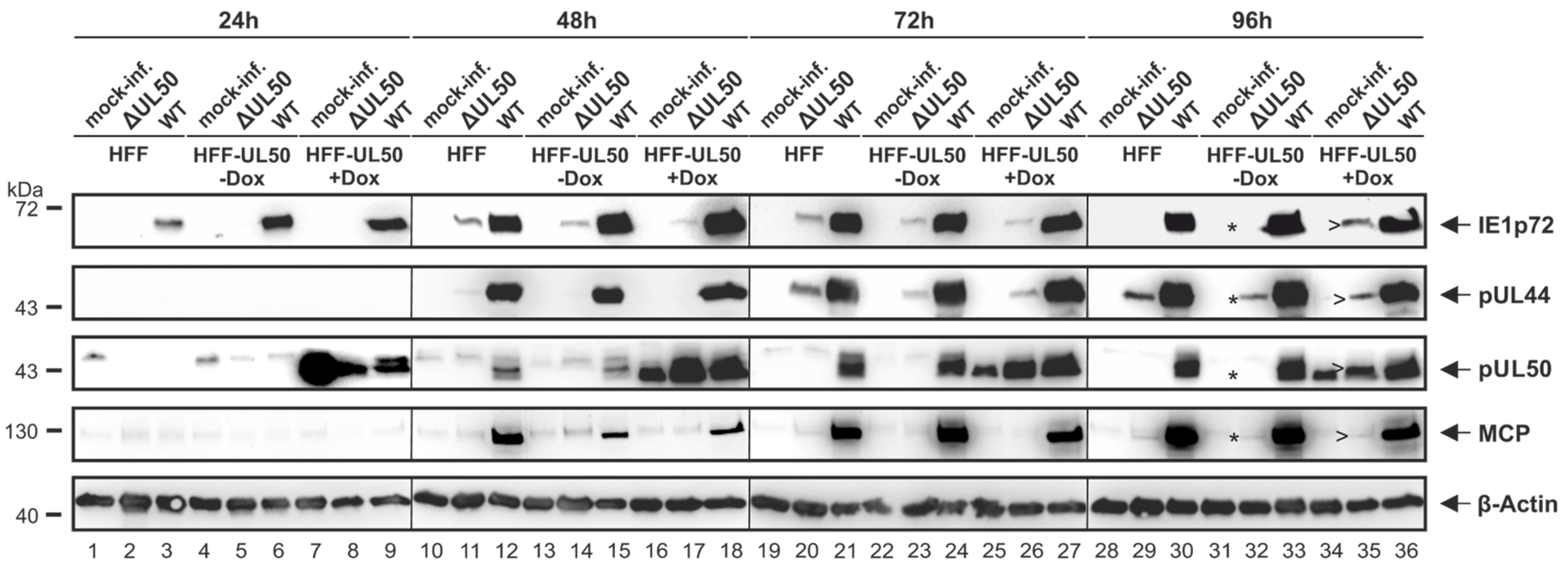
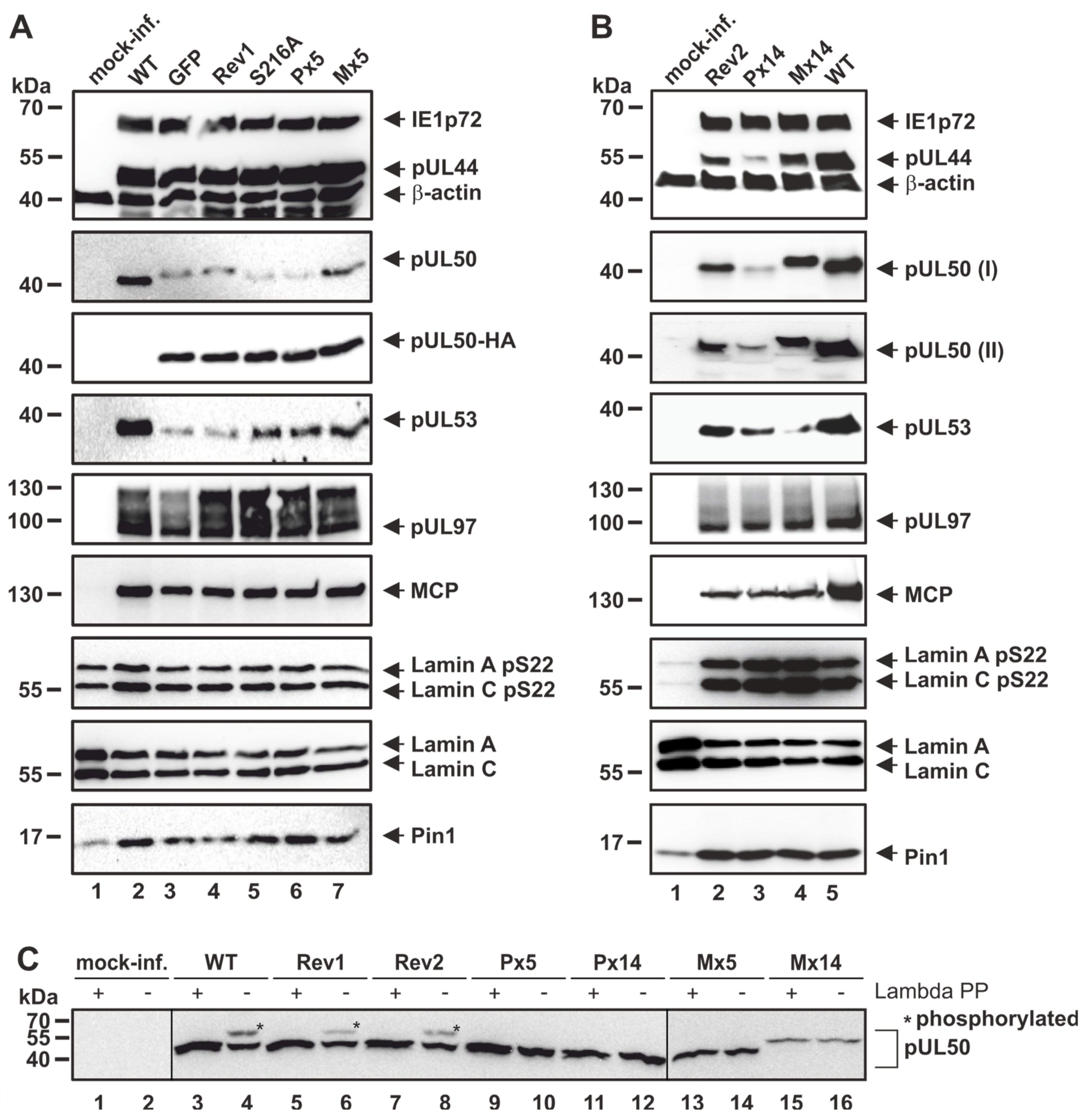
3.2. Patterns of Expression and Protein-Protein Interaction of ORF-UL50 Deletion and Phosphosite Mutants
3.3. Confocal Imaging of Mutant pUL50 Expressed by Plasmids or Recombinant HCMVs: Localization of Viral Proteins and Nuclear Lamina Components
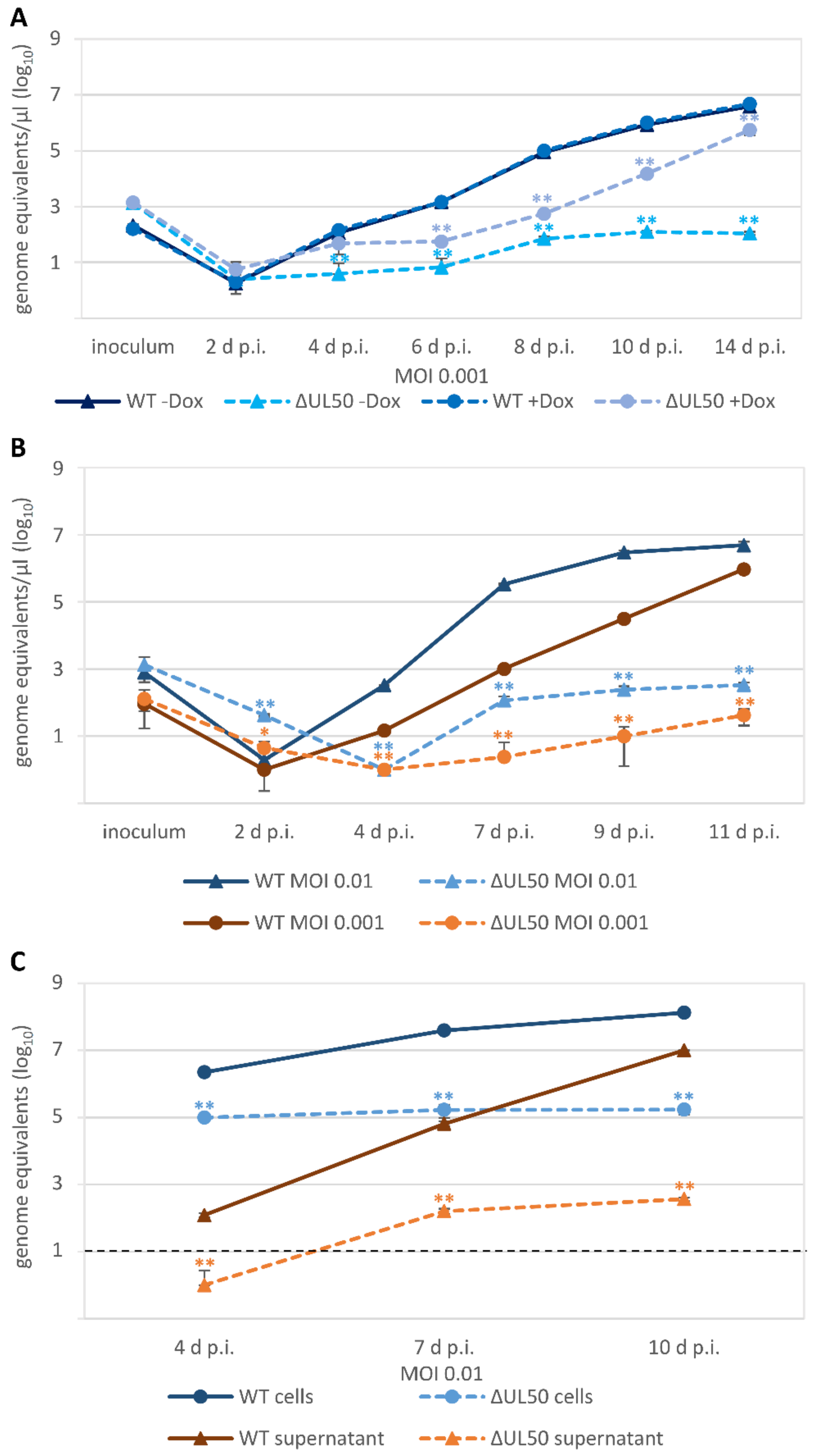
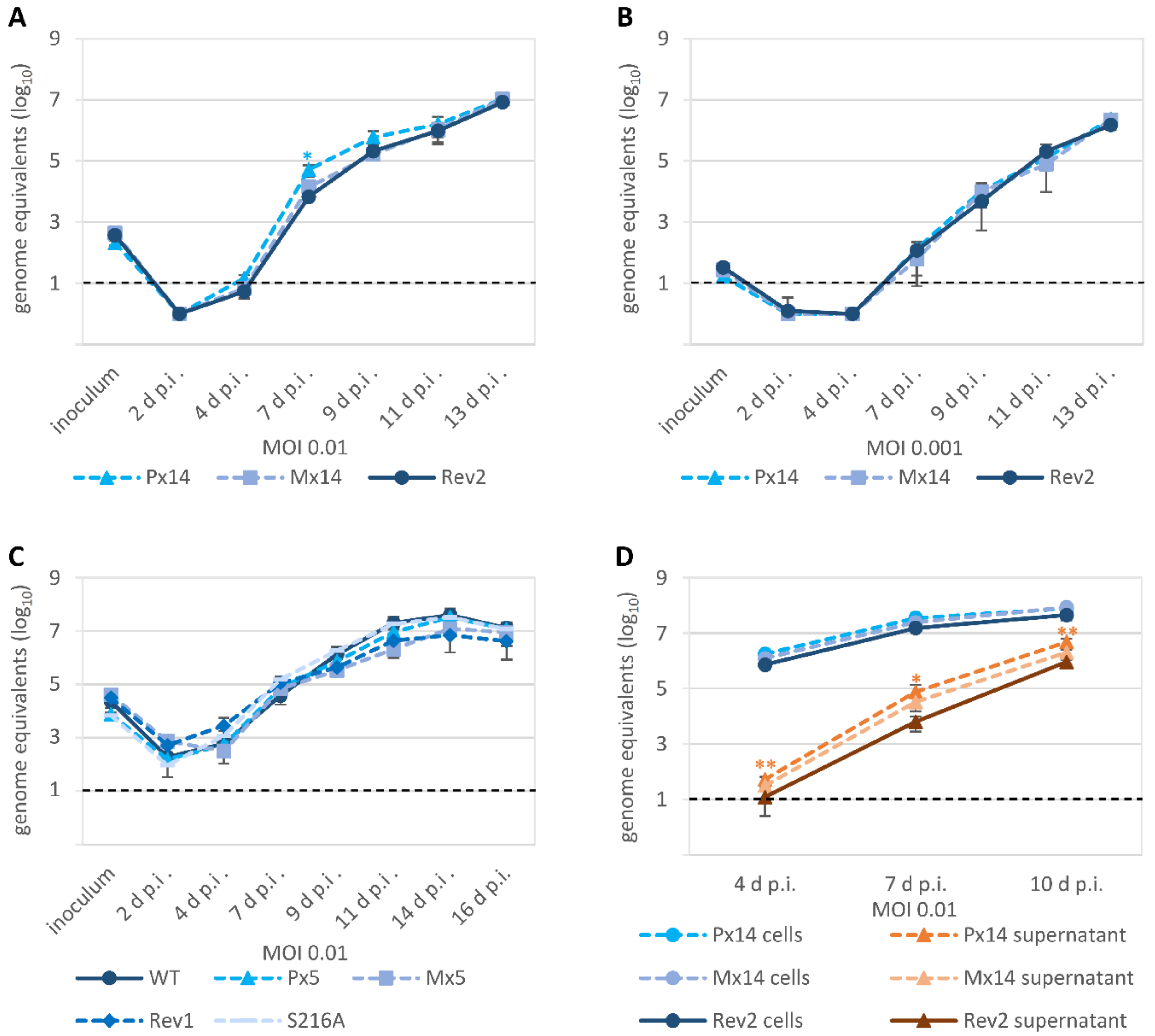
3.4. Characteristic Differences in the Replication Kinetics between Recombinant HCMVs Carrying ORF-UL50 Deletion or pUL50 Phosphosite Mutations
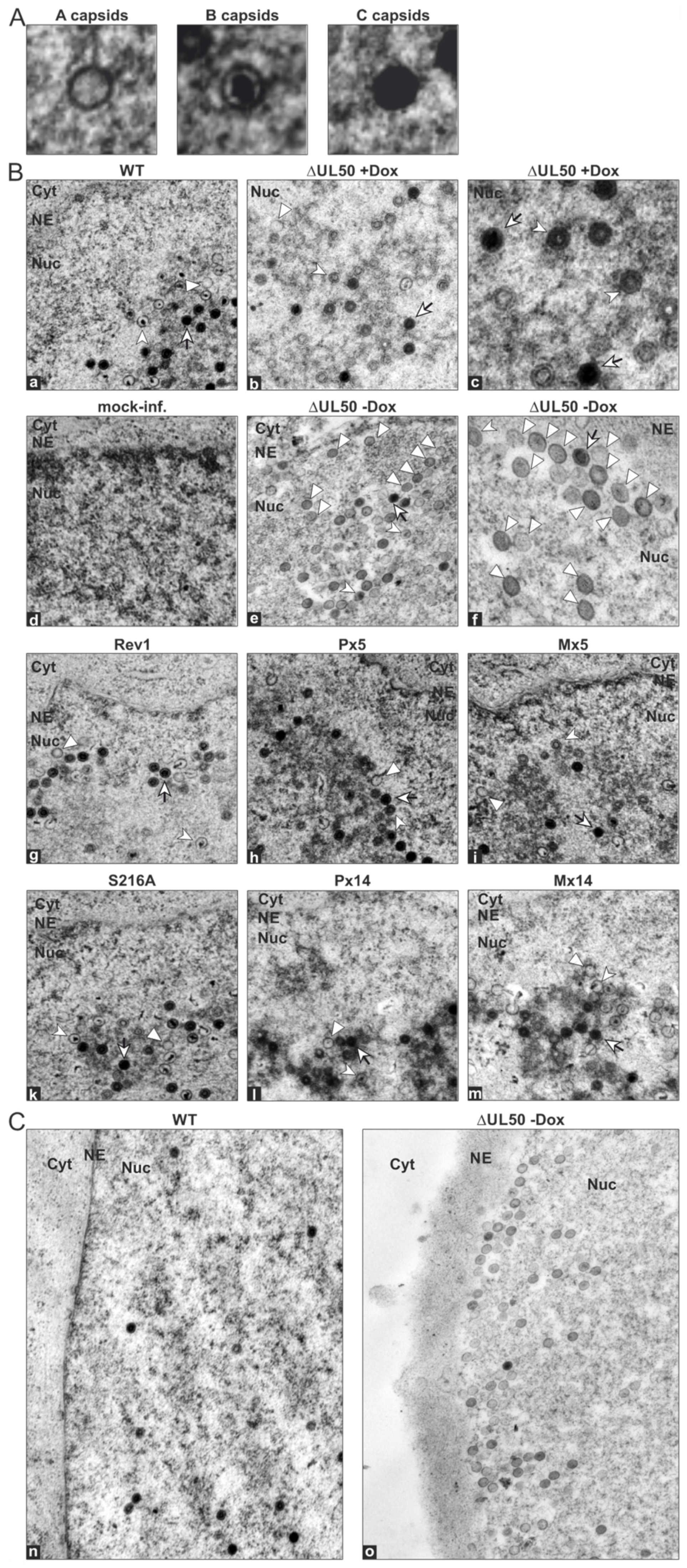
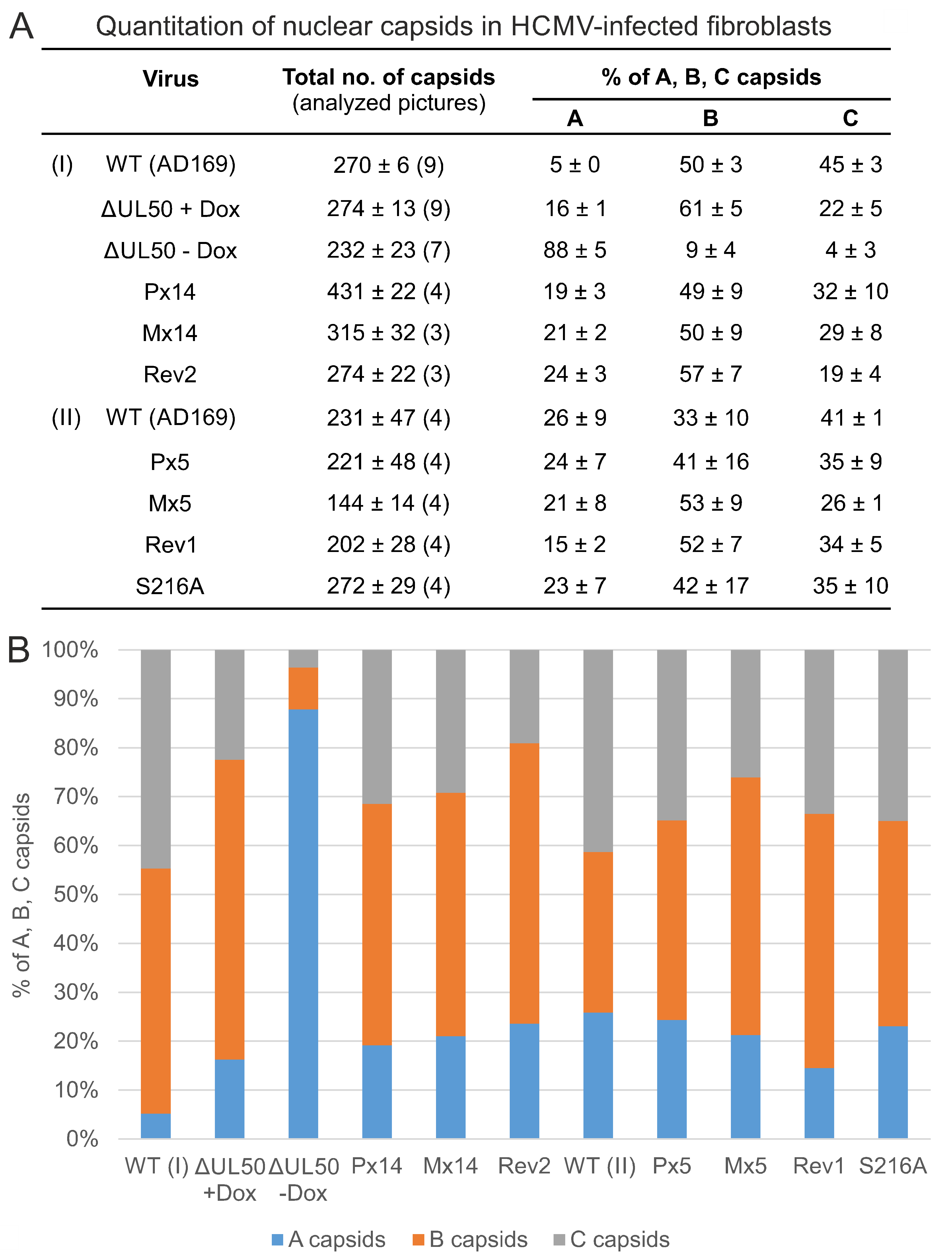
3.5. Electron Microscopic Analysis of HFFs Infected with the Recombinant HCMVs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marschall, M.; Muller, Y.A.; Diewald, B.; Sticht, H.; Milbradt, J. The human cytomegalovirus nuclear egress complex unites multiple functions: Recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Rev. Med. Virol. 2017, 27. [Google Scholar] [CrossRef] [PubMed]
- Hage, S.; Sonntag, E.; Borst, E.M.; Tannig, P.; Seyler, L.; Bauerle, T.; Bailer, S.M.; Lee, C.P.; Muller, R.; Wangen, C.; et al. Patterns of Autologous and Nonautologous Interactions Between Core Nuclear Egress Complex (NEC) Proteins of alpha-, beta- and gamma-Herpesviruses. Viruses 2020, 12, 303. [Google Scholar] [CrossRef] [PubMed]
- Marschall, M.; Häge, S.; Conrad, M.; Alkhashrom, S.; Kicuntod, J.; Schweininger, J.; Kriegel, M.; Lösing, J.; Tillmanns, J.; Neipel, F.; et al. Nuclear Egress Complexes of HCMV and Other Herpesviruses: Solving the Puzzle of Sequence Coevolution, Conserved Structures and Subfamily-Spanning Binding Properties. Viruses 2020, 12, 683. [Google Scholar] [CrossRef] [PubMed]
- Draganova, E.B.; Thorsen, M.K.; Heldwein, E.E. Nuclear Egress. Curr. Issues Mol. Biol. 2020, 41, 125–170. [Google Scholar] [CrossRef]
- Lye, M.F.; Wilkie, A.R.; Filman, D.J.; Hogle, J.M.; Coen, D.M. Getting to and through the inner nuclear membrane during herpesvirus nuclear egress. Curr. Opin. Cell Biol. 2017, 46, 9–16. [Google Scholar] [CrossRef]
- Milbradt, J.; Sonntag, E.; Wagner, S.; Strojan, H.; Wangen, C.; Lenac Rovis, T.; Lisnic, B.; Jonjic, S.; Sticht, H.; Britt, W.J.; et al. Human Cytomegalovirus Nuclear Capsids Associate with the Core Nuclear Egress Complex and the Viral Protein Kinase pUL97. Viruses 2018, 10, 35. [Google Scholar] [CrossRef]
- Sharma, M.; Bender, B.J.; Kamil, J.P.; Lye, M.F.; Pesola, J.M.; Reim, N.I.; Hogle, J.M.; Coen, D.M. Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. J. Virol. 2015, 89, 523–534. [Google Scholar] [CrossRef]
- Sonntag, E.; Milbradt, J.; Svrlanska, A.; Strojan, H.; Hage, S.; Kraut, A.; Hesse, A.M.; Amin, B.; Sonnewald, U.; Coute, Y.; et al. Protein kinases responsible for the phosphorylation of the nuclear egress core complex of human cytomegalovirus. J. Gen. Virol. 2017, 98, 2569–2581. [Google Scholar] [CrossRef]
- Mettenleiter, T.C.; Muller, F.; Granzow, H.; Klupp, B.G. The way out: What we know and do not know about herpesvirus nuclear egress. Cell Microbiol. 2013, 15, 170–178. [Google Scholar] [CrossRef]
- Bigalke, J.M.; Heldwein, E.E. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J. 2015, 34, 2921–2936. [Google Scholar] [CrossRef]
- Hagen, C.; Dent, K.C.; Zeev-Ben-Mordehai, T.; Grange, M.; Bosse, J.B.; Whittle, C.; Klupp, B.G.; Siebert, C.A.; Vasishtan, D.; Bauerlein, F.J.; et al. Structural Basis of Vesicle Formation at the Inner Nuclear Membrane. Cell 2015, 163, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Leigh, K.E.; Sharma, M.; Mansueto, M.S.; Boeszoermenyi, A.; Filman, D.J.; Hogle, J.M.; Wagner, G.; Coen, D.M.; Arthanari, H. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc. Natl. Acad. Sci. USA 2015, 112, 9010–9015. [Google Scholar] [CrossRef] [PubMed]
- Lye, M.F.; Sharma, M.; El Omari, K.; Filman, D.J.; Schuermann, J.P.; Hogle, J.M.; Coen, D.M. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J. 2015, 34, 2937–2952. [Google Scholar] [CrossRef] [PubMed]
- Walzer, S.A.; Egerer-Sieber, C.; Sticht, H.; Sevvana, M.; Hohl, K.; Milbradt, J.; Muller, Y.A.; Marschall, M. Crystal Structure of the Human Cytomegalovirus pUL50-pUL53 Core Nuclear Egress Complex Provides Insight into a Unique Assembly Scaffold for Virus-Host Protein Interactions. J. Biol. Chem. 2015, 290, 27452–27458. [Google Scholar] [CrossRef]
- Zeev-Ben-Mordehai, T.; Weberruss, M.; Lorenz, M.; Cheleski, J.; Hellberg, T.; Whittle, C.; El Omari, K.; Vasishtan, D.; Dent, K.C.; Harlos, K.; et al. Crystal Structure of the Herpesvirus Nuclear Egress Complex Provides Insights into Inner Nuclear Membrane Remodeling. Cell Rep. 2015, 13, 2645–2652. [Google Scholar] [CrossRef]
- Muller, Y.A.; Hage, S.; Alkhashrom, S.; Hollriegl, T.; Weigert, S.; Dolles, S.; Hof, K.; Walzer, S.A.; Egerer-Sieber, C.; Conrad, M.; et al. High-resolution crystal structures of two prototypical beta- and gamma-herpesviral nuclear egress complexes unravel the determinants of subfamily specificity. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Schnee, M.; Ruzsics, Z.; Bubeck, A.; Koszinowski, U.H. Common and specific properties of herpesvirus UL34/UL31 protein family members revealed by protein complementation assay. J. Virol. 2006, 80, 11658–11666. [Google Scholar] [CrossRef]
- Sonntag, E.; Hamilton, S.T.; Bahsi, H.; Wagner, S.; Jonjic, S.; Rawlinson, W.D.; Marschall, M.; Milbradt, J. Cytomegalovirus pUL50 is the multi-interacting determinant of the core nuclear egress complex (NEC) that recruits cellular accessory NEC components. J. Gen. Virol. 2016, 97, 1676–1685. [Google Scholar] [CrossRef]
- Häge, S.; Horsch, D.; Stilp, A.C.; Kicuntod, J.; Müller, R.; Hamilton, S.T.; Egilmezer, E.; Rawlinson, W.D.; Stamminger, T.; Sonntag, E.; et al. A quantitative nuclear egress assay to investigate the nucleocytoplasmic capsid release of human cytomegalovirus. J. Virol. Methods 2020, 283, 113909. [Google Scholar] [CrossRef]
- Klenovsek, K.; Weisel, F.; Schneider, A.; Appelt, U.; Jonjic, S.; Messerle, M.; Bradel-Tretheway, B.; Winkler, T.H.; Mach, M. Protection from CMV infection in immunodeficient hosts by adoptive transfer of memory B cells. Blood 2007, 110, 3472–3479. [Google Scholar] [CrossRef]
- Lorz, K.; Hofmann, H.; Berndt, A.; Tavalai, N.; Mueller, R.; Schlotzer-Schrehardt, U.; Stamminger, T. Deletion of open reading frame UL26 from the human cytomegalovirus genome results in reduced viral growth, which involves impaired stability of viral particles. J. Virol. 2006, 80, 5423–5434. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [PubMed]
- Milbradt, J.; Auerochs, S.; Marschall, M. Cytomegaloviral proteins pUL50 and pUL53 are associated with the nuclear lamina and interact with cellular protein kinase C. J. Gen. Virol. 2007, 88, 2642–2650. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Auerochs, S.; Sticht, H.; Marschall, M. Cytomegaloviral proteins that associate with the nuclear lamina: Components of a postulated nuclear egress complex. J. Gen. Virol. 2009, 90, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Marschall, M.; Marzi, A.; aus dem Siepen, P.; Jochmann, R.; Kalmer, M.; Auerochs, S.; Lischka, P.; Leis, M.; Stamminger, T. Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J. Biol. Chem. 2005, 280, 33357–33367. [Google Scholar] [CrossRef]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. 2010, 634, 421–430. [Google Scholar] [CrossRef]
- Schmeiser, C.; Borst, E.; Sticht, H.; Marschall, M.; Milbradt, J. The cytomegalovirus egress proteins pUL50 and pUL53 are translocated to the nuclear envelope through two distinct modes of nuclear import. J. Gen. Virol. 2013, 94, 2056–2069. [Google Scholar] [CrossRef]
- Webel, R.; Milbradt, J.; Auerochs, S.; Schregel, V.; Held, C.; Nöbauer, K.; Razzazi-Fazeli, E.; Jardin, C.; Wittenberg, T.; Sticht, H.; et al. Two isoforms of the protein kinase pUL97 of human cytomegalovirus are differentially regulated in their nuclear translocation. J. Gen. Virol. 2011, 92, 638–649. [Google Scholar] [CrossRef]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Takiyama, K.; Koike, T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell Proteom. 2006, 5, 749–757. [Google Scholar] [CrossRef]
- Sonntag, E.; Hahn, F.; Bertzbach, L.D.; Seyler, L.; Wangen, C.; Muller, R.; Tannig, P.; Grau, B.; Baumann, M.; Zent, E.; et al. In vivo proof-of-concept for two experimental antiviral drugs, both directed to cellular targets, using a murine cytomegalovirus model. Antivir. Res 2019, 161, 63–69. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 2012, 84, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Hutterer, C.; Bahsi, H.; Wagner, S.; Sonntag, E.; Horn, A.H.; Kaufer, B.B.; Mori, Y.; Sticht, H.; Fossen, T.; et al. The Prolyl Isomerase Pin1 Promotes the Herpesvirus-Induced Phosphorylation-Dependent Disassembly of the Nuclear Lamina Required for Nucleocytoplasmic Egress. PLoS Pathog. 2016, 12, e1005825. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Webel, R.; Auerochs, S.; Sticht, H.; Marschall, M. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 2010, 285, 13979–13989. [Google Scholar] [CrossRef] [PubMed]
- Schütz, M.; Thomas, M.; Wangen, C.; Wagner, S.; Rauschert, L.; Errerd, T.; Kießling, M.; Sticht, H.; Milbradt, J.; Marschall, M. The peptidyl-prolyl cis/trans isomerase Pin1 interacts with three early regulatory proteins of human cytomegalovirus. Virus Res. 2020, 285, 198023. [Google Scholar] [CrossRef]
- Krosky, P.M.; Baek, M.C.; Coen, D.M. The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J. Virol. 2003, 77, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, J.M.; Heldwein, E.E. Have NEC Coat, Will Travel: Structural Basis of Membrane Budding During Nuclear Egress in Herpesviruses. Adv. Virus Res. 2017, 97, 107–141. [Google Scholar] [CrossRef]
- Bailer, S.M. Venture from the Interior-Herpesvirus pUL31 Escorts Capsids from Nucleoplasmic Replication Compartments to Sites of Primary Envelopment at the Inner Nuclear Membrane. Cells 2017, 6, 46. [Google Scholar] [CrossRef]
- Hellberg, T.; Passvogel, L.; Schulz, K.S.; Klupp, B.G.; Mettenleiter, T.C. Nuclear Egress of Herpesviruses: The Prototypic Vesicular Nucleocytoplasmic Transport. Adv. Virus Res. 2016, 94, 81–140. [Google Scholar] [CrossRef]
- Mettenleiter, T.C. Breaching the Barrier-The Nuclear Envelope in Virus Infection. J. Mol. Biol. 2016, 428, 1949–1961. [Google Scholar] [CrossRef]
- Lee, C.P.; Chen, M.R. Escape of herpesviruses from the nucleus. Rev. Med. Virol. 2010, 20, 214–230. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef]
- Marschall, M.; Feichtinger, S.; Milbradt, J. Regulatory roles of protein kinases in cytomegalovirus replication. Adv. Virus Res. 2011, 80, 69–101. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, A.; Perlman, D.H.; Shenk, T.; Terry, L.J. Human cytomegalovirus pUL97 kinase induces global changes in the infected cell phosphoproteome. Proteomics 2015, 15, 2006–2022. [Google Scholar] [CrossRef] [PubMed]
- Steingruber, M.; Marschall, M. The Cytomegalovirus Protein Kinase pUL97: Host Interactions, Regulatory Mechanisms and Antiviral Drug Targeting. Microorganisms 2020, 8, 515. [Google Scholar] [CrossRef] [PubMed]
- Keating, J.A.; Striker, R. Phosphorylation events during viral infections provide potential therapeutic targets. Rev. Med. Virol. 2012, 22, 166–181. [Google Scholar] [CrossRef]
- Keck, F.; Ataey, P.; Amaya, M.; Bailey, C.; Narayanan, A. Phosphorylation of Single Stranded RNA Virus Proteins and Potential for Novel Therapeutic Strategies. Viruses 2015, 7, 5257–5273. [Google Scholar] [CrossRef]
- Radestock, B.; Morales, I.; Rahman, S.A.; Radau, S.; Glass, B.; Zahedi, R.P.; Muller, B.; Krausslich, H.G. Comprehensive mutational analysis reveals p6Gag phosphorylation to be dispensable for HIV-1 morphogenesis and replication. J. Virol. 2013, 87, 724–734. [Google Scholar] [CrossRef]
- Yu, M.; Summers, J. Multiple functions of capsid protein phosphorylation in duck hepatitis B virus replication. J. Virol. 1994, 68, 4341–4348. [Google Scholar] [CrossRef]
- Zhang, K.; Afroz, S.; Brownlie, R.; Snider, M.; van Drunen Littel-van den Hurk, S. Regulation and function of phosphorylation on VP8, the major tegument protein of bovine herpesvirus 1. J. Virol. 2015, 89, 4598–4611. [Google Scholar] [CrossRef]
- Mou, F.; Wills, E.; Baines, J.D. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J. Virol. 2009, 83, 5181–5191. [Google Scholar] [CrossRef] [PubMed]
- Klupp, B.G.; Hellberg, T.; Rönfeldt, S.; Franzke, K.; Fuchs, W.; Mettenleiter, T.C. Function of the Nonconserved N-Terminal Domain of Pseudorabies Virus pUL31 in Nuclear Egress. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Couté, Y.; Kraut, A.; Zimmermann, C.; Büscher, N.; Hesse, A.M.; Bruley, C.; De Andrea, M.; Wangen, C.; Hahn, F.; Marschall, M.; et al. Mass Spectrometry-Based Characterization of the Virion Proteome, Phosphoproteome, and Associated Kinase Activity of Human Cytomegalovirus. Microorganisms 2020, 8, 820. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Kim, Y.J.; Kim, Y.E.; Han, T.H.; Milbradt, J.; Marschall, M.; Ahn, J.H. Transmembrane Protein pUL50 of Human Cytomegalovirus Inhibits ISGylation by Downregulating UBE1L. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Lee, M.K.; Hyeon, S.; Ahn, J.H. The Human Cytomegalovirus Transmembrane Protein pUL50 Induces Loss of VCP/p97 and Is Regulated by a Small Isoform of pUL50. J. Virol. 2020, 94. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Häge, S.; Sonntag, E.; Svrlanska, A.; Borst, E.M.; Stilp, A.-C.; Horsch, D.; Müller, R.; Kropff, B.; Milbradt, J.; Stamminger, T.; et al. Phenotypical Characterization of the Nuclear Egress of Recombinant Cytomegaloviruses Reveals Defective Replication upon ORF-UL50 Deletion but Not pUL50 Phosphosite Mutation. Viruses 2021, 13, 165. https://doi.org/10.3390/v13020165
Häge S, Sonntag E, Svrlanska A, Borst EM, Stilp A-C, Horsch D, Müller R, Kropff B, Milbradt J, Stamminger T, et al. Phenotypical Characterization of the Nuclear Egress of Recombinant Cytomegaloviruses Reveals Defective Replication upon ORF-UL50 Deletion but Not pUL50 Phosphosite Mutation. Viruses. 2021; 13(2):165. https://doi.org/10.3390/v13020165
Chicago/Turabian StyleHäge, Sigrun, Eric Sonntag, Adriana Svrlanska, Eva Maria Borst, Anne-Charlotte Stilp, Deborah Horsch, Regina Müller, Barbara Kropff, Jens Milbradt, Thomas Stamminger, and et al. 2021. "Phenotypical Characterization of the Nuclear Egress of Recombinant Cytomegaloviruses Reveals Defective Replication upon ORF-UL50 Deletion but Not pUL50 Phosphosite Mutation" Viruses 13, no. 2: 165. https://doi.org/10.3390/v13020165
APA StyleHäge, S., Sonntag, E., Svrlanska, A., Borst, E. M., Stilp, A.-C., Horsch, D., Müller, R., Kropff, B., Milbradt, J., Stamminger, T., Schlötzer-Schrehardt, U., & Marschall, M. (2021). Phenotypical Characterization of the Nuclear Egress of Recombinant Cytomegaloviruses Reveals Defective Replication upon ORF-UL50 Deletion but Not pUL50 Phosphosite Mutation. Viruses, 13(2), 165. https://doi.org/10.3390/v13020165